
Abstract
In the present study, antagonistic activity of bacterial strain BS-Z15, was evaluated against Verticillium dahlia. The fermented broth of BS-Z15 inhibited the growth of Verticillium dahliae. The genome of strain BS-Z15 had a total size of 4,068,702 base pairs and contained 4318 genes, of which 4196 are coding sequences and 122 are non-coding RNA. Among these genes, nine genomic islands, 86 tRNAs, 13 sRNAs, and one prophage was determined. With the help of annotation databases, most unigene functions were identified. At the same time, genomic comparison between BS-Z15 and 12 Bacillus members showed that the genes of BS-Z15 were closely related to the Bacillus group, and were conserved between the two groups, including most of the genes associated with fungal antagonism. BS-Z15 contains genes involved in a variety of antagonistic mechanisms, including genes encoding or synthesizing mycosubtilin, chitinases (but not CHIA and CHIB), glycoside hydrolases, iron nutrients, and antibiosis. However, it only contained the complete mycosubtilin- and bacilibactin-related operators in the reported main antifungal gene cluster of B. subtilis. Mycosubtilin and bacilibactin may be the main active antifungal substance. Besides, some genes could encode products related to biofilm production, which may be related to the colonization ability of the strain in plant rhizospheres. The complete genome of B. subtilis BS-Z15 provided new insights into the potential metabolites it produces related to its biocontrol activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacillus species are well known to produce a variety of bioactive compounds with the potential for biotechnology applications, including antibiotics and enzymes that are widely used in environmental and biomedical applications (Banat et al. 2010). They are also used as biological control agents due to their ability to produce antifungal substances (Compant et al. 2005). Some Bacillus spp. have been identified as biosurfactant producers, including B. licheniformis, B. cereus, B. subtilis, B. thuringiensis, and B. pumilus (Joshi et al. 2012; Nakano et al. 1992; Pereira et al. 2013; Płaza et al. 2015).
The disease control activity exerted by members of Bacillus spp. is complex, and involves several mechanisms, including competition for nutrients (including iron), space, antibiosis, mycoparasitism, and biofilm formation (Pal et al. 2006). However, different Bacillus spp. contain different metabolic substances, different mechanisms, and exert different effects. It is known that B. cereus and B. thuringiensis are effective in fungal biocontrol through the production of chitinases and siderophores as antagonistic mechanisms (Hong et al. 2015; Hollensteiner et al. 2016), or the production of parasporal crystal proteins that can act as an insecticide. B. subtilis secretes various bioactive compounds that have potent commercial importance, such as lipopeptides (LPs) (Jacques et al. 2010). Most LPs of Bacillus described to date are classified into three main families: surfactins, iturins, and fengycins (Mongkolthanaruk 2012).
In previous studies, B. subtilis BS-Z15 was isolated from the rhizosphere soil of cotton grown in Xinjiang, which can inhibit cotton Verticillium wilt and thus protect cotton (Zhao et al. 2017). BS-Z15 also had a strong inhibitory effect on many fungi, such as Candida albicans and Aspergillus flavus (Zhao et al. 2018). This has generated interest in the BS-Z15 strain, especially as it is an effective antagonistic activity. In the present study, we explored the effect of BS-Z15 metabolites on the growth and germination of Verticillium dahliae. To further develop BS-Z15 strain and provide a research basis, the whole genome of BS-Z15 was sequenced to explore its antifungal mechanisms. The genome of BS-Z15 was compared with those of 12 related Bacillus spp., and the common and unique genetic components of BS-Z15 were analyzed. Here, we describe some of the genes encoding the biological control activity of the BS-Z15 genome, and investigate the genetic potential to control fungal pathogens and other biocontrol agents among the other strains belonging to this taxonomic group. It is expected that BS-Z15 could be used to control Verticillium Wilt in cotton and fungal diseases of other crops or other pathogenic fungi in humans.
Materials and methods
Microbial strains and culture medium
BS-Z15 was isolated from the rhizosphere of cotton by Zhao et al. 2017. Verticillium dahliae 991 (V. dahliae 991) was provided by Xinjiang Academy of Agricultural Sciences.
Extraction of the crude antifungal substance
The pH of the sterile filtrate of BS-Z15 broth was adjusted to 1.5 using concentrated HCl and stored at 4 °C overnight. The precipitate of 500 mL sterile filtrate was harvested by centrifugation at 4 °C and 12,000 rpm for 30 min, and 50 mL 80% acetone was added to remove insoluble impurities. The supernatant was evaporated under vacuum to remove acetone. The remnant was collected as a crude antifungal substance (Zhao et al. 2018).
Stability of crude antifungal substance
We used the perforated-plate method to evaluate the effects of the crude antifungal substance on V. dahliae mycelial growth. We cultured V. dahliae in a Petri dish at 28 °C. Compare the antimicrobial situation after 3 days and 2 months.
Effect on spore germination and growth of Verticillium dahliae
We added 100 mL liquid Czapek medium containing crude active substance (0 mg/ml, 0.05 mg/ml, 0.2 mg/ml, 0.5 mg/ml, and 1 mg/ml) to 250 mL conical flasks, and added 1 mL V. dahliae spore suspension (~ 107 spores). After culture for 0, 2, 4, 6, 8, 10, 24, 36, or 48 h in darkness at 25 °C with shaking at 180 rpm, the OD600 value was determined. In addition, after 0.5, 1, 2, 6 h, we transferred 100 µL broth to solid Czapek medium and incubated in the dark at 25 °C for 48 h to assess the germination and growth of V. dahliae. Among them, after 0.5 h, we transferred 100 µL broth to solid Czapek medium and incubated in the dark at 25 °C for 2, 3, 4, 5, 6, 7 days to assess the germination and growth of V. dahliae (Zhao et al. 2017).
Genome sequencing and assembly
Genomic DNA was extracted using the CTAB-PEG method (Huang et al. 2018). The harvested DNA was detected by agarose gel electrophoresis and quantified by Qubit. Whole-genome sequencing was performed on the Illumina HiSeq PE150 platform. DNA was A-tailed, ligated to paired-end adaptors and PCR-amplified with a 350 bp insert and a mate-pair library with an insert size of 6 kb were used to construct the gene library at the Beijing Novogene Bioinformatics Technology Co., Ltd. Illumina PCR adapter reads and low-quality reads from the paired-end and mate-pair library were filtered by the quality control step using ripe compling pipeline. All good-quality paired reads were assembled using SOAPdenovo (http://soap.genomics.org.cn/soapdenovo.html) into a number of scaffolds. Next, the filter reads were processed by gap-closing (Li et al. 2010, 2008).
Genome component prediction
Genome component prediction includes the prediction of coding genes, repetitive sequences, non-coding RNA, genome islands, transposons, prophage, and clustered regularly interspaced short palindromic repeat (CRISPR) sequences. We proceeded as follows: (1) For bacteria, we used the GeneMarkS program to retrieve the related coding genes (Besemer et al. 2001). (2) The interspersed repetitive sequences were predicted using RepeatMasker (http://www.repeatmasker.org/). The tandem repeats were analyzed by Tandem Repeats Finder. (3) tRNA genes were predicted by tRNAscan-SE (Benson 1999; Lowe and Eddy 1997). rRNA genes were analyzed by rRNAmmer (Lagesen et al. 2007). Small nuclear RNAs (snRNAs) were predicted by BLAST against the Rfam database (Gardner et al. 2008; Eric et al. 2009). (4) The IslandPath-DIOMB program was used to predict genomics islands, and transposon PSI was used to predict transposons based on the homologous blast method. PHAST was used for prophage prediction (http://phast.wishartlab.com/), and CRISPRFinder was used for CRISPR identification (Ibtissem et al. 2007; William et al. 2003).
Gene function annotation
We used seven databases to predict gene functions; these were Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups of proteins (COG), Non-Redundant Protein Database (NR), and Transporter Classification Database (TCDB) (Ashburner et al. 2000; Kanehisa et al. 2004; Tatusov et al. 2003; Li et al. 2002; Saier et al. 2013). A whole-genome BLAST search (E value < 1e-5, minimal alignment length percentage > 40%) was performed against the databases listed above (Altschul et al. 1990). The signal peptide prediction tool SignalP was used to annotate whether the protein sequence contains a transmembrane structure, and the TMHMM tool was used to annotate whether the protein sequence contains a transmembrane structure. Finally, a protein containing a signal peptide structure and not containing a transmembrane structure was selected as a secreted protein. We used the Pathogen-Host Interactions (PHI) database (Martin et al. 2015) (Pathogen Host Interactions), VFDB (Chen et al. 2012) (Virulence Factors of Pathogenic Bacteria), ARDB (Liu and Pop et al. 2009) (Antibiotic Resistance Genes Database) to perform the above analyses, the genes interacting with the pathogen were analyzed.
Comparative genomic analysis
In this study, the phylogenetic tree of B. subtilis BS-Z15 and the other twelve Bacillus species were constructed using whole genome-wide sequences, and the closest strain was selected using FastANI (v1.32, fragLen selected 1000 bp, other parameters default) (Jiménez et al. 2013) to compare genomic affinities. Orthologous protein prediction was performed using Proteinortho (v6, with default parameters) (Lechner et al. 2011). Among the predicted orthologous gene clusters, 2733 highly conserved single-copy gene clusters were chosen and aligned using MUSCLE (v3.8.31_i86linux64, with default parameters) (Edgar 2004). To remove divergence and ambiguously aligned blocks from the alignment, Gblocks (v0.91b) (Talavera et al. 2007) was employed under the default parameter setting. Trimmed alignments of orthologous sequences were concatenated using a Perl script FASconCAT (v1.02, with default parameters) (Kück et al. 2010), and a maximum likelihood phylogenetic tree was created using the Dayhoff model in TREE-PUZZLE v5.3.rc16 (Schmidt et al. 2002). The tree was visualized using Figtree v1.42 (http://tree.bio.ed.ac.uk/software/figtree). Protein sequences of the other twelve species presented in the comparative analysis were all acquired from the Bacillus Comparative Database (May 2019 (http://www.broadinstitute.org/). Proteomes were classified into proteolytic enzyme families by performing a batch Blast search against the MEROPS protease database (release 9.13) (Rawlings and Morton 2008), and carbohydrate-active enzymes were classified using a HMMER (v3.1b1, with default parameters) scan against the profiles compiled with dbCAN release 4.0 (Yin et al. 2012) based on the CAZy database (Cantarel et al. 2009). Statistical comparisons of carbohydrate-active enzymes between B. subtilis BS-Z15 and the twelve other Bacillus species were made using the Wilcoxon rank-sum test in the R platform.
Results and discussion
Stability of crude antifungal substance and its effect on Verticillium dahliae germination
The amount of germination spores expressed by light absorption at 600 nm. After culture for 10–20 h in the absence of the crude antifungal substance, V. dahliae spores germinated and the biomass rapidly increased (Fig. 1). After culture for 20–36 h, the number of cells increased, albeit at a lower rate, and most spores germinated and produced new hyphae. After culturing for 48 h, the biomass and the growth rate increased. However, the biomass did not increase after 48 h in medium containing 0 mg/ml, 0.05 mg/ml, 0.2 mg/ml, 0.5 mg/ml, and 1 mg/ml crude antifungal substance. Therefore, the substance at 0.05 mg/ml or greater concentration inhibited the germination of V. dahliae spores.

Germination of V. dahliae spores according to B. subtilis BS-Z15 antifungal substance concentration and treatment duration
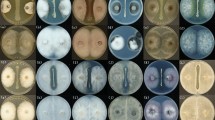
It takes 3 days to obtain the crude antifungal substance from fermentation broth to extraction, and second, the general outbreak of cotton yellow wilt occurs from cotton seedling stage to cotton boll stage, which is nearly two months in terms of time. Therefore, we selected two time points, 3 days and 2 months, for our study. In the perforated plate method, the bacteriostatic circle of the experiment time of 2 days was clear and transparent (Fig. 2a), and after being placed for 2 months, it can be found that the size and transparency of the bacteriostatic circle have not changed significantly (Fig. 2b), indicating that the antibacterial activity of the substance is stable for a long time. V. dahliae spores were cultured on a solid medium after treatment for 0.5 h with the substance. The magnitude of inhibition of spore germination increased with increasing substance concentration and treatment duration (Fig. 2c). In the presence of 1 mg/ml substance, few spores germinated, and growth was insufficient to form visible colonies after several days. In addition, the substance increased melanin secretion by mycelia. Therefore, the substance-induced aging of mycelia increased with increasing substance concentration.

Effects of different concentrations of B. subtilis BS-Z15 antifungal substances on the growth of Verticillium dahliae at different times. a Inhibition area after 3 days of test. b Inhibition area after 2 months of test. c germination of V. dahliae Kleb spores according to BS-Z15 antifungal substance concentration and treatment duration, the concentration increased from bottom to top and treatment duration from left to right
Genome features of BS-Z15
The BS-Z15 genome was sequenced using an Illumina HiSeq platform. The complete sequences for the whole-genome shotgun projects have been deposited at GenBank under the accession number QOCJ00000000, BioSample SAMN09582630 and BioProject PRJNA479656. The whole genome of BS-Z15 consisted of 4,068,702 base pairs (bp) with a GC content of 43.96%, 4196 predicted coding sequences (CDSs; genes), and 122 non-coding RNAs. The final assembly contained 16 contigs with a total size of 4,068,612 bp and an N50 contig length of 681,277 bp. The largest contig assembly measured 1,613,004 bp, with seven scaffolds with an N50 contig length of 2,150,442 bp. The tRNA, rRNA, and sRNA genes were identified using the tRNAscanSE, RNAmmer, and Rfam databases, respectively. A total of 4318 genes, nine genomic islands (GIs), 86 tRNAs, 13 sRNAs, and one prophage were determined (Table S1).
Genomic function annotation
To further understand the function of genes in B. subtilis BS-Z15, we performed gene function predictions in some commonly used databases (Table S2). The results showed that there were 2889 genes annotated in the GO database, 2889 genes annotated in the KEGG database, and 2889 genes annotated in the COG database. At the same time, 89 secreted proteins were also predicted.
Among them, 2889 CDSs were related to the GO functional categories database (Fig. S1), According to the different functions of the transcriptional text annotation, it was further divided into three categories: molecular function (MF), cellular component (CC), and biological process (BP). In the MF category, the genes were mainly assigned to “catalytic activity” (1635) and “binding” (1366). In the CC category, “cell” (1128) and “cell part” (1128) contained the highest number of genes. In the BP category, “cellular process” (1659) and “metabolic process” (1703) contained the highest number of genes. 3135 CDSs were related to the COGs functional categories database. A total of 3601 genes were assigned to 26 COG functional classifications (Fig. S2). From these, “General function” prediction contained only 351 genes, accounting for 9.7% of the total. This was followed by “Transcription”, which mapped to 9% of genes in the COG database. However, “RNA processing and modification” mapped to only one gene, which was for chromatin. Among all the genes, the function of 6.4% differential genes was unknown, but these may be the undiscovered genes in B. subtilis BS-Z15. 2494 CDSs were related to the KEGG pathway categories database, we found that 2014 genes were annotated in 24 metabolic pathways in the KEGG database (Fig. S3). The most common were “Carbohydrate metabolism,” followed by “Amino acid metabolism,” and “Metabolism of cofactors and vitamins,” with 226, 196, and 103 genes, respectively, annotated into these pathways. This indicates their important roles in the life cycle of B. subtilis BS-Z15.
We mapped reads using Circos software (Martin et al. 2009) for the assembled genomic sequences of the sequenced samples of BS-Z15, combined with the predicted results of the coding genes, as seen in Fig. S4 for the distribution of CDSs, COGs, KEGG, GO, and ncRNA.
Genomic properties of the B. subtilis group
We selected 12 similar Bacillus strains by 16S rDNA sequence of blast B. subtilis BS-Z15 on NCBI (B. subtilis ALBA01, B. subtilis MD-5, B. spizizenii DV1-B-1, B. subtilis strain 516, B. subtilis BJ3-2, B. spizizenii RFWG1A3, B. spizizenii RFWG1A4, B. spizizenii RFWG5B15, B. spizizenii W23, B. subtilis Y3, B. subtilis 168, and B. subtilis delta6), and the genomic information between them was compared. The genome size and number of predicted genes in BS-Z15 were slightly lower but similar to B. spizizenii RFWG1A3, and all strains had a similar GC-content percentage (43–47%; see Table S3). Although they share similar evolutionary trees, there remained some differences, namely in genome size, GC content, gene numbers, number of CDSs, and number of RNA genes.
Whole-genome single-copy orthologous protein sequencing
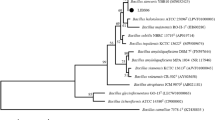
In this study, we used the entire dataset of concatenated single-copy orthologous proteins to build the phylogenetic tree (Fig. 3). Using the Perl script Proteinortho, after aligning the homologous protein sequences of 13 strains of Bacillus, 2733 highly conserved single-copy homologous genes from all strains were obtained. We constructed a maximum likelihood tree based on the homologous protein sequences of these 13 strains of Bacillus, and selected B. spizizenii DV1-B-1 as the exogenous group.

A maximum likelihood phylogenomic tree was inferred on the basis of 2733 concatenated orthologous single-copy amino acid sequences of the twelve Bacillus genomes (B. subtilis BS-Z15, B. spizizenii DV1-B-1, B. subtilis strain 516, B. subtilis ALBA01, B. subtilis MD-5, B. subtilis BJ3-2, B. spizizenii RFWG1A3, B. spizizenii RFWG1A4, B. spizizenii RFWG5B15, B. spizizenii W23, B. subtilis Y3, B. subtilis 168) using the Dayhoff model in TREE-PUZZLE
The results showed that BS-Z15 had the highest similarity with B. subtilis ALBA01 strain with ANI comparison result of 98.36 and belonged to Bacillus subtilis group. It was reported that B. subtilis ALBA01 was isolated from onion rhizosphere soil and had strong antagonistic activity against the soil fungal pathogen Setophoma terrestris (Albarracín et al. 2016). There was also a high similarity with B. subtilis BJ3-2 and MD-5, which have a strong inhibitory effect on fungi. B. subtilis BJ3-2 is a strain isolated from fermented lobster sauce. B. subtilis MD-5 was isolated from sediments in the Pacific Ocean and produced a macromolecular antimicrobial protein that was active against Staphylococcus aureus (Wu et al. 2018).
Carbohydrate-active enZYmes (CAZymes)
Secreted CAZymes are crucial for fungal biological activity. Using the common CAZy annotation pipeline (see Methods section) for genomic analysis of fungi, which was also adopted to perform similar analyses in previous studies, we explored the B. subtilis BS-Z15 genome for genes coding carbohydrate-active enzymes and carbohydrate-binding modules (CBMs).
We identified 157 putative CAZy-coding genes in the BS-Z15 genome of B. subtilis belonging to 55 families and 35 putative CBM-coding genes belonging to nine families (Table 1). These proteins containing the CAZy domain are considered to be candidate proteins involved in enzymatic degradation of plant polysaccharides. Of the 157 CAZy-coding genes, 48.4% were associated with PPD, while the proportion of PDD-related proteins in the GH and PL groups were 69.7% and 7.8%, respectively, and 15 of them were predicted as secreted proteins according to the SignalP analysis. Eight of them contain carbohydrate-binding domains, four belong to the glycoside hydrolase family, two belong to the polysaccharide lyase family, and one belongs to the sugar esterase family. Antagonistic bacteria degrade the cell wall components of pathogenic bacteria by secreting cell wall degrading enzymes, which include chitinase, glucanase, cellulase, xylanase, pectinase, and protease (Tseng et al. 2008). These PPD-related CAZy proteins may be involved in the antagonism of BS-Z15 to Verticillium dahliae and other fungi.
The heatmap based on family classification suggested that B. subtilis BS-Z15, B. spizizenii RFWG1A3, and B. subtilis ALBA01 tended to have similar degrading carbohydrates. Additionally, the CBM50 subfamily of molecules was present in high numbers in the B. subtilis BS-Z15 genome (Fig. 4).

Comparison of the CAZymes identified in the genomes of the selected fungi using hierarchical clustering. The fungi analysed along with B. subtilis BS-Z15 were B. spizizenii DV1-B-1, B. subtilis strain 516, B. subtilis ALBA01, B. subtilis MD-5, B. subtilis BJ3-2, B. spizizenii RFWG1A3, B. spizizenii RFWG1A4, B. spizizenii RFWG5B15, B. spizizenii W23, B. subtilis Y3, and B. subtilis 168. The enzyme families are represented by their classes (GH: glycoside hydrolases, GT:glycosyltransferases, PL: polysaccharide lyases, CE: carbohydrate esterases, and CBM: chitin binding modules) and the family numbers from the HMM predictions based on the carbohydrate-active enzyme database. The abundance levels of the different enzymes within a family are represented by a colour scale from the lowest (dark blue) to the highest occurrences (dark red) per species
Genes involved in antagonism
We compared some of their antagonistic genes, which focused on 47 genes related to biofilm production, siderophores (bacillibactin), extracellular enzymes (glycoside hydrolase, chitinase, and chitosanase), and antibiotics (surfactin, fengycin, and iturin) (Table 2).
The results showed that the 13 strains had consistent antifungal-related gene clusters. CHIA, CHIB, SrfB, FENa, FeNb, ItuA, ItuB, and ItuC were not found in the strains, indicating that the antifungal properties of these strains were not produced by the expression products of these genes. Among them, IituA–C are key enzyme-coding genes for iturin biosynthesis.
The iturin family of antibiotics, represented by iturin A, is commonly studied for its biocontrol activity (Arrebola et al. 2010). The iturin A operon spans a region > 38 kb in length and is composed of four open reading frames; ituD, ituA, ituB, and ituC. The ituD gene encodes a putative malonyl coenzyme A transacylase, whose disruption results in a specific deficiency in iturin A production. Iturin A specifically shows strong antibiotic activity with a broad antifungal spectrum, making it an ideal potential biological control agent that could reduce the use of chemical pesticides in agriculture (Hsieh et al. 2008). B. subtilis reportedly produces many antifungal peptide antibiotics, of which the iturin class exhibits powerful antifungal activities. Based on the analysis of the resistance genes described above, we determined that the main substance produced by BS-Z15 that inhibited V. dahlia did not belong to the iturin class because BS-Z15 lacks a complete operator for the synthesis of iturin. Lytic enzymes are produced by most bacteria, and they hydrolyze a wide variety of polymeric compounds, including chitin, proteins, cellulose, lipids, glucans, and the major components of the fungal cell wall, which suppresses phytopathogen growth (David 2004). BS-Z15 possesses one gene that encodes chitosanase, which may be required for fungal growth inhibition. Chitinase was previously identified from Bacillus as an important protein that potentially played a key role in fungal growth inhibition (Douriet-Gámez et al. 2018). However, we found no CHIA or CHIB in BS-Z15 and the 12 other selected Bacillus species. This result differs from previous reports (Chang et al. 2010; Saber et al. 2015).
Other antifungal-related genes, such as rmld, TasA-like 2, and SrfAA, were found in several genomes of the 12 strains, but not in BS-Z15. The coding product of srfAA is a key enzyme for the synthesis of surfactin, which also requires the expression of four structural genes, and the srfA operon contains four genes (srfAA, srfAB, srfAC, and srfAD) that act as a key biocontrol mechanism (Li et al. 2014). Based on nucleotide sequence and genetic analyses, the sfrA operon is believed to encode three enzymes (EIA, E1B, and E2) that catalyze the incorporation of the surfactin substrate amino acids. Among them, srfAA encodes ElA, the subunit that incorporates Glu, Leu, and D-Leu. SrfAB encodes E1B, which incorporates Val, Asp, and D-Leu, and SrfAC encodes E2, which incorporates Leu (Arrebola et al. 2010). SrfAD encodes a thioesterase-like protein that has high homology to the grsT gene product involved in the biosynthesis of gramicidin S (Tsuge et al. 2001). These four structural genes are relatively conserved, but BS-Z15 lacks the srfAA gene. The results show that surfactin did not underlie BS-Z15 resistance to fungi.
The BS-Z15 genome contained several antifungal-related genes, such as FenF, RpoN, and SfP, which were not present in most of the other 12 species. These genes and their coding products may play important roles in the antifungal activities of BS-Z15. Among them, FenF, plays an important role in the synthesis of fengycin and mycosubtilin. However, other fengycin synthetase genes, such as FENa and FeNb, were not found in BS-Z15, suggesting that fengycin is not the antifungal substance produced by BS-Z15. It is worth noting that the phosphopantetheinyl transferase SFP may be closely linked to the colonization of bacteria in the host (Li et al. 2014). The comparison of the 12 strains suggests that SFP may contribute to resistance in BS-Z15.
Biofilm formation is the first step of endophytic bacteria in the colonization of roots (Gao et al. 2015). In BS-Z15, we identified that some sipW-tasA operon genes (SipW and TasA-like 1) and regulatory genes sinR and sinI could be determinants for V. dahlia biocontrol mechanisms involving cell–cell communication via quorum sensing and the production of antifungal metabolites. Once B. subtilis colonizes the roots, it forms a stable and extensive biofilm and produces surfactin, which could protect the plant from pathogenic organisms.
The 13 strains listed in Table 2 contain many antifungal-related genes, including DhaA, DhaB, DhaE, and DhaF, which are related to the synthesis of bacilibactin; glycoside, which is related endoglucana; srfAB, srfAC, srfAD, and srfB, which are related to the synthesis of surfactin; mycA, mycB, and mycC, which are related to the synthesis of mycosubtilin; SipW, TasA-like 1, SinR, and SinI, which are related to the synthesis of biofilm; and other transcription factor genes, such as ComA, and ComP. It has been suggested that these resistance-related genes play an important role in the antifungal action of Bacillus spp., including BS-Z15. Among the antifungal-related factors, the most complete synthesis-related genes present are for bacilibactin and mycosubtilin. It was reported that bacillibactin (a catechol-based siderophore) plays a key role in antagonism and has been found in several species (Chu et al. 2010). Bacillibactin is encoded by the operon dhbACEBF (dhbA, dhbC, dhbE, dhbB, and dhbF), which is negatively controlled by the central iron regulatory protein Fur and is de-repressed upon iron limitation. The BS-Z15 genome contains all of the genes associated with siderophore synthesis, suggesting that this antagonistic mechanism may be important against fungal pathogens in BS-Z15. In addition, the coding genes of mycosubtilin synthesis-related enzymes are all present in BS-Z15, indicating that mycosubtilin is likely the main active substance involved in the antifungal activity of BS-Z15. It has been reported that mycosubtilin has strong antifungal activity, but it is not widely distributed in B. subtilis (Nasir and Besson 2012). Its molecular structure is similar to that of iturin, and it is a cyclic lipopeptide, but the amino acid sequence of the cyclic peptide chain and the number of carbons in the fatty acid chain differ (Genest et al. 1987).
Data accessibility
NCBI gene bank. www.ncbi.nlm.nih.gov/biosample/?term=SAMN09582630. www.ncbi.nlm.nih.gov/bioproject /?term = PRJNA479656. www.ncbi.nlm.nih.gov/nuccore/QOCJ00000000.
References
Albarracín Orio AG, Tobares RA, Ducasse DA, Smania AM (2016) Draft genome sequence of Bacillus Onion Setophoma terrestris. Genome Announc 4:e00455-e1416
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Arrebola E, Jacobs R, Korsten L (2010) Iturin A is the principal inhibitor in the biocontrol activity of Bacillus amyloliquefaciens PPCB004 against postharvest fungal pathogens. J Appl Microbiol 108:386–395
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM (2000) Gene Ontology: tool for the unification of biology. Nat Genet 25:25
Banat IM, Andrea F, Isabella G, Giuseppina B, Martinotti MG, Letizia F, Smyth TJ, Roger M (2010) Microbial biosurfactants production, applications and future potential. Appl Microbiol Biotechnol 87:427–444
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res 29:2607–2618
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res 37:D233–D238
Chang WT, Chen ML, Wang SL (2010) An antifungal chitinase produced by Bacillus subtilis using chitin waste as a carbon source. World J Microbiol Biotechnol 26:945–950
Chen LH, Xiong ZH, Sun LL, Yang J, Jin Q (2012) VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res 40:D641–D645
Chu BC, Garcia-Herrero A, Johanson TH, Krewulak KD, Lau CK, Peacock RS, Slavinskaya Z, Vogel HJ (2010) Siderophore uptake in bacteria and the battle for iron with the host; a bird’s eye view. Biometals 23:601–611
Compant S, Duffy BJ, Clement C, Barka EA (2005) Use of plant growth-promoting bacteria for biocontrol of plant diseases: principles, mechanisms of action, and future prospects. Appl Environ Microbiol 71:4951–4959
David JA (2004) Fungal cell wall chitinases and glucanases. Microbiology 150:2029–2035
Douriet-Gámez NR, Maldonado-Mendoza IE, Ibarra-Laclette E, Blom J, Calderón-Vázquez CL (2018) Genomic analysis of Bacillus sp. Strain B25, a biocontrol agent of maize pathogen Fusarium verticillioides. Curr Microbiol 75:1–9
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Eric PN, Diana LK, Sean RE (2009) Infernal 1.0: inference of RNA alignments. Bioinformatics 25:1335–1337
Gao T, Foulston L, Chai Y, Wang Q, Losick R (2015) Alternative modes of biofilm formation by plant-associated Bacillus cereus. Microbiologyopen 4:452–464
Gardner PP, Daub J, Tate JG, Nawrocki EP, Kolbe DL, Lindgreen S, Wilkinson AC, Finn RD, Griffiths-Jones S, Eddy SR, Bateman A (2008) Rfam: updates to the RNA families database. Nucleic Acids Res 37:D136-140
Genest M, Marion D, Caille A, Ptak M (1987) Modelling and refinement of the conformation of mycosubtilin in solution from two-dimensional NMR data. Eur J Biochem 169:389–398
Hollensteiner J, Wemheuer F, Harting R, Kolarzyk AM, Diaz Valerio SM, Poehlein A, Brzuszkiewicz EB, Nesemann K, Braus-Stromeyer SA, Braus GH, Daniel R, Liesegang H (2016) Bacillus thuringiensis and Bacillus weihenstephanensis Inhibit the Growth of Phytopathogenic Verticillium Species. Front Microbiol 7:2171
Hong N, Siquan Z, Xu Q, Xiaowen S, Shan Z, Xiuyun Z, Ziniu Y, Lin L (2015) Molecular docking and site-directed mutagenesis of a Bacillus thuringiensis Chitinase to improve chitinolytic, synergistic lepidopteran-larvicidal and nematicidal activities. Int J Biol Sci 11:304–315
Hsieh FC, Lin TC, Meng M, Kao SS (2008) Comparing methods for identifying Bacillus strains capable of producing the antifungal lipopeptide iturin A. Curr Microbiol 56:1–5
Huang X, Duan N, Xu H, Xie TN, Xue YR, Liu CH (2018) Ctab-peg dna extraction from fungi with high contents of polysaccharides. Mol Biol 52:621–628
Ibtissem G, Gilles V, Christine P (2007) CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52-57
Jacques P (2010) Surfactin and Other Lipopeptides from Bacillus spp. Springer-Verlag, Berlin
Jiménez G, Urdiain M, Cifuentes A, López-López A, Blanch AR, Tamames J, Kämpfer P, Kolstø AB, Ramón D, Martínez JF, Codoñer FM, Rosselló-Móra R (2013) Description of Bacillus toyonensis sp. nov., a novel species of the Bacillus cereus group, and pairwise genome comparisons of the species of the group by means of ANI calculations. Syst Appl Microbiol 6:383–391
Joshi SJ, Suthar H, Yadav AK, Hingurao K, Nerurkar A (2012) Occurrence of Biosurfactant Producing Bacillus spp. in Diverse Habitats. ISRN Biotechnol 2013:652340
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Res 32:D277-280
Kück P, Meusemann K (2010) FASconCAT: convenient handling of data matrices. Mol Phylogenet Evol 56:1115–1118
Lagesen K, Hallin P, Rodland E, Staerfeldt H, Rognes T, Ussery D (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ (2011) Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinf 12:124–132
Li W, Jaroszewski L, Godzik A (2002) Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18:77–82
Li RQ, Li YR, Karsten K, Wang J (2008) SOAP: short oligonucleotide alignment program. Bioinformatics 24:713–714
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J (2010) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20:265–272
Li B, Li Q, Xu Z, Zhang N, Shen Q, Zhang R (2014) Responses of beneficial Bacillus Amyloliquefaciens SQR9 to different soilborne fungal pathogens through the alteration of antifungal compounds production. Front Microbiol 5:636
Liu B, Pop M (2009) ARDB–antibiotic resistance genes database. Nucleic Acids Res 37:D443–D447
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Martin K, Jacqueline S, Inanç B, Joseph C, Randy G, Doug H, Jones JS, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Martin U, Rashmi P, Arathi R, Irvine AG, Helder P, Hammond-Kosack KE (2015) The pathogen-host interactions database (phi-base): additions and future developments. Nucleic Acids Res 43:D645–D655
Mongkolthanaruk W (2012) Classification of Bacillus beneficial substances related to plants, humans and animals. J Microbiol Biotechnol 22:1597–1604
Nakano MM, Corbell N, Besson J, Zuber P (1992) Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol Gen Genet 232:313–321
Nasir MN, Besson F (2012) Interactions of the antifungal mycosubtilin with ergosterol-containing interfacial monolayers. Biochim Biophys Acta Bioenerg 1818:1302–1308
Pal KK, Mc Spadden Gardener B (2006) Biological control of plant pathogens. Plant Health Instructor 67:1–25
Pereira JFB, Gudiña EJ, Costa R, Rui V, Teixeira JA, Coutinho JAP, Rodrigues LR (2013) Optimization and characterization of biosurfactant production by Bacillus subtilis isolates towards microbial enhanced oil recovery applications. Fuel 111:259–268
Płaza G, Chojniak J, Rudnicka K, Paraszkiewicz K, Bernat P (2015) Detection of biosurfactants in Bacillus species: genes and products identification. J Appl Microbiol 119:1023–1034
Rawlings ND, Morton FR (2008) The MEROPS batch BLAST: a tool to detect peptidases and their nonpeptidase homologues in a genome. Biochimie 90:243–259
Saber WIA, Ghoneem KM, Alaskar AA, Rashad YM, Rashad EM (2015) Chitinase production by Bacillus subtilis ATCC 11774 and its effect on biocontrol of Rhizoctonia disease of potato. Acta Biol Hung 66:436–448
Saier MH Jr, Reddy VS, Tamang DG, Västermark Å (2013) The transporter classification database. Nucleic Acids Res 42:D251–D258
Schmidt HA, Strimmer K, Vingron M, von Haeseler A (2002) TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18:502–504
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA (2003) The COG database: an updated version includes eukaryotes. BMC Bioinf 4:376–386
Tseng SC, Liu SY, Yang HH, Lo CT, Peng KC (2008) Proteomic study of biocontrol mechanisms of Trichoderma harzianum ETS 323 in response to Rhizoctonia solani. J Agric Food Chem 56:6914–6922
Tsuge K, Akiyama T, Shoda M (2001) Cloning, sequencing, and characterization of the iturin A operon. J Bacteriol 183:6265–6273
William H, Ivan W, Steven JJ, Fiona SLB (2003) IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19:418–420
Wu Y, Guo W, Zhao J, Ding L, Chen X (2018) Isolation and identification of a novel LCI like antibacterial protein from Bacillus sp. MD-5 reveals its potential application in controlling Staphylococcus aureus in food industry. Food Control 89:142–149
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451
Zhao JJ, Zeng WJ, Li YH, Ge FW, Du Y, Yuan LL, Zhao QQ, Wang M, Xie HT, Bai RX (2017) Isolation and identification of antagonistic Bacillus spp. against Verticillium dahliae: the antibacterial properties of two strains. J Beijing Norm Univ Nat Sci 3:294–300
Zhao QQ, Wu MJ, Dong YY, Wang QY, Lin RR, Zhao HH, Zhao HX (2018) Antifungal activity of BS-Z15 metabolites and its safety in mice. Nat Prod Res Dev 30:1608–1613
Acknowledgements
We are thankful to Dr. Li Xiaorong (Chinese Academy of Agricultural Sciences) for providing Verticillium dahliae strain.
Funding
This research was supported by the Open project of Key Laboratory in Xinjiang Uygur Autonomous Region (2020D04010), the National Natural Science Foundations of China (U1703112) and Innovation Project for Graduate Students in Xinjiang normal university (XSY202002011).
Author information
Authors and Affiliations
Contributions
Z-YC, XA and NM have contributed equally to this work. H-XZ, J-YL and H-PZ conceived of and designed the experiments. Z-YC, NM and XA performed the experiments and wrote the articles. M-JW, Q-LY and X-RL helped to performed the experiments and collected the data, RW and R-RL participated in the statistical analysis. H-CN helped with the chart processing. H-XZ and W-JZ contributed to manuscript discussion and revision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests. All authors declare that they have no conflict of interest.
Ethical statement
This article does not contain any studies related to human participants or animals.
Consent to participate
All authors agree to participate.
Consent for publication
All authors agree to publication.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Fig. S1
Terms of enrichment of genes in B. subtilis BS-Z15 in the GO database. Y-axis indicates the go functional classification on the sample annotation; the upper x-axis indicates the number of genes on the annotation; the lower x-axis indicates the percentage of the number of genes on the annotation to all coding genes. (TIF 10656 KB)
Fig. S2
Annotation of genes in B. subtilis BS-Z15 in the COG protein database. X-axis indicates the type of function; Y-axis indicates the number of genes on the annotation (TIF 5724 KB)
Fig. S3
Enrichment results of B. subtilis BS-Z15 genes according to the KEGG pathway database. X-axis indicates the type of function; Y-axis indicates the number of genes on the annotation. (TIF 8033 KB)
Fig. S4
The whole genome of B. subtilis BS-Z15. The outermost circle shows the position coordinates of the genomic sequence. From the outside inwards, coding genes and gene function annotations (including the annotation results showing the distribution of CDSs, COGs, KEGG, GO, and ncRNA) are listed. (TIF 16501 KB)
Rights and permissions
About this article

Cite this article
Chen, ZY., Abuduaini, X., Mamat, N. et al. Genome sequencing and functional annotation of Bacillus sp. strain BS-Z15 isolated from cotton rhizosphere soil having antagonistic activity against Verticillium dahliae. Arch Microbiol 203, 1565–1575 (2021). https://doi.org/10.1007/s00203-020-02149-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-020-02149-7