
Abstract
Wilt (Fusarium oxysporum f. sp. lentis; Fol) is one of the major diseases of lentil worldwide. Two hundred and thirty-five isolates of the pathogen collected from 8 states of India showed substantial variations in morphological characters such as colony texture and pattern, pigmentation and growth rate. The isolates were grouped as slow (47 isolates), medium (118 isolates) and fast (70 isolates) growing. The macroconidia and microconidia (3.0–77.5 × 1.3–8.8 µm for macroconidia and 1.8–22.5 × 0.8–8.0 µm for microconidia for length × width) were variable in size and considering the morphological features, the populations were grouped into 12 categories. Seventy representative isolates based on their morphological variability and place of origin were selected for further study. A set of 10 differential genotypes was identified for virulence analysis and based on virulence patterns on these 10 genotypes, 70 Fol isolates were grouped into 7 races. Random amplified polymorphic DNA (RAPD), universal rice primers (URPs), inter simple sequence repeats (ISSR) and sequence-related amplified polymorphism (SRAP) were used for genetic diversity analysis. URPs, ISSR and SRAP markers gave 100% polymorphism while RAPD gave 98.9% polymorphism. The isolates were grouped into seven clusters at genetic similarities ranging from 21 to 80% using unweighted paired group method with arithmetic average analysis. The major clusters include the populations from northern and central regions of India in distinct groups. All these three markers proved suitable for diversity analysis, but their combined use was better to resolve the area specific grouping of the isolates. The sequences of rDNA ITS and TEF-1α genes of the representative isolates were analysed. Phylogenetic analysis of ITS region grouped the isolates into two major clades representing various races. In TEF-1α analysis, the isolates were grouped into two major clades with 28 isolates into one clade and 4 remaining isolates in another clade. The molecular groups partially correspond to the lentil growing regions of the isolates and races of the pathogen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lentil (Lens culinaris Medikus subsp. culinaris) is an annual winter rainfed pulse crop growing throughout northern and central India. It is a valuable food having high protein content 19.5–36.4% (Tickoo et al. 2005) and its straw is used as fodder for animal (Sarker and Erskine 2006). Cultivation of lentil improves soil fertility by the ability to fix atmospheric nitrogen and carbon sequestration (Sarker and Erskine 2006). Various biotic factors including wilt affect the yield of lentil (Muehlbauer et al. 2006). The wilt caused by Fusarium oxysporum Schl f. sp. lentis (Vasudeva & Srinivasan) Gordon (Fol) significantly affects the productivity of lentil worldwide (Vasudeva and Srinivasan 1952; Bhalla et al. 1992). Fol can affect plants at any growing stage starting from the seedling to the reproductive stage (Tickoo et al. 2005). Growing resistant cultivars is a cost-effective practice for the management of Fusarium wilt (Bayaa et al. 1995; Kraft et al. 2000).
The knowledge about pathogenic and genetic diversity is important for designing effective management strategies of plant diseases. Earlier to this, Pouralibaba et al. (2016) and Hiremani and Dubey (2018) analysed Fol populations for race identification but either Indian populations were not included or some of the genotypes are not giving clear-cut differential reactions which needs further modification in the differential set. Molecular markers are an important tool for elucidating the genetic diversity of the pathogen and in classifying genes of economic importance. Various molecular markers have been used for the analysis of molecular diversity in Fusarium species such as inter simple sequence repeats (ISSR) (Dubey and Singh 2008; Dubey et al. 2012a) and random amplified polymorphic DNA (RAPD) (Dubey and Singh 2008; Honnareddy and Dubey 2006; Dubey et al. 2012b). Genetic characterization by RAPD analysis were used for the isolates of Fusarium oxysporum f. sp. lentis (Datta et al. 2009, 2011). Sequence-related amplified polymorphism (SRAP) markers (Li and Quiros 2001) were also used to determine genetic variability in F. poae (Dinolfo et al. 2015), Rhizoctonia solani (Tripathi and Dubey 2015), Colletotrichum capsici (Kumar et al. 2020) and Diaporthe spp. (Rajput et al. 2021). Universal rice primers (URPs) were also applied to study genetic diversity of phytopathogenic fungi (Kang et al. 2002; Aggarwal et al. 2010; Kumar et al. 2018, 2020; Singh et al. 2021) including F. oxysporum f. sp. ciceris (Dubey et al. 2012a). Other molecular markers including restriction fragment length polymorphism (RFLP) (Sharma et al. 2009), simple sequence repeats (SSR) (Dubey and Singh 2008) and amplified fragment length polymorphism (AFLP) (Sivaramakrishnan et al. 2002) were used for diversity analysis. Comprehensive study was not undertaken to use all these markers together to analyse the diversity of Fol populations representing almost all lentil growing diverse climatic areas of the country.
Therefore, keeping the above points in a view, the present study was aimed to understand the morphological and molecular variability among the Indian populations of Fol and to identify a set of lentil genotypes to determine the prevalence of Fol races based on virulence analysis.
Materials and methods
Collection, isolation and identification of the pathogen
Three hundred samples of lentil plants showing characteristic wilt symptoms were collected from various lentil growing areas of India and processed for isolation of Fol. Finally, 235 pathogenic isolates representing 102 isolates from Madhya Pradesh, 54 isolates from Uttar Pradesh, 49 isolates from Bihar, 13 isolates from Jharkhand, 8 isolates from Rajasthan, 7 isolates from Chhattisgarh and one each isolate from Gujarat and Delhi state were isolated. Thus, the collected isolates represent 8 major lentil growing states of India. Single-spore culture technique was employed to obtain pure culture and the culture was maintained by transferring periodically to PDA (potato dextrose agar; High Media, India) slants at 4 °C (Belabid and Fortas 2002). Morphological characters such as colony growth, pigmentation and especially size and shape of conidia were observed under the calibrated compound microscope for identification of the pathogen (Booth 1997). The pathogenicity of the identified isolates was tested on highly susceptible lentil cultivar K-75.
Morphological variability
Two hundred and thirty-five Fol isolates were grown on PDA medium to determine cultural and morphological variability. Morphological characters such as colony growth, pigmentation and size and shape of conidia were observed under the calibrated compound microscope (Booth 1997). The colony diameter was measured on PDA medium poured (15 mL/plate) in Petri dishes (90 mm) in three replications (Lilly and Burnett 1951). The inoculated plates were incubated at 28 ± 1 °C under 12-h alternate light and dark period for 6 days. Colony characters such as growth pattern and pigmentation were recorded. The isolates were grouped into 3 categories based on colony diameter as slow (up to 10 mm/day), medium (> 10 to 12 mm/day) and fast (> 12 mm/day) growing (Dubey et al. 2010). The size of microconidia and macroconidia (50 conidia for each isolate) was also measured through calibrated compound microscope and the mean was recorded for final observation. The isolates were grouped in different categories considering all these morphological features individually as well as in combination to reduce the number of the isolates representing all the features and location for virulence analysis. The representative isolates (70) from these groups were selected for virulence analysis.
Standardization of differentials
One hundred fourteen genotypes of lentil, collected from the Division of Plant Breeding and Genetics, IARI, New Delhi, India and ICRISAT, Hyderabad, India, were evaluated in net house against highly pathogenic isolate (FLDL1) of the pathogen. Fifteen seeds of each cultivar were sown in 15-cm diameter surface sterilized plastic pots (0.1% mercuric chloride) filled with 2 kg sterilized soil (1.0% formalin for 15 days) and inoculated with 15-day-old culture multiplied on sorghum grains (10 g/kg soil) at 4 days before sowing (Dubey and Singh 2008). Un-inoculated pots were also maintained as control for comparison. The experiment was conducted in completely randomized block design (CRD) with 114 treatments (114 genotypes) in two replications. Seed germination was recorded 15 days after sowing. The wilted plants were counted at 15-day interval up to maturity of the crop and percent wilt incidence was calculated on the basis of total number of wilted plants out of germinated plants. The wilt reaction was categorized as resistant (0–10%), moderately resistant (> 10 to 20%), susceptible (> 20 to 50%) and highly susceptible (> 50%) (Haware and Nene 1982). The environmental parameters, namely, maximum temperature from 12 to 32 °C, minimum temperature from 1 to 18 °C, mean temperature from 3.4 to 24.9 °C, maximum relative humidity from 72 to 100%, minimum relative humidity from 29 to 96%, mean relative humidity from 51 to 97% and mean sunshine duration 4.4 h with a range of 0–9.5 h, were prevalent during the crop period.
Virulence analysis for race profiling
The pot experiments were conducted in two consecutive crop seasons during 2014–2015 and 2015–2016 in net house. Based on the evaluation of 114 germplasm, 10 germplasm/cultivars of lentil, namely PL406 (DPL35 × EC 157,634/382), L4076 (PL 234 × PL 639), NDL1 (Precoz × L 9–12), DPL15 (PL 406 × L 4076), L4147 (L3875xP4 × PKVL), Vipasa (local selection), Sehore74-3 (local selection from MP, India), LC284-1206/12 (breeding line), JL3 (landrace selection from Sagar, India) and Vidhohar Local (local wilt susceptible), showing different degree of resistant and susceptible reactions with different genetic pedigrees were selected and they were used as differential genotypes. Seeds (15 seeds/pot) of these cultivars were sown in pots (15-cm diameter) filled with sterilized soil and inoculated with the 70 representative isolates of the pathogen separately in both the years. The environmental parameters, viz., maximum temperature from 12.5 to 35.2 °C, minimum temperature from 0.9 to 20.2 °C, mean temperature from 7.5 to 27.7 °C, maximum relative humidity from 71 to 100%, minimum relative humidity from 23 to 100%, mean relative humidity from 56 to 99% and mean sunshine duration 4.9 h with a range of 0–10.3 h, were prevalent during 2014–2015 crop period. During the 2nd crop season 2015–2016, maximum temperature from 13.9 to 35.9 °C, minimum temperature from 0.7 to 17.5 °C, mean temperature from 8.2 to 26.7 °C, maximum relative humidity from 66 to 98%, minimum relative humidity from 30 to 88%, mean relative humidity from 57 to 93% and mean sunshine duration 4.3 h with a range of 0–10.3 h were prevalent. The other experimental details were kept same as mentioned in the screening of the genotypes. The disease reactions were graded only in two categories as resistant (0–20% wilt) and susceptible (> 20% wilt) for race differentiation (Haware and Nene 1982; Dubey et al. 2012a).
DNA extraction
Seventy representative isolates of Fol belonging to 7 races purified by single-spore culture representing eight major lentil growing states of India were included in the present study (Table 1). Seventy isolates used for the study were representative populations of eight major lentil growing states of India covering differential Fol races and morphological groups. DNA was extracted from mycelial mat of Fol, multiplied on potato dextrose broth in shaker incubator (120 rpm) for 7 days at 28 ± 1 °C by using modified cetyl trimethyl ammonium bromide (CTAB) method (Dubey and Singh 2008; Murray and Thompson 1980). DNA was dissolved in TE (10 mM Tris-hydrochloric acid and 1 mM sodium EDTA, pH-8) buffer and stored at − 20 °C. The quality and quantity of the extracted DNA were measured using nanodrop spectrophotometer.
RAPD, URP and ISSR analysis
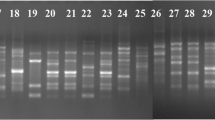
Thirteen RAPD, eleven URP and ten ISSR primers (Dubey and Singh 2008; Dubey et al. 2012a) were used to determine the genetic diversity within the Indian populations of Fol (70 isolates) representing 8 states and 7 races. Various concentrations of DNA (25, 50 and 75 ng), dNTPs (0.2, 0.4 and 0.6 mM), MgCl2 (1.5, 2.5, 3.5 mM) and primers (5, 10 and 15 pmol) were evaluated for good amplification as per the protocol described (Cobb and Clarkson 1994). The PCR mixture (25 µL) consisted of 50 ng template DNA, 1.0 U Taq polymerase and 0.6 mM of each dNTPs (Bangalore Genei, India) and 10 pmol of primer in 1 × reaction buffer for RAPD, URP and ISSR analysis. The concentration of MgCl2 for RAPD and URP was 2.5 mM and for ISSR it was 3.5 mM. The PCR for RAPD and URP was done at 94 °C for 5-min initial denaturation followed by 40 cycles for RAPD and 35 cycles for URP of denaturation at 94 °C for 1 min, annealing at 35 °C (RAPD)/55 °C (URP) for 1 min and extension at 72 °C for 2 min with a final extension at 72 °C for 5 min. For ISSR, the PCR was done at 94 °C for 4-min initial denaturation followed by 35 cycles of denaturation at 94 °C for 30 s, and extension at 72 °C for 2 min with a final extension at 72 °C for 10 min. Appropriate annealing temperatures (Table 8) for 30 s were used for each primer. Electrophoresis on agarose gel (1.2%) in 1 × TBE buffer was used to analyse the amplified products. The gels were stained with ethidium bromide and photographed under UV light using gel documentation system (Bio-Rad, India). A 100-bp plus DNA ladder (Genetix, India) was used as marker. All the experiments were repeated twice and the primers giving consistent and scorable amplifications were analysed.
SRAP analysis
Genetic diversity of 70 isolates of Fol was analysed using 20 sequence-related amplified polymorphism (SRAP) primers (Li and Quiros 2001). The PCR mixture (25 µL) consisted of 50 ng template DNA, 1.0 U Taq polymerase, 2.0 mM MgCl2, 0.4 mM of each dNTPs (Bangalore Genei, India) and 7.5 pmol of each primer in 1 × reaction buffer. The PCR for the first five cycles was done at 94 °C for 1 min, 35 °C for 1 min and 72 °C for 1 min for denaturation, annealing and extension, respectively with initial denaturation at 94 °C for 5 min and then the annealing temperature was raised to 50 °C for another 35 cycles. Final extension was done at 72 °C for 1 min. Amplified products were determined by electrophoresis using 2% agarose gel (1 × TBE buffer) along with a 100-bp plus DNA ladder. The gels were stained with ethidium bromide and photographed under UV light by using gel documentation system (Bio-Rad, India). Primers giving consistent and scorable amplifications were analysed.
ITS and TEF-1α amplification and sequencing
Area and race representative 32 isolates of the pathogen were selected for ITS and TEF-1α gene analysis. The ITS (ITS1 + 5.8 s + ITS2) regions of rDNA were amplified using universal primers ITS1 and ITS4 (White et al. 1990). ITS amplifications were performed in a total volume of 25 μL containing 10 × PCR buffer (100 mM Tris–HCl, 500 mM KCl, 0.8% (v/v) Nonidet P40), 10 mM dNTPs, 1 U Taq DNA polymerase, 0.2 µM of each primer and 25 ηg of fungal genomic DNA. The amplifications were performed using a Thermal Cycler (GenePro PCR, Bioer, New Hampshire, USA), conditions included an initial denaturation step at 94 °C for 4 min and cycling conditions were 94 °C for 45 s, 56 °C for 45 s, 72 °C for 1 min (32 cycles), followed by a final extension at 72 °C for 5 min. TEF-1α region of the selected isolates was amplified using EF1 and EF2 primers (O’Donnell et al. 2008). TEF-1α amplifications were performed in a total volume of 25 μL containing 10 × PCR buffer (100 mM Tris–HCl, 500 mM KCl, 0.8% (v/v) Nonidet P40), 10 mM dNTPs, 1 U Taq DNA polymerase, 0.2 µM of each primer and 25 ηg of fungal genomic DNA. The amplifications were performed using a Thermal Cycler (GenePro PCR, Bioer, New Hampshire, USA), conditions included an initial denaturation step at 94 °C for 4 min and cycling conditions were 94 °C for 45 s, 60 °C for 45 s, 72 °C for 1 min (35 cycles), followed by a final extension at 72 °C for 8 min. All the amplified PCR products were determined using 1.5% agarose electrophoresis for 2.0 h in 1 × TBE buffer at 80 V. The gels were stained with ethidium bromide and photographed using a gel documentation system (AlphaImager® Corporation, California, USA). The PCR products were purified using GeneJET™ PCR Purification Kit (Fermentas, Germany) following the manufacturer’s instructions. The purified PCR products were sequenced in the automated sequencer at the Eurofins Laboratory, Bangalore, India.
Data analysis
Completely randomized design (CRD) was followed for the experiments related to morphological studies and racial profiling and data were analysed statistically in CRD (Gomez and Gomez 1984) using Windostat version 7.0 (Indostat Services, Hyderabad, India). The statistical significance was assessed at p < 0.05 and Fisher’s protected least significant difference (LSD) was computed only when ANOVA showed significant differences. For genetic diversity, amplified products of template DNA by using RAPD, URP and ISSR primers that could be scored unequivocally for their presence (1) or absence (0) were included in the analysis. The binary matrices using numerical taxonomy and multivariate analysis system (NTSYS-PC) were analysed. NTSYS-PC assists to calculate a phylogenetic tree that uses the neighbour-joining or unweighted pair-group method with averaging (UPGMA) methods for constructing dendrograms. Dendrogram based on the scoring of 70 isolates was generated using SAHN clustering programme utilizing Jaccard’s similarity coefficients through UPGMA (Rohlf 1998). Consensus sequences for both ITS and TEF-1α of 32 isolates were compiled into a FASTA file format and aligned using Clustal W (Thompson et al. 1994). Phylogenetic tree construction using neighbour-joining method and analysis were conducted using the PAUP* (Phylogenetic Analysis Using Parsimony* and other methods) version 4.0a152 software (Swofford 2002). Statistical support was calculated from 1000 bootstrap replicates. Tree length, consistency index (CI), retention index (RI) and the rescaled consistency index (RC) values were also calculated.
Results
Morphological variability
Altogether 235 isolates of Fol isolated from eight states of India, viz. Madhya Pradesh, Uttar Pradesh, Bihar, Jharkhand, Chhattisgarh, Rajasthan, Delhi and Gujarat, showed substantial variability in their morphological characters such as texture, pigmentation, growth rate and size of conidia. All the isolates tested for pathogenicity on highly susceptible lentil cultivar K-75 showed sudden drooping and drying of leaves expressing typical wilt symptoms and satisfied the Koch’s postulates. One hundred forty-three Fol isolates showed fluffy mycelial growth, whereas the remaining 92 isolates showed appressed growth in culture. The colony was variable in pigmentation from white to violet/purple to creamy/pinkish (Supplementary Table S1). Based on the growth rate, 47 isolates were slow growing, 118 isolates medium growing and 70 isolates were fast growing (Table 2). The isolates were also variable in respect of their conidial dimension and septation. Micrometry revealed that the average size of macroconidia varied from 3.0–77.5 µm × 1.3–8.8 µm with 1 to 8 septa per conidia and the microconidia from 1.8–22.5 × 0.8–8 µm (Supplementary Table S2). Among 235 isolates, 175 isolates had macroconidia with an average length of > 12 µm and the remaining 60 isolates had macroconidia with an average length up to 12 µm (Supplementary Table S3). All the isolates were further grouped on the basis of colony characters, texture, growth rate and average length of macroconidia into 12 groups. The majority of the isolates came into medium growth rate along with fluffy growth and size of macroconidia greater than 12 µm, whereas less numbers of isolates showed fast growth along with appressed growth and size of macroconidia up to 12 µm (Table 3). The isolates representing 12 groups were further selected for virulence analysis.
Standardization of differentials
Out of 114 genotypes screened, 17 genotypes, namely, DPL-21, DPL-15, EC-1, Haryana M1, ILL-2581, L-4640, L-7818, LC-279–1237, LL-931, NDL-1, P-8109, PL-4, PL-406, PL-5, Vipasha, L-5227 and RLG-157, showed less than 10% wilt incidence and are considered resistant against the pathogen. Fifteen genotypes, namely, L-4648, L-5126, L-5228, L-9–12, L-5253, LC-292–1544, MC-6, P-2117, PL-101, PL-72–2, RL-1, Sehore-71–3, L-5228, LC-284–1206/12 and LL-1266, showed > 10 to 20% wilt incidence and are considered moderately resistant. The rest of the genotypes showed susceptible to highly susceptible reactions (Supplementary Table S4). The ten genotypes, namely, PL406, L4076, NDL1, DPL15, L4147, Vipasa, Sehore74-3, LC284-1206/12, JL3 and Vidhohar Local showing different levels of disease reactions, were selected for virulence analysis to determine the races of the pathogen.
Virulence analysis for race profiling
The wilt incidence ranged from 0 to 100% on a set of 10 lentil differential genotypes against 70 representative isolates originated from different states during crop seasons (Supplementary Tables S5 and S6). The corresponding reactions against each genotype were considered into resistant and susceptible. During both the years similar reactions were observed, therefore combined table for both the years has been given in Table 4. Based on the reactions, the isolates were grouped into 7 races. The first race consisted of 6 isolates from Madhya Pradesh differentiated by genotypes L4076 and Vipasa with resistant and susceptible reactions, respectively. The second race consisted of 5 isolates from Madhya Pradesh, 3 isolates from Chhattisgarh, 2 isolates from Uttar Pradesh and 1 isolate from Rajasthan differentiated by genotypes JL3 and NDL1 with resistant and susceptible reactions, respectively. The genotypes NDL1 and JL3, which differentiated 4 isolates from Madhya Pradesh, 2 isolates from Bihar, 2 isolates from Rajasthan and one isolate from Gujarat by showing resistant and susceptible reactions, respectively were named race 3. The differential genotypes for the fourth race were Sehore 74–3 and LC284-1206/12 which differentiated 7 isolates from Madhya Pradesh, 3 isolates of Uttar Pradesh and one isolate each from Bihar and Delhi by showing resistance and susceptible reactions, respectively. The genotypes Vipasa as resistant and PL406 as susceptible reactions differentiated race 5 consisting of the isolates one each from Bihar and Chhattisgarh and 3 from Jharkhand. Race 6 was differentiated by genotypes Vipasa and L4076 by showing resistant and susceptible reactions, respectively against 8 isolates from Uttar Pradesh, 5 isolates from Bihar, 2 isolates each from Jharkhand and Madhya Pradesh and one isolate each from Rajasthan and Chhattisgarh. The genotypes Sehore 74–3 and JL3, which differentiated 3 isolates from Madhya Pradesh, 2 isolates each from Jharkhand and Uttar Pradesh and one isolate from Bihar by showing resistant and susceptible reactions, respectively were considered race 7. In all the 70 Fol isolates were categorized into seven races of the pathogen based on the differential reactions (Table 5) and have been mapped in India (Fig. 1).

Map showing distribution patter of races of Fusarium oxysporum f. sp. lentis in different states of India
RAPD analysis
Amplification of 70 isolates of the pathogens with 13 RAPD-PCR primers produced 7–20 bands in the range of 0.3–5.0 kb (Supplementary Fig. S1a; Table 6). The polymorphism levels obtained with 186 DNA fragments were 98.9%. The 70 isolates of the pathogen were grouped into seven clusters at 22% Jaccard’s similarity coefficient using UPGMA analysis (Supplementary Fig. S2). There were two major clusters and five minor clusters formed by RAPD analysis. The first cluster had 27 isolates representing 6 races except race 5, originating from three states, namely, Madhya Pradesh, Uttar Pradesh and Bihar. Thirty isolates originated from different states, namely, Uttar Pradesh, Bihar, Jharkhand, Chhattisgarh, Rajasthan, Delhi and Gujarat representing 6 races except race 1 which were grouped into the second cluster. The third cluster consisted of 4 isolates, originating from two states, namely, Madhya Pradesh and Uttar Pradesh representing 3 races, namely races 1, 4 and 6. The fourth cluster consisted of 5 isolates originating from two states, namely, Madhya Pradesh and Bihar representing 3 races including races 1, 2 and 4. The fifth cluster consisted of 2 isolates, each originated from Uttar Pradesh representing races 2 and 7. The sixth and seventh clusters had single isolate each originating from Uttar Pradesh and representing races 4 and 6.
URP analysis
Polymerase chain reaction-based amplifications of 70 isolates of the pathogens with 11 URP primers produced 13–20 bands in the range of 0.1–3.0 kb (Supplementary Fig. S1b; Table 7) with 100% polymorphism. The isolates of the pathogen were grouped into seven clusters at 21% similarity coefficient using UPGMA analysis (Supplementary Fig. S3). URP analysis formed 2 major and 5 minor clusters depending on the number of isolates in each clusters. The first cluster consisted of 28 isolates representing 6 races except race 5, originating from two states, namely, Madhya Pradesh and Uttar Pradesh. The second cluster consisted of 5 isolates of 4 races, namely races 1–3 and 7 originating from two different states, namely, Madhya Pradesh and Uttar Pradesh. The third cluster consisted of 29 isolates representing 6 races except race 1, originating from different states, namely, Bihar, Jharkhand, Uttar Pradesh, Chhattisgarh, Rajasthan, Delhi and Gujarat. The fourth cluster consisted of 2 isolates of race 4 originating from different states, namely, Uttar Pradesh and Bihar. The fifth cluster consisted of 3 isolates of race 6 from Uttar Pradesh and Bihar. The sixth cluster consisted of single isolate representing race 6 and the seventh cluster consisted of 2 isolates of race 1 from Madhya Pradesh.
ISSR analysis
The PCR amplification of 70 isolates of the pathogens with 10 ISSR primers produced 8–13 bands in the range of 0.1–3.0 kb (Supplementary Fig. S1c; Table 8). The level of polymorphism obtained with 106 DNA fragments was 100%. The isolates of the pathogen were grouped into seven clusters at 50% similarity coefficient using UPGMA analysis (Supplementary Fig. S4). ISSR analysis formed 2 major clusters and 5 minor clusters. The first cluster consisted of 32 isolates representing 6 races except race 5, originating from two states, namely, Madhya Pradesh and Uttar Pradesh. The second cluster consisted of 31 isolates representing 6 races except race 1, originating from Uttar Pradesh, Bihar, Jharkhand, Chhattisgarh, Rajasthan and Delhi. The third cluster consisted of 2 isolates both from race 6 originating from Uttar Pradesh. The fourth cluster had a single isolate of Gujarat representing race 3. The fifth cluster consisted of 2 isolates from Madhya Pradesh representing races 1 and 4. The sixth and seventh clusters had a single isolate each from Uttar Pradesh and Madhya Pradesh representing races 4 and 1, respectively.
SRAP analysis
The PCR amplification of the isolates of the pathogens with 20 SRAP primers produced 8–17 bands in the range of 0.1–3.0 kb (Supplementary Fig. S1d; Table 9). The level of polymorphism obtained with 206 DNA fragments was 100%. The isolates of the pathogen were grouped into seven clusters at 42% similarity coefficient using UPGMA analysis (Supplementary Fig. S5). The first cluster consisted of 44 isolates representing all 7 races, originating from different states, namely, Madhya, Uttar Pradesh, Bihar, Jharkhand, Rajasthan, Chhattisgarh, Delhi and Gujarat. The second cluster consisted of 8 isolates representing races 2, 3, 5 and 6, originating from different states, namely, Madhya Pradesh, Jharkhand, Chhattisgarh and Rajasthan. The third cluster consisted of 3 isolates representing races 2, 6 and 7, originating from Uttar Pradesh and Chhattisgarh. The fifth cluster consisted of 7 isolates representing 5 races mainly 2–5 and 7, originating from Madhya Pradesh, Uttar Pradesh and Bihar. The sixth cluster consists of 6 isolates representing 4 races, namely races 1, 3, 4 and 6, originating from Madhya Pradesh, Uttar Pradesh and Bihar. The fourth and seventh cluster had a single isolate from Rajasthan and Madhya Pradesh representing races 3 and 1, respectively.
Combined analysis of RAPD, URP, ISSR and SRAP
The combined analysis of the 70 isolates of the pathogen representing seven races, using RAPD, URP, ISSR and SRAP, grouped them into seven clusters at 37% similarity coefficient using UPGMA analysis (Fig. 2). The first cluster consisted of 25 isolates representing all 7 races, originating from 4 states, namely, Madhya Pradesh Bihar, Jharkhand and Uttar Pradesh. The second cluster consisted of 26 isolates representing all 7 races, originating from different states, namely, Uttar Pradesh, Bihar, Jharkhand, Chhattisgarh, Rajasthan, Delhi and Gujarat. The third cluster consisted of 4 isolates representing races 2, 6 and 7, from Uttar Pradesh, Bihar and Chhattisgarh. The fifth cluster consisted of 7 isolates representing 4 races, namely races 1, 2, 6 and 7, originating Madhya Pradesh, Uttar Pradesh and Bihar. The sixth cluster consisted of 6 isolates representing races 1, 2, 4 and 7, from 3 states, namely, Madhya Pradesh, Uttar Pradesh and Bihar. The fourth and seventh cluster consisted of a single isolate in each from Uttar Pradesh of race 6.

Dendrogram derived from combined analysis of 13 random amplified polymorphic DNA (RAPD) primers, 11 universal rice primer (URP) primers, 10 inter simple sequence repeat (ISSR) primers and 20 sequence-related amplified polymorphism (SRAP) primers of 70 isolates of F. oxysporum f. sp. lentis by unweighted paired group method with arithmetic average analysis (UPGMA). The bottom scale is the percentage of Jaccard’s similarity coefficient. Vertical scale representing numbers (Fol 1–70) and state of origin of the isolates (FLMP–Madhya Pradesh, FLUP–Uttar Pradesh, FLBR–Bihar, FLJH–Jharkhand, FLRJ–Rajasthan, FLCG–Chhattisgarh, FLDL–Delhi and FLGJ–Gujarat)
ITS sequence analysis
Amplification products of approximately 550 bp were obtained in all 32 representative isolates of Fol and all the aligned ITS sequences (ITS1 + 5.8 s + ITS2 region) deposited at NCBI GenBank with the accession numbers KY678270-KY678301 (Table 1). The dataset used for ITS analysis contained 541 characters, 441 characters were conserved sites, 100 were variable sites, 73 were parsimony informative and 27 characters were singleton change. The isolates clustered into two major phylogenetic groups. The first group contains 19 isolates and 13 isolates in the second group (Fig. 3). Within the first group, isolates from all the races grouped into three sub-groups and race 6 present in all the three sub-groups. In the second group, race 6 separately grouped as sub-group with high bootstrap value (99%). Race 4 and race 7 form separate sub-group, whereas races 2, 3, 5 and 7 belong to different geographical locations sub-grouped separately.

Molecular phylogenetic analyses of rDNA ITS sequences of Fusarium oxysporum f. sp. lentis by neighbour-joining method. The analyses involve 32 nucleotide sequences and there were 541 characters in this analysis. All the bootstrap values (1000 replicates) were indicated above and below internodes of the tree. The scale bar denotes 0.01 substitution per nucleotide position. CI consistency index, RI retention index, RC rescaled consistency index. Evolutionary analyses were conducted in PAUP version 4.0a152
TEF-1α sequence analysis
The PCR amplification of 32 different race and area representative isolates of Fol using TEF-1α gene-specific primers produced approximately 710 bp amplicon. The aligned TEF-1α sequences of those isolates were deposited at NCBI GenBank with the accession numbers KY852314–KY852345 (Table 1). The dataset used for TEF-1α analysis contained 709 characters, 477 characters were conserved sites, 232 were variable sites, 183 were parsimony informative and 49 characters were singleton change. Phylogenetic analysis clustered into two major phylogenetic groups, twenty-eight isolates were clustered into the first group and 4 isolates in the second group (Fig. 4). Within the first group, the isolates from all the races clustered into three sub-groups with race 7 in all the three sub-groups and race 2 clustered as separate sub-group (with high bootstrap value 99%). In the second group, race 7 (FLUP39) from Uttar Pradesh grouped with race 4 and collected from Bihar (FLBR43), Uttar Pradesh (FLUP49) and Madhya Pradesh (FLMP96) belongs to different geographical locations which grouped separately as sub-group.

Molecular phylogenetic analyses of TEF-1α sequences of Fusarium oxysporum f. sp. lentis by neighbour-joining method. The analyses involve 32 nucleotide sequences and there were 709 characters in this analysis. All the bootstrap values (1000 replicates) were indicated above and below internodes of the tree. The scale bar denotes 0.02 substitution per nucleotide position. CI consistency index, RI retention index, RC rescaled consistency index. Evolutionary analyses were conducted in PAUP version 4.0a152
Discussion
The populations of F. oxysporum f. sp. lentis collected from all the major lentil growing areas of the India were highly variable in respect of their morphological characters. Considering the morphology and cultural characters, 235 isolates of the pathogen were categorized into 12 groups. Forty-two isolates showed fast growth rate, large macroconidia and fluffy colony growth. It was followed by the similar number of isolates (42) characterized as medium growing, large macroconidia and fluffy growth. Forty-one isolates with similar growth rate and macroconidia showed appressed growth. Only 47 isolates were slow growing with the majority having > 12 µm macroconidia. Thus, the majority of the isolates were medium growing. The present results could not establish any correlations among pigmentation, growth rate and size of conidia of the isolates originating from different parts of country. However, Belabid et al. (2004) reported great variability in the morphological characters of thirty-two Fol isolates belonging to single race from the northwest Algeria.
Screening of lentil genotypes against Fol clearly indicated the presence of low level of resistance among them. Out of 114, only 17 genotypes were found resistant and 15 moderately resistant against the pathogen. The rest of the genotypes were susceptible to highly susceptible. Ten genotypes, namely PL406, L4076, NDL1, DPL15, L4147, Vipasa, Sehore74-3, LC284-1206/12, JL3 and Vidhohar local variable in genetic background as well as in level of resistance and susceptibility, were selected as differential set for race profiling in the present study. The virulence analysis showed existence of high degree of variability among the pathogen. Based on the reactions on differential genotypes, the isolates of Fol were categorized into seven races and differential genotypes for each race were identified. Except race 5, all the six races were prevalent in Madhya Pradesh, a major lentil growing state of India. Race 1 was only present in Madhya Pradesh. Out of 70 isolates representing various parts of the country, 19 isolates representing six states of India were categorized as race 6 followed by 12 isolates as race 4 representing 4 states and 11 isolates as race 2 originated from 4 states of India. Four races, namely, 2, 4, 6 and 7, were present in Uttar Pradesh, whereas 5 races, namely, 3, 4, 5, 6 and 7, were present in Bihar State of India. This clearly indicated that the states having maximum area under lentil production and diversity in respect of varieties under cultivation showed the highest number of races. The pair of differential genotypes provided resistant and susceptible reaction identified for each race to avoid the overlapping reaction for different isolates under each race. The place of origin of the isolates and morphological features did not correspond to the races of the pathogen. The present findings are in accordance with the earlier observations in lentil (Belabid et al. 2004) and chickpea (Dubey et al. 2012a).
The variability among Fol isolates with regard to aggressiveness (Naimuddin and Chaudhary 2009) as well as pathogenicity has been observed (Bayaa et al. 1994; Erskine and Bayaa 1996; Datta et al. 2011; Pouralibaba et al. 2015). The isolates of Fol were grouped into 6 clusters on the basis of typical wilt symptoms of Fol on single cultivar using computer software (Naimuddin and Chaudhary 2009). Pouralibaba et al. (2016) determined the pathotypes among 52 Fol isolates originated from Iran, Syria and Algeria using 4 accessions of lentil as a putative differential set and identified 7 pathotypes. Only 28 lentil accessions were evaluated for designing of the differentials and only 4 accessions may not be sufficient for differentiation of large number of isolates. The isolates of the pathogen from India, which is considered a major lentil growing country worldwide, were not included in the study. The present findings are in accordance with the results obtained by Pouralibaba et al. (2016) that the pathotypes did not correspond to the geographical origin of the isolates. In India, Datta et al. (2011) collected 100 isolates of Fol from 4 states and included only 15 isolates (majority of them from Uttar Pradesh and Bihar and one each from Tripura and West Bengal) in the study. They observed molecular diversity among 15 isolates of Fol using molecular markers and pathogenicity but failed to report about the races of Fol. They have not used a set of lentil cultivars for diversity analysis to identify races of Indian population of the pathogen. Furthermore, an attempt was made to standardize the differential cultivars to determine the races and grouped 50 Indian isolates of the pathogen into 8 races (Hiremani and Dubey 2018). In contrast to the present study, Belabid et al. (2004) reported that the Fol isolates were homogeneous without any variation in virulence, but they observed differences in aggressiveness indicated variability, which might be differentiated by using the cultivars having variable genetic background. They also observed no apparent correlation with geographical origin or aggressiveness of isolates. The differential cultivars used by Hiremani and Dubey (2018) need modification by replacing some of the genotypes in existing set with appropriate genotypes to avoid overlapping reactions against the isolates originating from the same locations. Thus, earlier used 5 genotypes (PL4, PL101, DPL62, K75 and L6813) were replaced with 5 new genotypes (PL406, L4076, NDL1, DPL15 and Vipasa). In the present study, by replacing these 5 genotypes with 5 new, clear-cut grouping patterns were obtained and 70 isolates were grouped into 7 races.
In the present study, four molecular markers, namely, RAPD, URP, ISSR and SRAP, were used for the analysis of genetic diversity of Fol populations representing 7 races collected from different geographical regions of India. These molecular markers showed very good amplification of various plant pathogenic fungi (Dubey and Singh 2008; Zhou et al. 2009; Hiremani and Dubey 2019). Prior to this study, RAPD markers showed 22% genetic similarity between the isolates of Fol and grouped them into five clusters based on geographical origin barring few exceptions. There was no apparent correlation with geographical origin or aggressiveness of Fol (Belabid et al. 2004). In the present study, RAPD markers also provided good amplification and grouped the Fol populations into 7 clusters. Another important molecular marker, URPs proved more appropriate for genetic diversity analysis of Fol isolates. The isolates of Fol showed 21% genetic similarity with 100% polymorphism between themselves using URP markers and grouped them into 7 clusters. Earlier, URPs have been proved more superior over other markers for genetic diversity analysis of Rhizoctonia solani (Dubey et al. 2012b), Bipolaris oryzae (Kandan et al. 2015) and Diaporthe spp. (Rajput et al. 2021). ISSR markers have been extensively used for various applications as conservation, molecular taxonomy and analysis of the population structure and genetic variation of F. oxysporum (Baysal et al. 2010). The results of the present study in corroborate with earlier report of Yuan et al. (2013) that the ISSR markers were unable to make a clear distinction within and between the isolates of Fol from different provinces. Earlier, Dubey and Singh (2008) while working on 64 isolates of F. oxysporum f. sp. ciceris reported the suitability of ISSR and RAPD markers for genetic diversity of the pathogen. Similarly, Kumar et al. (2018) have reported ISSR markers more efficient than URP markers in diversity analysis of Curvularia lunata. SRAP is another PCR-based marker which is simple, efficient, repetitive and co-dominant (Li and Quiros 2001). It has been widely applied in genetic diversity studies of different fungal species (Tripathi and Dubey 2015; Kumar et al. 2020; Rajput et al. 2021). Among all the markers used in the present study, SRAP provided 42% genetic similarity between Fol isolates and grouped them into 7 major clusters. The present study was in agreement with previous report of Dinolfo et al. (2015) showing SRAP as informative molecular marker to study the genetic diversity analysis of F. poae. The present SRAP results are in concordance with Tripathi and Dubey (2015) who reported that the molecular diversity of R. solani based on SRAP marker did not correspond to the agro-ecological regions and crops of the origin of the isolates.
Combined analysis of all the four markers grouping 70 isolates into seven clusters with 37% genetic similarity clearly indicated that the clustering partially corresponds to their geographical origin or virulence. The present results are in agreement with Kandan et al. (2015) who reported with the exception of URP marker data, ISSR and RAPD molecular marker pooled data were found to be more informative in genetic diversity analysis of Bipolaris oryzae, B. sorghicola, B. holmi and B. tetramera (Kandan et al. 2016; Singh et al. 2021). Nourollahi and Madahjalali (2017) reported a low level of genetic variability and genetic distance within and between populations of Fol using microsatellite SSR markers from Iran. Al-Husien et al. (2017) reported the high genetic diversity of Fol within population than among geographical location of Syria using RAPD, ISSR and SSR markers. However, phylogenetic analysis of Fol using TEF-1 alpha gene and diversity analysis using URP and SRAP marker altogether of Fol has not been reported so far.
Phylogenetic analysis of rDNA ITS and TEF-1α sequence analyses did not show correlation with other variables like geographical location, and virulence. Grouping of these isolates from different locations showed that geographic location or virulence of the isolates did not decide their grouping pattern, clearly suggesting that existence of few degrees of divergence within the isolates and lack of relationship between isolates and geographic locations may be due to movement of the pathogen in nature through infected seeds and soils. However, in ITS analysis, presence of isolates that belong to race 6 in both clusters indicates the evolutionary relationship of this pathogen with other races that belong to various geographical locations and virulence patterns. In TEF-1α analysis, Fol isolates representing 7 races originating from different places have very close relationship with each other. However, the isolates that belong to race 7 grouped in most of the major and minor clusters indicating the evolutionary relationship of this pathogen with other races originated from different location in India. These results are in congruence with earlier reports of F. verticillioides (Rocha et al. 2011), F. oxysporum f. sp. ciceris (Dubey et al. 2012a) and Sclerotinia sclerotiorum (Mandal and Dubey 2012) showing no correlation within the isolates and their origin of location.
Conclusions
The present study clearly revealed the diversity of F. oxysporum f. sp. lentis populations collected from different parts of India that could be a milestone for further development of race-specific resistant varieties of lentil. Furthermore, analysis of large number of isolates with a set of genotypes to standardize new differentials revealed clear-cut grouping of the present pathogenic population of the pathogen. The molecular markers, namely RAPD, URP, ISSR and SRAP used in the study, could be very efficiently used for diversity analysis of Fol populations. The ITS and TEF-1α analysis provided phylogenetic relationships among the Indian populations of the pathogen. The present findings may help to understand the virulence performance of the Fol populations which is helpful in devising efficient disease resistance strategies.
Data availability
The data generated and analysed during the current study are given as supplementary files.
References
Aggarwal R, Tripathi A, Yadav A (2010) Pathogenic and genetic variability in Tilletia indica monosporidial culture lines using universal rice primer-PCR. Eur J Plant Pathol 128:333–342
Al-Husien NH, Hamwieh A, Ahmed S, Bayaa B (2017) Genetic diversity of Fusarium oxysporum f. sp. lentis population affecting lentil in Syria. J Phytopathol 165:306–312
Bayaa B, Erskine W, Abbas A (1994) Evaluating different methods for screening lentil germplasm for resistance to lentil wilt caused by Fusarium oxysporum f. sp. lentis. Arab J Plant Prot 12:83–91
Bayaa B, Erskine W, Hamdi A (1995) Evaluation of a wild lentil collection for resistance to vascular wilt. Genet Resour Crop Evol 42:231–235
Baysal Ö, Siragusa M, Gümrükcü E, Zengin S, Carimi F, Sajeva M, da Silva TJA (2010) Molecular characterization of Fusarium oxysporum f. sp. melongena by ISSR and RAPD markers on eggplant. Biochem Genet 48:524–537
Belabid L, Baum M, Fortas Z, Bouznad ZI (2004) Pathogenic and genetic characterization of Algerian isolates of Fusarium oxysporum f. sp. lentis by RAPD and AFLP analysis. Afr J Biotech 3:25–31
Belabid L, Fortas Z (2002) Virulence and vegetative compatibility of Algerian isolates of Fusarium oxysporum f. sp. lentis. Phytopathol Mediter 41:179–187
Bhalla MK, Nozoolillo C, Schneider E (1992) Observation on the responses of lentil root cells to hypha of Fusarium oxysporum. J Phytopathol 135:335–341
Booth C (1997) The genus Fusarium. Commonwealth Mycological Institute, Kew, Surrey, England
Cobb BD, Clarkson JM (1994) A simple procedure for optimizing the polymerase chain reaction (PCR) using modified Taguchi methods. Nucleic Acids Res 22:3801–3805
Datta S, Choudhary RG, Shamim MD, Singh RK, Dhar V (2011) Molecular diversity in Indian isolates of Fusarium oxysporum f. sp. lentis inciting wilt disease in lentil. Afr J Biotech 10:7314–7323
Datta S, Choudhary RG, Rai R, Singh RK, Dhar V (2009) Molecular diversity and pathotype analyses of Fusarium oxysporum f. sp. lentis population from north eastern gangetic plain region of India. SABRAO J Breed Genet 41:1–11
Dinolfo MI, Castanares E, Stenglein SA (2015) SRAP as an informative molecular marker to study the Fusarium poae genetic variability. J Phytopathol 163:657–663
Dubey SC, Priyanka K, Singh V, Singh B (2012a) Race profiling and molecular diversity analysis of Fusarium oxysporum f. sp. ciceris causing wilt in chickpea. J Phytopathol 160:576–587
Dubey SC, Singh SR (2008) Virulence analysis and oligonucleotide fingerprinting to detect diversity among Indian isolates of Fusarium oxysporum f. sp. ciceris causing chickpea wilt. Mycopathology 165:389–406
Dubey SC, Singh SR, Singh B (2010) Morphological and pathogenic variability of Indian isolates of Fusarium oxysporum f. sp. ciceris causing chickpea wilt. Arch Phytopathol Plant Prot 43:174–190
Dubey SC, Tripathi A, Upadhyay BK (2012b) Molecular diversity analysis of Rhizoctonia solani isolates infecting various pulse crops in different agro-ecological regions of India. Folia Microbiol 57:513–524
Erskine W, Bayaa B (1996) Yield loss, incidence and inoculum density associated with vascular wilt of lentil. Phytopathol Mediter 36:24–32
Gomez KA, Gomez AA (1984) Statistical procedures for agricultural research, 2nd edn. John Wiley and Sons, Singapore
Haware MP, Nene YL (1982) Races of Fusarium oxysporum f. sp. ciceri. Plant Dis 66:809–810
Hiremani NS, Dubey SC (2018) Race profiling of Fusarium oxysporum f. sp. lentis causing wilt in lentil. Crop Prot 108:23–30
Hiremani NS, Dubey SC (2019) Genetic diversity of Fusarium oxysporum f. sp. lentis populations causing wilt of lentil in India. Indian Phytopathol 72(4):1–7
Honnareddy N, Dubey SC (2006) Pathogenic and molecular characterization of Indian isolates of Fusarium oxysporum f. sp. ciceris causing chickpea wilt. Curr Sci 91:661–666
Kandan A, Akhtar J, Singh B, Dixit D, Chand D, Roy A, Rajkumar S, Aggarwal PC (2015) Molecular diversity of Bipolaris oryzae infecting Oryza sativa in India. Phytoparasitica 43:5–14
Kandan A, Akhtar J, Singh B, Pal D, Chand D, Rajkumar S, Agarwal PC (2016) Genetic diversity analysis of fungal pathogen Bipolaris sorghicola infecting Sorghum bicolor in India. J Environ Biol 37:1323–1330
Kang HW, Park DS, Go SJ, Eun MY (2002) Fingerprinting of diverse genomes using PCR with universal rice primers generated from repetitive sequence of Korean weedy rice. Mol Cells 13:281–287
Kraft JM, Haware MP, Halila H, Bayaa B (2000) Soilborne diseases and their control. In: Knight R (ed) Proceedings of the third international food legumes research conference on Linking research and marketing opportunities for pulses in the 21st century. Kluwer Academic Publishers, The Netherlands, pp 457–466
Kumar P, Akhtar J, Kandan A, Singh B, Kiran R, Nair K, Dubey SC (2018) Efficacy of URP and ISSR markers to determine diversity of indigenous and exotic isolates of Curvularia lunata. Indian Phytopathol 71:235–242
Kumar P, Dubey SC, Akhtar J, Kiran R, Nair K, Bhati H (2020) Genetic diversity and population structure analysis of Colletotrichum capsici isolates using SRAP and URP markers. Indian J Plant Prot 48:64–73
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Lilly VG, Burnett HL (1951) Physiology of fungi. McGraw Hill Book Co. Inc., New York, USA
Mandal AK, Dubey SC (2012) Genetic diversity analysis of Sclerotinia sclerotiorum causing stem rot in chickpea using RAPD, ITS-RFLP, ITS sequencing and mycelial compatibility grouping. World J Microbiol Biotechnol 28:1849–1855
Muehlbauer FJ, Cho S, Sarker A, Mc Phee KE, Coyne CJ, Rajesh PN, Ford R (2006) Application of biotechnology in breeding lentil for resistance to biotic and abiotic stress. Euphytica 147:149–165
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight DNA. Nucleic Acids Res 8:4321–4325
Naimuddin R, Chaudhary RG (2009) Pathogenic variability in isolates of Fusarium oxysporum f. sp. lentis. Trends Biosci 2:50–52
Nourollahi K, Madahjalali M (2017) Analysis of population genetic structure of Iranian Fusarium oxysporum f. sp. lentis isolates using microsatellite markers. Australas Plant Pathol 46:35–42
O’Donnell K, Sutton DA, Fothergill A, Mc Carthy D, Rinaldi MG, Brandt ME, Zhang N, Geiser DM (2008) Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J Clin Microbiol 46:2477–2490
Pouralibaba HR, Rubiales D, Fondevilla S (2015) Identification of resistance to Fusarium oxysporum f. sp. lentis in Spanish germplasm. Eur J Plant Pathol 143:399–405
Pouralibaba HR, Rubiales D, Fondevilla S (2016) Identification of pathotypes in Fusarium oxysporum f. sp. lentis. Eur J Plant Pathol 144:539–549
Rajput LS, Kumar S, Bhati H, Nair K, Akhtar J, Kumar P, Dubey SC (2021) Diversity assessment of indigenous and exotic Diaporthe species associated with various crops using ISSR, URP and SRAP markers. Indian Phytopathol 74:615–624. https://doi.org/10.1007/s42360-020-00313-z
Rocha LDO, Reis GM, Silva VND, Braghini R, Teixeira MMG, Correa B (2011) Molecular characterization and fumonisin production by Fusarium verticillioides isolated from corn grains of different geographic origins in Brazil. Int J Food Microbiol 145:9–21
Rohlf FJ (1998) NTSYS-pc: numerical taxonomy and multivariate analysis system. Version 2.02. Setauket, NY, Exeter Software. 47 Route 25A, Suite 2 Setauket, New York
Sarker A, Erskine W (2006) Recent progress in the ancient lentil. J Agric Sci 144:19–29
Sharma M, Varshney RK, Rao JN, Kannan S, Hoisington D, Pande S (2009) Genetic diversity in Indian isolates of Fusarium oxysporum f. sp. ciceris, chickpea wilt pathogen. Afr J Biotech 8:1016–1023
Singh S, Sharma S, Ali A, Kandan A, Kumar P, Akhtar J (2021) Morpho-molecular characterization of Bipolaris and Exserohilum spp. infecting various agricultral crops. Int J Agric Appl Sci 2:110–117
Sivaramakrishnan S, Kannan S, Singh SD (2002) Genetic variability of Fusarium wilt pathogen isolates of chickpea (Cicer arietinum L.) assessed by molecular markers. Mycopathologia 155:171–178
Swofford DL (2002) PAUP*. Phylogenetic analysis using parsimony (*and other methods), Version 4. Sinauer Associates, Sunderland, Massachusetts
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weigh matrix choice. Nucleic Acids Res 22:4673–4680
Tickoo JL, Sharma B, Mishra SK, Dikshit HK (2005) Lentil (Lens culinaris) in India: present status and future perspectives. Indian J Agric Res 75:539–562
Tripathi A, Dubey SC (2015) Sequence-related amplified polymorphism-PCR analysis for genetic diversity in Rhizoctonia solani populations infecting pulse crops in different agro-ecological regions of India. Plant Pathol J 14:234–241
Vasudeva RS, Srinivasan KV (1952) Studies on the wilt disease of lentil (Lens esculenta Moench). Indian Phytopathol 5:23–32
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand D, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications. Academic Press, San Diego
Yuan L, Mi N, Liu S, Zhang H, Li Z (2013) Genetic diversity and structure of the Fusarium oxysporum f. sp. lini populations on linseed (Linum usitatissimum) in China. Phytoparasitica 41:391–401
Zhou Q, Chang K, Hwang SF, Strelkov SE, Gossen BD, Chen YY (2009) Pathogenicity and genetic diversity of Rhizoctonia solani isolates from lupin and other crops in Alberta, Canada. Can J Plant Pathol 31:340–347
Acknowledgements
The authors gratefully acknowledge the Director, ICAR-National Bureau of Plant Genetic Resources for the administrative and technical support.
Funding
This study was financially supported by the Department of Science and Technology, Government of India, New Delhi, India (F. No. SERB/SR/SO/PS/83/2012 dated 10–07-2013).
Author information
Authors and Affiliations
Contributions
SCD conceptualized, designed and supervised the experiments and prepare the manuscript; VDS and VKP performed the experiments and analysed the data; JA and AK reviewed and edited the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dubey, S.C., Sharma, V.D., Kumar Prajapati, V. et al. Phenotypic variability, race profiling and molecular diversity analysis of Indian populations of Fusarium oxysporum f. sp. lentis causing lentil wilt. Folia Microbiol 67, 757–775 (2022). https://doi.org/10.1007/s12223-022-00975-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12223-022-00975-4