
Abstract
Fusarium oxysporum f. melongenae is a major soil-borne pathogen of eggplant (Solanum melongena). ISSR and RAPD markers were used to characterize Fusarium oxysporum f. melongenae isolates collected from eggplant fields in southern Turkey. Those isolates were not pathogenic to tomato. Pathogens were identified by their morphology, and their identity was confirmed by PCR amplification using the specific primer PF02-3. The isolates were classified into groups on the basis of ISSR and RAPD fingerprints, which showed a level of genetic specificity and diversity not previously identified in Fusarium oxysporum f. melongenae, suggesting that genetic differences are related to the pathogen in the Mediterranean region. The primers selected to characterize Fusarium oxysporum f. melongenae may be used to determine genetic differences and pathogen virulence. This study is the first to characterize eggplant F.oxysporum species using ISSR and RAPD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Eggplant (Solanum melongena) has been produced in greenhouses, polytunnels, and fields in the southern Turkish provinces of Antalya, Adana, and Mersin since 2002. The major plant disease in eggplant production areas is fusarium wilt, caused by Fusarium oxysporum f. melongenae (FOM). Diseased plants exhibit leaf chlorosis as a slight vein clearing on the outer leaflets, followed by yellowing and drooping of leaves, then vascular discoloration of the stem, and finally death of the aboveground plant parts. This disease also occurs in Europe in both greenhouse and open-field cultivation (Urrutia Herrada et al. 2004; Altinok 2005). Strategies to control this soil-borne disease have been based, especially in greenhouse cultivation, on soil treatments with methyl bromide, but this compound has officially been phased out in the European Union, and phaseout in Turkey began in 2008 (Pizano and Banks 2008).
Fusarium oxysporum was initially described by Matuo and Ishigami (1958) from a S. melongena (Solanaceae) plant suffering from a vascular wilt disease. In plant inoculation studies, the recovered isolate could not infect other Solanaceae species (including Lycopersicon esculentum, Nicotiana tabacum, Solanum tuberosum, and Capsicum annuum). This forma specialis (f. sp.) was recently reported from Turkey on S. melongena and described by Altinok (2005) as Fusarium oxysporum f. sp. melongenae. The occurrence of F. oxysporum on S. melongena has also been reported in China (Tai 1979), Korea (Cho and Shin 2004), the USSR (Richardson 1990), Fiji (Firman 1972), Kenya (Nattrass 1961), Greece (Holevas et al. 2000), Italy (Richardson 1990), the Netherlands (Steekelenburg 1976), and the United States (Alfieri et al. 1994). A high degree of variability within F. oxysporum has been associated with the complexity of the species (Baayen et al. 2000). Molecular methods such as polymerase chain reaction, inter-simple sequence repeat (ISSR), and randomly amplified polymorphic DNA (RAPD) primers are now widely used in the study of fungal taxonomy (Lamboy 1994; Dubey and Singh 2008). Analyses of the relationship of S. melongenae to other forms and formae speciales within the F. oxysporum species, and information on variation within formae speciales, are not available; the pathogen is consistently cited as a forma specialis of FOM (e.g., Altinok 2005; Mutlu et al. 2008), though Cline (2009) has stated that this designation is incorrect.
A rapid and reliable method of evaluating pathogen virulence is important if eggplant breeders are to develop resistant cultivars and effectively control disease. Characterization of pathogenic variation in a pathogen population is time-consuming and does not provide information on the genetic relatedness of the evaluated strain. Cultivar resistance varies by location, so knowledge of the genetic variability of pathogen populations is required for eggplant-growing areas.
The aim of this study was to determine specific ISSR and RAPD primers and to evaluate genetic diversity of the pathogen and investigate whether any correlation existed between its genetic diversity and its virulence.
Materials and Methods
Plant Material
Five-week-old eggplant seedlings (cv. “Pala”) with four fully expanded leaves were used for all experiments. Plants were grown in pots in a soil mix containing sand, perlite, and peat compost (1:2:3) in a greenhouse at 25 ± 5°C with 68–80% relative humidity. The soil mix also contained a slow-release fertilizer (14–12–14, N–P–K). Light was supplemented by a single 1,000 W sodium vapor lamp during a 16 h photoperiod.
Pathogen Used in Experiments and Pure Cultures
Although 50 isolates of different soil-borne pathogens were collected during surveys carried out in this Mediterranean region, only 10 were identified as F. oxysporum (Table 1, Fig. 1). Pathogen isolates were tested on susceptible seedlings of eggplant (Solanum melongena cv. “Pala”) at the six-leaf stage. Wounded roots were submerged for 10 min in a conidial suspension (5 × 106 conidia per ml in sterile H2O), while control plants were dipped in sterile tap water. All of the tested isolates were pathogenic to eggplant. Symptoms observed on inoculated plants were similar to those observed in climatic chambers after inoculating the plants with a reference isolate obtained from Italy. The test plants showed leaf chlorosis and necrosis and later died. Control plants had no symptoms, and two other members of Solanaceae (Solanum lycopersicum, Capsicum annuum) tested with the same method displayed no symptoms.

Region of intensive eggplant production in southwestern Turkey, where the pathogens were collected. Collection sites are listed in Table 1
Fusarium oxysporum was successfully reisolated from the stems of the inoculated eggplants in order to satisfy Koch’s postulates (according to Altinok 2005). Reisolation was done from the discolored stem vascular tissue. Necrotic tissue fragments were surface-sterilized (2% NaOCl) and plated on potato dextrose agar (PDA). Fusariumoxysporum was the only fungus isolated, and it grew from most fragments.
Culture characteristics and micromorphology were investigated using PDA and carnation leaf agar (Leslie and Summerell 2006). Pathogenicity and virulence tests were performed twice for each isolate using a root-dip inoculation method (modified from Biles and Martyn 1989) with pot experiments in a controlled greenhouse at 25 ± 5°C with 68–80% relative humidity. The level of resistance induced in seedlings against FOM was evaluated 30 days after inoculation using a 0–3 arbitrary scale according to Zink and Gubber (1986). A mean disease severity index (DSI) was calculated from each treatment by adding the ratings for 30 plants (two replicates of 15 plants for each isolate) and expressing the value as a percentage using the following formula: DSI = [∑(rating no. × no. of plants in rating) × 100]/(total no. of plants × highest rating)]. Least significant difference (LSD) analysis was conducted on DSI values obtained from four replications.
Molecular Studies: Pathogen Identification by PF02-3 Primer
Fusarium oxysporum isolates were confirmed by amplification using a specific primer, PF02-3 (Edel et al. 2000). A reference FOM isolate obtained from Dr. G. L. Rotino (Research Institute for Vegetable Crops, Montanaso Lombardo, Italy) was used as a positive control in PCR amplifications.
Genomic DNA was extracted from mycelium as described by Lodhi et al. (1994). The mycelia were collected and ground in a mortar with liquid nitrogen. DNA was quantified by measuring OD260 as described by Sambrook et al. (1989). Each amplification was performed in a 25 μl reaction volume containing 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 800 μM dNTP (dATP:dTTP:dCTP:dGTP in a 1:1:1:1 ratio), 0.25 μM primer PF02-3, 1 U Platinum Taq polymerase (Invitrogen, Life Technologies), and 15 ng template DNA. Amplification was performed under the following cycle program: initial denaturation step for 5 min at 94°C, followed by 35 cycles at 94°C for 60 s (denaturation), 58.5°C for 30 s (annealing), and 72°C for 60 s (extension), followed by a final extension step at 72°C for 6 min.
ISSR and RAPD Analysis
Six ISSR primers and two RAPD primers were used in PCR reactions. The ISSR primers were (ACC)6CC, CCA(TGA)5TG, CCA(TG)7T, GCA(AC)7, (CA)8RG, and (AG)8YT[15], provided by Dr. S. Lucretti of the Biotechnology and Agriculture Division, ENEA C.R. Casaccia, Rome; and the RAPD primers were OPI09 (Jimenez-Gasco et al. 2001) and P5 (Vakalounakis and Fragkiadakis 1999). Primers were purchased from MWG Biotech AG, Ebersberg, Germany.
Each amplification with ISSR primers was performed in a 25 μl reaction volume containing 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 2 mM MgCl2, 800 μM dNTP (dATP:dTTP:dCTP:dGTP in a 1:1:1:1 ratio), 0.5 μM of each primer, 1 U Platinum Taq polymerase (Invitrogen, Life Technologies), and 15 ng template DNA. The amplification was performed in a 96-well GeneAmp PCR System 9700 thermocycler (Applied Biosystems) equipped with a Hot Bonnet under the following cycle program: initial denaturation step for 4 min at 94°C, followed by 36 cycles at 94°C for 30 s (denaturation), 46–56°C for 45 s (annealing), and 72°C for 120 s (extension), followed by a final extension step at 72°C for 7 min. DNA amplification reactions with RAPD primers were performed in a 25 μl reaction volume containing 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 3 mM MgCl2, 800 μM dNTP (dATP:dTTP:dCTP:dGTP in a 1:1:1:1 ratio), 0.4 μM of each primer, 1 U Platinum Taq polymerase, and 15 ng template DNA. The amplification was performed in a 98-well GeneAmp PCR System 9700 thermocycler equipped with a Hot Bonnet under the following cycle program: initial denaturation step for 90 s at 94°C, followed by 40 cycles at 94°C for 1 min (denaturation), 35°C for 2 min (annealing), and 72°C for 2 min (extension), followed by a final extension step at 72°C for 10 min. PCR-amplified DNA fragments were visualized as described above. PCR-amplified DNA fragments were separated on a 1.5% agarose gel containing 1× TBE (45 mM Tris–borate, 1 mM EDTA) and 0.5 μg/ml aqueous solution of ethidium bromide. About 25 μl of the reaction products with 5 μl of loading buffer (0.25% bromophenol blue, 40% w/v sucrose) was loaded per well, and the gel was run for 6 h at 100 V. The gel was then visualized with a UV transilluminator at 300 nm. To confirm the reproducibility of the banding patterns, the PCR experiments were repeated twice.
Data Analysis
Amplified bands from each primer were scored as present (1) or absent (0). Only those bands showing consistently were considered; smeared and weak bands were excluded from the analysis. Dice’s coefficient of similarity (D ij ) was determined between each pair of strains. Dice’s coefficient has been recommended for the evaluation of genetic similarities when using RAPD data (Lamboy 1994). Genetic dissimilarity (GD) was calculated as GD = 1 − D ij . The estimates of similarity between strains were used for cluster analysis by the unweighted pair group method with arithmetic average (UPGMA; Sneath and Sokal 1973) using the NTsys package version 2.02 for Windows (Rohlf 1994). A principal coordinate analysis (PCA) was also carried out, and a two-dimensional point diagram was obtained. Additional statistics were computed to estimate the grade of polymorphism among strains studied. The allele frequency (p i) for each primer and among all primers was calculated for each sample and among all samples as p i = 1 − √(1 − F b), where F b was the band frequency. The percentage of polymorphism (Pp) was given as the number of polymorphic loci per number of total loci, regardless of allele frequencies. Loci and total genetic diversity (h and H t, respectively; Siegfried 2000) and Shannon’s index (S; Gryta et al. 2005) were used to summarize the data for molecular markers and their standard deviations. All calculations and analyses were conducted using Popgene version 1.31 (Yeh et al. 1999). The Pearson product-moment correlation coefficient (r) was calculated between the genetic and virulence distance matrices. Virulence distances (VD ij ) between strains i and j were calculated as VD = |DS i − DS j |, where DS is disease severity.
To estimate species fingerprinting potential of the primers used, the number of identified haplotypes was calculated for each primer, and the differentiation coefficient (δ) (Gregorius 1987) was measured as δ = (1 – ∑p 2i )[N/(N − 1)], where p i was the haplotype frequency and N was the total number of samples.
Results
In Vivo Studies
Fusarium oxysporum isolates from inoculated plants showed characteristic three- to five-septate, sickle-shaped macroconidia, with a foot-shaped basal cell, ellipsoid microconidia borne in false heads or on short monophialides, and chlamydospores were visible in culture. A typical cream-colored colony developed on PDA, with purple pigmentation on the reverse side (Leslie and Summerell 2006). The characteristic morphological properties of fungal isolates were compared with a reference culture of FOM. Afterward, pot experiments were conducted to compare the virulence of isolates. In experiments carried out under controlled conditions, the most virulent isolates 30 days after inoculation were F9, F17, F21, and F11 (Table 2).
Molecular Studies
PCR amplification primed with the PF02-03 primer showed that these isolates were F. oxysporum, because of common bands amplified in the reference isolate (Fig. 2). The specific, expected band was visible at around 70 bp.

PCR results for 10 FOM isolates by amplification with PF02-3 primer specific for Fusarium oxysporum. The amplification provided confirmation of the isolates as F. oxysporum, in addition to morphological characterization. The first sample on the gel is the reference positive control isolate obtained from Italy
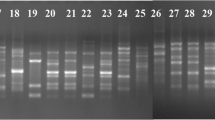
Six ISSR and two RAPD primers were used to evaluate 10 Turkish F. oxysporum samples, and 113 well-resolved bands were observed. The amplified fragments ranged in size from 200 bp [primer OPI09 and (CA)8RG] to 2.5 Kb [primer (ACC)6CC, (AG)8YT, and P5]. The number of ISSR bands obtained with each primer varied from 6 [primer GCA(AC)7] to 23 [primer CCA(TGA)5TG] (Fig. 3), with an average of 14 bands per primer. Of the 113 amplified bands, 77 (Pp = 85%) were polymorphic. Of the screened primers used to analyze isolates, the most polymorphic loci amplified were obtained from the ISSR primers CCA(TGA)5TG and (ACC)6CC (100 and 95%, respectively), and the least were from ISSR primers GCA(AC)7 and (CA)8RG (67 and 63%, respectively). Primer CCA(TGA)5TG had the highest number of polymorphic loci (according to Lodhi et al. 1994), and higher values were obtained for its 500 bp (h = 0.5) and 1,350 bp (h = 0.5) amplified bands (Fig. 3).

ISSR profiles amplified from DNA of FOM isolates analyzed using primer CCA(TGA)5TG. M, 123 bp DNA ladder
High values of the differentiation coefficient were observed for all primers, with an average value of 0.93. ISSR primer CCA(TGA)5TG was able to discriminate all strains (δ = 1). Minimum and maximum band size, number of total amplified bands, percentage of polymorphic loci, the number of haplotypes, and the differentiation coefficient of the primers indicate high diversity among the studied strains (Table 3).
The polymorphisms identified were used to generate a genetic dissimilarity and similarity matrix (Table 4). The highest genetic dissimilarity value was identified between strains E16 and F11 (0.591), whereas the lowest value was identified between strains 683 and 719 (0.106). A high genetic dissimilarity average estimate was calculated (0.358 ± 0.124).
Dice’s similarity measure was used to carry out a cluster analysis and to generate a dendrogram representing the relationships among selected strains (Fig. 4). This dendrogram contained four main branching nodes, with all strains well separated. At node 1, strain E16 appeared strongly separated from other strains (D E16 = 0.53). At node 2, all strains could be clearly assigned into two different groups (1 and 2) with a genetic similarity of 0.60. Group 1 appeared to divide at node 3 into two subgroups containing 683, 719, E18, and F17 strains (subgroup 1A) and E7 and E8 strains (subgroup 1B). At node 4, group 2 showed F9 and F11 strains to be more tightly clustered than with F21 (D (F21 – F9/F11) = 0.64). However, genetic differences cannot be linked to geographic distances among strains.

UPGMA dendrogram of the analyzed FOM isolates obtained from Dice’s coefficient similarity matrix using ISSR and RAPD marker data
Similar diversity relationships were obtained with PCA. The point diagram obtained (Fig. 5) showed a clustering of six strains (683, 719, E18, F17, E7, and E8) separated from strain E16 and from another separate cluster including strains F21, F9, and F11. The first dimension was able to discriminate 68.5% of differences, while the second discriminated 8.46% (cumulative percentage 76.96). Total Nei’s genetic diversity (H t) was 0.28 ± 0.03. An additional measure of genetic variation was obtained using Shannon’s index (S), a genetic diversity index suitable when dominant markers are used. The S value was 0.42 ± 0.22 for all the analyzed strains. Both values were very high, indicating a high level of genetic diversity among the analyzed strains.

Principal coordinate analysis of FOM isolates obtained from Dice’s coefficient similarity matrix using ISSR and RAPD marker data
These results were compared with greenhouse experiments that followed pathogen invasion on plants. There was a strong correlation between symptom development and pathogen discrimination, according to UPGMA analysis. In fact, the cluster of strains F9, F11, and F21 showed very high disease severity values (100, 85, and 94%, respectively). The only strain that had high disease severity (100%) and was grouped with the other strains was F17. A high Pearson product-moment correlation coefficient was obtained (r = 0.57) by comparing the genetic and virulence distance matrices. In addition, the primers employed led to discrimination of pathogens based on virulence observed in experiments in vivo.
Discussion
This study indicates that the RAPD and ISSR primers tested can be used to evaluate and discriminate FOM virulence. These primers can also be used to map pathogens and their distribution in problematic eggplant production fields since they are able to make distinctions within a small number of types isolated from different districts. Our study shows that the markers we employed can be successful tools for determining F. oxysporum f. melongenae virulence, not just in Turkey but also in other countries where fusarium wilt disease causes severe problems in eggplant cultivation areas. In addition, the molecular markers tested in this study can be powerful tools in characterizing population structure. Pathogen virulence corresponds to findings we obtained both in pot experiments and in molecular studies. Molecular markers have been used to compare genetic relationships with pathogenicity in many groups of fungi (O’Donnell et al. 1999; Waalwijk et al. 1996; Baayen and Gams 1998). Although amplified fragment length polymorphisms (AFLPs) are used to characterize Fusarium species (Voigt et al. 1995), no studies have characterized eggplant F. oxysporum species using ISSR and RAPD. AFLP has also been used to differentiate pathogenic isolates from nonpathogenic F. oxysporum (Baayen et al. 2000), and the present study, through its use of molecular markers, will assist in further studies of such previously reported Fusarium species. Genetic differences between pathogenic and nonpathogenic strains of F. oxysporum determined on conifers were previously examined using the gene encoding histone H3, and a few genetic differences between high- and low-virulence or nonpathogenic isolates were found (Chiocchetti et al. 1999). Previous results obtained on F. oxysporum f. sp. ciceris suggested that molecular genetic techniques, including ISSR and RAPD, could be used to separate phenotypes (Bayraktar et al. 2007). Therefore, ISSR and RAPD primers can be used to compare the phylogenetic relationships of virulent FOM isolates on eggplant within the F. oxysporum species complex, as shown in our study. Since our study focuses on Turkey, it might be worthwhile to indicate the geographic sources of the strains/isolates of F. oxysporum. Even though we collected more than 50 isolates from different problematic fields in several regional districts in Turkey, only 10 showed the properties of F. oxysporum in our analysis. These strains can be considered representative of the population, according to the survey we carried out in the region. This number of isolates can be assessed as insufficient, however. A few previous studies also involved small numbers of pathogens, but those studies compared pathogen isolates collected in different states or on different continents (Mes et al. 1999; Cai et al. 2003). Finally, each band in the ISSR data is interpreted as representing a separate locus. Within a strain this conclusion is sound. However, comparisons between strains are more difficult; these loci may have multiple alleles, and the simple presence/absence analysis of the bands can be insufficient, since bands of different sizes may be allelic. However, these techniques and tools can also determine if these populations are clonal, which may aid in disease management. Development of a genetic marker system for distinguishing between virulent and nonpathogenic isolates would be useful for managing fusarium wilt disease in eggplant seedlings and other crops (O’Donnell et al. 1999). Even if conventional methods are preferred as an easier way to compare results, sensitive molecular markers such as those obtained through ISSR or RAPD could allow for more cost-effective detection and quantification of pathogens than can be obtained using AFLP (Dubey and Singh 2008). The techniques employed in the present study can also be used to improve plant-resistant varieties in breeding programs by assisting in the selection of the most virulent strains of pathogens when screening resistant varieties. Population levels and dissemination of highly virulent isolates could be estimated for different locations, and thereby prediction of disease potential for future growing seasons could be improved.
We believe that this study has achieved significant progress toward developing simple and effective molecular markers for detecting and quantifying virulent pathogen populations within the cultivation of eggplant. In further studies, the amplified fragments that permitted discrimination will be investigated by sequence analysis and comparison with known databases on genes conferring pathogen virulence.
References
Alfieri SA Jr, Langdon KR, Kimbrough JW, El-Gholl NE, Wehburg C (1994) Diseases and disorders of plants in Florida. Florida Department of Agriculture and Consumer Services, Division of Plant Industry. Bulletin 14:1114
Altinok HH (2005) First report of fusarium wilt of eggplant caused by Fusarium oxysporum f. sp. melongenae in Turkey. Plant Pathol 54:577
Baayen RP, Gams W (1998) The Elegans fusaria causing wilt disease of carnation. I. Taxonomy. Netherlands. J Plant Pathol 94:273–278
Baayen RP, O’Donnell K, Bonants PJM, Cigelnik E, Kroon LPNM, Roebroeck EJA, Waalwijk C (2000) Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathology 90:891
Bayraktar H, Dolar FS, Maden S (2007) Use of RAPD and ISSR markers in detection of genetic variation and population structure among Fusarium oxysporum f. sp. ciceris isolates on chickpea in Turkey. J Phytopathol 156:146–154
Biles CL, Martyn RD (1989) Local and systemic resistance induced in watermelons by formae speciales of Fusarium oxysporum. Phytopathology 79:856–860
Cai G, Gale LR, Schneider RW, Kistler HC, Davis RM, Elias KS, Miyao EM (2003) Origin of race 3 of Fusarium oxysporum f. sp. lycopersici at a single site in California. Phytopathology 93:1014–1022
Chiocchetti A, Ghignone S, Minuto A, Gullino ML, Garibaldi A, Migheli Q (1999) Identification of Fusarium oxysporum f. sp. basilici isolated from soil, basil seed, and plants by RAPD analysis. Plant Dis 83:576–581
Cho WD, Shin HD (2004) List of plant diseases in Korea, 4th edn. Korean Society of Plant Pathology, Suwon
Cline E (2009) Fusarium oxysporum f. melongenae. Systematic Mycology and Microbiology Laboratory, ARS, USDA. http://nt.ars-grin.gov/sbmlweb/OnlineResources/FungiOnline.cfm
Dubey SC, Singh SR (2008) Virulence analysis and oligonucleotide fingerprinting to detect diversity among Indian isolates of Fusarium oxysporum f. sp. ciceris causing chickpea wilt. Mycopathologia 165:389–406
Edel V, Steinberg C, Gautheron N, Alabouvette C (2000) Ribosomal DNA-targeted oligonucleotide probe and PCR assay specific for Fusarium oxysporum. Mycol Res 104:518–526
Firman ID (1972) A list of fungi and plant parasitic bacteria, viruses and nematodes in Fiji. Phytopathol Pap 15:1–36
Gregorius HR (1987) Selection in plant populations of effectively infinite size. VI. Overlapping generations. Theor Popul Biol 31:75–90
Gryta H, Carriconde F, Charcosset JY, Jargeat P, Gardes M (2005) Population dynamics of the ectomycorrhizal fungal species Tricholoma populinum and Tricholoma scalpturatum associated with black poplar under differing environmental conditions. Environ Microbiol 8:773–786
Holevas CD, Chitzanidis A, Pappas AC, Tzamos EC, Elena K, Psallidas PG, Alivizatos AS, Panagopoulos CG, Kyriakopoulou PE, Bem FP, Lascaris DN, Velissariou DE, Vloutoglou I, Analytis SC, Paplomatas EJ, Aspromougos JS, Varveri C (2000) Disease agents of cultivated plants observed in Greece from 1981 to 1990. Benaki Phytopathol Inst, Kifissia, Athens 19:1–96
Jimenez-Gasco MM, Perez-Artes E, Jimenez-Diaz RM (2001) Identification of pathogenic races 0, 1B/C, 5, and 6 of Fusarium oxysporum f. sp. ciceris with random amplified polymorphic DNA (RAPD). Eur J Plant Pathol 107:237–248
Lamboy WF (1994) Computing genetic similarity coefficients from RAPD data: the effects of PCR artifacts. Genome Res 4:31–37
Leslie JF, Summerell BA (2006) Fusarium laboratory workshops: a recent history. Mycotoxin Res 22:73–74
Lodhi MA, Ye GN, Weeden NF, Reisch BI (1994) A simple and efficient method for DNA extraction from grapevine cultivars, Vitis species and Ampelopsis. Plant Mol Biol Rep 12:6–13
Matuo T, Ishigami K (1958) On the wilt of Solanum melongena L. and its causal fungus Fusarium oxysporum f. melongenae n. f. Ann Phytopathol Soc Jpn 23:189–192
Mes JJ, Weststeijn EA, Herlaar F, Lambalk JJM, Wijbrandi J, Haring MA, Cornelissen BJC (1999) Biological and molecular characterization of Fusarium oxysporum f. sp. lycopersici divides race 1 isolates into separate virulence groups. Phytopathology 89:156–160
Mutlu N, Boyacı FH, Göçmen M, Abak K (2008) Development of SRAP, SRAP-RGA, RAPD and SCAR markers linked with a fusarium wilt resistance gene in eggplant. Theor Appl Genet 117:1303–1312
Nattrass RM (1961) Host lists of Kenya fungi and bacteria. Mycol Pap 81:1–46
O’Donnell K, Gherbawy Y, Schweigkofler W, Adler A, Prillinger H (1999) Phylogenetic analyses of DNA sequence and RAPD data compared in Fusarium oxysporum and related species from maize. J Phytopathol 147:445–452
Pizano M, Banks J (eds) (2008) Phasing out methyl bromide in Turkey. Final report
Richardson MJ (1990) An annotated list of seed-borne diseases, 4th edn. International Seed Testing Association, Zurich
Rohlf FJ (1994) NTsys: Numerical taxonomy and multivariate analysis system, version 2.02. State University of New York, Stony Brook
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Siegfried LK (2000) Accurate gene diversity estimates from amplified fragment length polymorphism (AFLP) markers. Mol Ecol 9:1241–1245
Sneath PH, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. W. H. Freeman, San Francisco
Steekelenburg NAM (1976) Fusarium wilt of eggplant in the Netherlands. Neth J Plant Pathol 82:191–192
Tai FL (1979) Sylloge fungorum sinicorum. Science Press, Academia Sinica, Peking
Urrutia Herrada MT, Gómez Garcia VM, Marquinaa TJ (2004) La fusariosis vascular de la berenjena en Almería. Boletín de Sanidad Vegetal, Plagas 30:85–92
Vakalounakis DJ, Fragkiadakis GA (1999) Genetic diversity of Fusarium oxysporum isolates from cucumber: Differentiation by pathogenicity, vegetative compatibility, and RAPD fingerprinting. Phytopathology 89:161–168
Voigt K, Schleier S, Bruckner B (1995) Genetic variability in Gibberella fujikuroi and some related species of the genus Fusarium based on random amplification of polymorphic DNA (RAPD). Curr Genet 27:528–535
Waalwijk C, Baayen RP, De Koning JRA, Gams W (1996) Ribosomal DNA analyses challenge the status of Fusarium sections Liseola and Elegans. Sydowia 48:90–104
Yeh FC, Yang RC, Boyle T (1999) Popgene, Version 1.31. Centre for International Forestry Research and University of Alberta. Edmonton, Alberta, Canada
Zink FW, Gubber WD (1986) Inheritance of resistance to races 0 and 2 of Fusarium oxysporum f sp. melonis of gynoecious muskmelon. Plant Dis 70:676–678
Acknowledgments
The authors wish to thank Dr. Francesco Mercati and Dr. Paolo Ruisi for their assistance with molecular studies carried out at the CNR Institute of Plant Genetics; Palermo, Italy; student F. Akay in the BATEM Molecular Biology Laboratory; technical assistant N. Karatekin for helping with studies carried out in climatic rooms; and Dr. F. Boyacı of BATEM for providing the Fusarium oxysporum f. melongenae reference isolate obtained from Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baysal, Ö., Siragusa, M., Gümrükcü, E. et al. Molecular Characterization of Fusarium oxysporum f. melongenae by ISSR and RAPD Markers on Eggplant. Biochem Genet 48, 524–537 (2010). https://doi.org/10.1007/s10528-010-9336-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-010-9336-1