
Abstract
Lentil (Lens culinaris Medik.) is an important widely cultivated food legume crop in Iran. Wilt disease caused by Fusarium oxysporum f.sp. lentis has been observed with high losses in some of cultivated areas during recent years. Simple sequence repeat (SSR) were used to determine the genetic structure, and estimate genetic diversity in 101 of Fusarium oxysporum f. sp. lentis (FOL) isolates from five counties in Ilam provinces of western Iran (Sarableh, Sirvan, Ivan, Badreh, Darehshahr). A set of five microsatellite primer pairs revealed a total of 10 alleles each locus across the five populations; the number of alleles varied from 6 to 9 for each population. A low level of genetic variability was observed among FOL isolates in the region. Genetic diversity was low (H = 0.234) within populations with corresponding high average gene flow and low genetic distances between populations. The smallest genetic distance was observed between isolates from Sirvan and Badreh. Observed allele number (Na) and effective numbers (Ne) of alleles were higher in Ivan in comparison to other populations. The number of (H) and (I) were also higher in Ivan (H = 0.301; I = 0.442). The total gene diversity (Ht) and gene diversities between subpopulation (Hs) were estimated 0.267 and 0.233, respectively. Gene diversity attributable to differentiation among populations (Gst) was 0.126, while gene flow (Nm) was 3.439. Cluster analysis based on UPGMA showed the lowest genetic distance between Sirvan and Badrah, than between Sarableh and Sirvan. The dendrogram indicated a clear break between the population from Darehshahr and the four remaining populations. Results from this study will be useful in breeding for Crown and root rot resistant cultivars and developing necessary control measures.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lentil (Lens culinaris Medikus sub sp. culinaris) was among the first crops domesticated and has become an important food legume crop in the farming and food systems of many countries globally (Sarker and Erskine 2006). Vascular wilt caused by Fusarium oxysporum Schlecht. Emend. Snyder and Hansen 1940 f. sp. lentis Vasudeva and Srinivasan (FOL) is one of the most important biological constraints to productivity of lentil worldwide except Australia (Bhalla et al. 1992; Beniwal et al. 1993; Tosi and Cappelli 2001). General symptoms at the seedling stage include seed rot and sudden drooping more like wilting and damping off (Khare 1980). The disease may cause complete crop loss under favorable conditions, and can be the major limiting factor for lentil cultivation in certain areas (Chaudhary and Amarjit 2002). Identification of Fusarium according to morphological and pathological characteristics is time consuming, labor intensive and subject to varying environmental or cultural growth conditions during the experiments (Woo et al. 1996). Molecular biology techniques such as RAPD-PCR and SSR overcome all limitations and provide additional information for fungal characterization (Ouellet and Seifert 1993). Molecular techniques based on the polymerase chain reaction (PCR) have been used as a tool in genetic mapping, molecular taxonomy, evolutionary studies, and diagnosis of several fungal species (Williams et al. 1990; Clulow et al. 1991; Nasir and Hoppe 1991; Welsh et al. 1991 and McDonald 1997). F. oxysporum f. sp. lentis (FOL) is a soil borne pathogen; chlamydospores can survive either dormant form or saprophytic in soil for several years without a suitable host (Yadav et al. 2007). FOL isolates exhibit great variability in morphology and aggressiveness (Abbas 1995; Belabid et al. 2004). Control of FOL infection in the field is difficult because the pathogen can survive for a long time in the form of mycelium in infected plant debris or in the form of chlamydospores in soil (Haware et al. 1996; Agrios 1997). Fusarium wilt disease can be managed by the use of resistance cultivars (Jalali and Chand 1992). Knowledge of the amount and distribution of genetic diversity between and within pathogen populations is a prerequisite for the development of resistant cultivars. Identification of diversity by morphological characters is highly variable in Fusarium isolates, these characters are influenced by cultural conditions. In recent years, numerous DNA based methods have been increasingly used to study variability in pathogenic Fusarium population (Kiprop et al. 2002; Sivaramakrishnan et al. 2002a, b; Belabid et al. 2004). SSRs provides a powerful tool for taxonomic and population genetic studies (Britz et al. 2002). Alleles vary according to the number of repeat units present but other mutations have also been shown to be responsible for allele length variation in SSR analysis (Burgess et al. 2001; Slippers et al. 2004b). SSRs have also been used in other fungal species including Sclerotinia sclerotiorum (Sirjusingh and Kohn 2001), Rhizoctonia. solani (Mwang Òmbe et al. 2007) and Ascochyta rabiei (Nourollahi et al. 2011) because of the high resolution that they provide (Jarne and Lagoda 1996; Barnes et al. 2005; Enjalbert et al. 2005). Numerous researchers have already worked on molecular variation of different F. oxysporum formae specials (Baayen et al. 2000; O’Donnell 2000; Abd-Elsalam et al. 2004; Bogale et al. 2006; Groenewald et al. 2006; Stewart et al. 2006). ISSR marker has been used for determining of genetic variations between several populations of F. oxysporum f. sp. cicer (Bayraktar and Dolar 2008). SSR markers distinguished the four races of F. oxysporum ciceri causing varied levels of wilting with differential host cultivars (Barve et al. 2001). Bogale et al. (2005) showed that polymorphism revealed with 8 SSR markers should be sufficient for study of genetic diversity in F. oxysporum complex. Most studies of F. oxysporum have focused on the plant pathogenic isolates (Mohammadi et al. 2004; Pasquali et al. 2003). Fungi with a high level of genetic variation can be difficult to control, they adapt more quickly to any control measure, such as resistant cultivar. Therefor knowledge of the genetic diversity of FOL has contributed to the development of disease control strategies (Kistler 2001), this study was conducted to assess the genetic diversity of Iranian FOL isolates in lentil farms using SSR marker.
Materials and methods
Fungal isolates and pathogenicity test
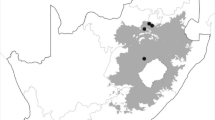
Lentil plants with wilt symptoms and brown or black lesions on root were randomly collected in 2012–13 from five different regions in Ilam province of the western Iran. For the purposes of this study, isolates of each region was considered as a population (Fig. 1; Table 1). The regions are different in altitude and climate, and separated by substantial mountain ranges. Diseased samples were cut into 2- to 5-mm-long pieces, surface sterilized by dipping into domestic bleach solution (5% NaOCl) for 2–3 min, washed three times with sterile distilled water, dried with sterile filter paper and plated on potato dextrose agar (PDA). Samples were incubated for three days in an incubator at 25 °C with a 12-h photoperiod to induce production of conidia. The fungus was isolated and purified using the hyphal tip and single spored method (Hawker 1950). One hundred one FOL isolates were initially identified according to their morphological and microscopic characters as described by (Jens et al. 1991; Nelson et al. 1983; Barnett and Hunter 1972; Leslie et al. 2006). Identified isolates were stored for short time on SNA at 4 °C, but for perennial time stored in tube containing sand at 4 °C. Detail of the FOL isolates are presented in Table 1. The pathogenicity of FOL isolates were tested in the greenhouse on wilt susceptible local cultivar artificially inoculated. Lentil plants were grown in pots containing sand-maize meal and 10% w/w fungus inoculums (Miller 1946). Control plants were grown in a comparable mixture of non-infested sand- maize meal medium with autoclaved soil.

Geographical origins of the five FOL populations in western Iran used in this study
DNA extraction and SSRs analyses
To obtain the mycelia mass, liquid cultures were initiated by adding 2–4 mm 2 pieces of filter paper to 250-mL Erlenmeyer flasks containing 100 mL PDB medium (potato dextrose broth). Flasks were incubated at room temperature approximately 25 °C on a rotary shaker for 6–8 days. Mycelium was collected by filtration through sterile filter paper with a vacuum funnel. Mycelia were harvested, frozen and stored at −20 °C. DNA was extracted using a modified hexadecyl trimethyl-ammonium bromide (CTAB) procedure (Doyle and Doyle 1987). Genomic DNA was isolated from single spore culture of each isolates (Murray and Thompson 1980). Mycelia were ground in liquid nitrogen and suspended in 2% CTAB extraction buffer (1.4 M NaCl, 0.1 M Tris-HCl, pH 8.0, 20 mM EDTA, 0.2% β-mercaptoethanol). Samples were treated with 5 units RNAse at 37 °C for 30 min., and then extracted with chloroformisoamyl alcohol 24:1 (v/v). DNA in the supernatant was precipitated with isopropanol, rinsed with ethanol, and adjusted to a final concentration of 20 ng/μl in TE (pH 7.4). The quality of the extracted DNA was visually checked on 0.8% agarose gels.
A set of five locus-specific primers pair for SSRs (Table 2) described by (Bogale et al. 2005) were selected. Primer aliquots for each marker were prepared by mixing equimolar amounts of appropriate forward and reverse primer in 1× TE (1 mM EDTA, 10 mM Tris–HCl, pH 8.0) and used for the amplification of individual microsatellite loci. PCR amplification was performed in a 25 μl reaction volume containing 2.5 μl of 10X PCR Buffer, 1.5 mM MgCl2, 0.2 mM of dNTPs mix (100 mM of each dNTPs), 1 μl of each forward and reversed primer, 0.6 U of Taq polymerase with 25 ng of template DNA. Amplification was performed using Biometra thermal cycler (USA), PCR conditions for SSR were as Follows; the PCR programmed had one initial denaturation step at 94 °C for 4 min Followed by 35 cycles of 94 °C for 30 s, annealing for 30 s (appropriate annealing temperature were used for each primers set, Table 2) and 72 °C for 5 min. The thermal cycles were terminated by a final extension of 10 min at 72 °C. Amplified products were resolved in 2.0% agarose gel at 60 V cm-1 using in Tris Boric Acid EDTA (1X TBE) buffer and stained with DNA Safe Stain at 0.5 mg ml- and photographed under UV Trans laminator with Gel Doc. Intas. A 1kbp ladder (Gene Ruler TM, Fermantas,) was used as a molecular size standard.
Molecular analysis of the data
Data analyses of populations were defined according to the geographic locations. The bands generated by SSR primers that were repeatable and clearly visible with a high intensity were scored manually for the presence (1) or absence (0) of bands in each isolate. The pair-wise distance among the isolates was calculated from the binary matrix using the simple mismatch coefficient (Sneath and Sokal 1973) that is recommended for haploid fungi (Kosman and Leonard 2005). Genetic similarity between pairs was estimated using Jaccard’s similarity coefficient. Similarity coefficients were used for the construction of UPGMA (Unweighted Pair Group Method with Arithmetic Average) dendrogram (Rolhf 1990). For each primer pair, the polymorphic information content (PIC), marker index (MI) was calculated. The polymorphic information content (PIC) was calculated using PICi = 2fi (1-fi), where i is the information of marker I, fi is the frequency of the amplified allele (presence of fragments) and (1 – fi) is the frequency of the null alleles (Roldan-Ruiz et al. 2000). The genetic variation was measured in terms of genetic diversity and was computed by averaging PIC estimates over all loci (Nei and Li 1979). The marker index (MI) was calculated by MI = PIC x EMR, where EMR is the “effective multiplex relationship” given by the product of the total number of fragments per primer (n) and the fraction of polymorphic fragments (β) (Varshney et al. 2007).
Genotypic diversity (H) among isolates was estimated from allelic frequencies using the eq. H = 1 - Σ xi2, where, xi is the frequency of the ith allele (Nei 1973). The coefficient of population subdivision (GST) was computed as (Ht – Hs)/Ht, where, Ht is the total genetic diversity and Hs is the average gene diversity over all subgroups (Nei 1973). the allele frequencies at polymorphic loci, the Nm values (effective migration rate), and the genetic identity among populations for characterize genetic variation, observed number of alleles (Na), effective number of alleles (Ne), Nei’s gene diversity (He) and Shannon’s information index (I) were calculated in both origin sites and subspecies levels. Mean values of gene diversity in total populations (Ht), gene diversity between populations (Hs), proportion of gene diversity attributable to differentiation among populations (Gst) and estimate of gene flow from Gst (Nm) were obtained across loci (McDermott and McDonald 1993). Relationships of the populations were estimated from the SSR data using the UPGMA (Unweighted Pair Group Method with Arithmetic Mean), clustering method on the basis of Nei’s (1978) unbiased genetic distance, Based on SSR data, Analysis of molecular variance (AMOVA) tests were performed to assess population variance among and within populations and Principal coordinate analysis (PCA) was performed to evaluate the genetic differences among isolates within populations too, all above calculations were performed using POPGENE ver. 1.31 (Yeh et al. 1999) and Gen Alex ver. 6.5 (Peakall and Smouse 2006).
Results
Distribution of alleles at polymorphic SSR loci
Five pair SSR primers were used for amplification loci of 101 FOL isolates. A total of 28 alleles were produced by SSR primers with an average of 7 alleles in all populations. All isolates amplified a single band ranging from 100 to 1000 bp.
Three microsatellite loci were polymorphic when considered over all isolates. The average of allelic variability per locus was the highest (0.245) in Darehshahr population and the lowest in Sarableh with 0.156 alleles. The allele’s frequency is the highest in Ivan with nine and lowest in Sarableh and Sirvan with six alleles (Table 3).
A summary of the genetic diversity data of five microsatellite loci in populations is given in Table 3. Observed allele number (Na =1.70) and effective numbers (Ne = 1.533) of alleles were higher in Ivan compared to other populations. The number of (H) and (I) were also higher in Ivan (H = 0.301; I = 0.442) but lower values were estimated for Sarableh (H = 0.156; I = 0.252).
Primers information
On basis of microsatellite data, the polymorphic information content (PIC) varied from 0.287 (primer MB13) to 0.491 (primers MB18), with an average of 0.424, which reflects the informative content of the primers used. EMR (effective multiplex relationship) varied from 100 to 300. The marker index (MI), which incorporates the informative content of the marker (PIC), the number of fragments per primer pair and the fraction of polymorphic fragments (EMR), varied from 1.49 (MB11) to 8.96 (MB5) (Table 2).
Genetic variability of populations
The average genetic distance was calculated among the five populations. Nei’s pairwise genetic distances between the populations varied from 0.007 to 0.123. The lowest genetic distance was found between Badreh and Sirvan, while the highest genetic distance was revealed between Sarableh and Darahshahr (Table 4). Cluster analysis (UPGMA) was used to produce a dendrogram showing the genetic relationships between the populations based on the SSR data, the dendrogram showed a distinction between the Darehshahr population and the four remaining populations (Fig. 2).

Dendrogram of genetic relationships between each FOL county constructed by UPGMA
The total gene diversity (Ht) and gene diversities between subpopulations (Hs) were estimated to be 0.267 and 0.233, respectively. Gene diversity attributable to differentiation among populations (Gst) was 0.126, while gene flow (Nm) was 3.439 (data not shown). The genetic similarity varied from 88% to 99%, in isolates with different geographical origins, the most similar isolates were F33 with F36, and F32 with F84. The cophenetic correlation indicated a high level of accuracy between the original data of the genetic distance matrix and the data represented in the dendrogram (data not shown). A dendrogram based on UPGMA analysis showed three major groups containing isolates with different origins. Group I, II and III. Group I, with F43, F44, F45 showing broad molecular differences in relation to those of FOP isolates. Group II consisted of Different isolates such as: F2, 10, 31, 34, 42, 50, 63, 80, 87, 01, 8, 35, 61, 65, 85, 62, 68, 33, 36, 20, 30, 46, 64, 81, 82, 83, 32, 84, 88, 80, 00, 02, 28, 60, 66, 70, 58, 86, 47, 48. Group III contains other isolates except Group I and II.
The AMOVA of genetic variation in FOL populations revealed that 11% of the variance occurred among populations and 89% within populations (Table 5).
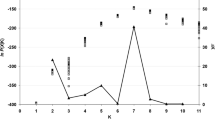
PCA using SSR data showed the genetic differences among isolates within populations and gene flow between different populations (Fig. 3), this suggests the geographical regions play important role for the formation of populations. PCA allows for visualizing the patterns of genetic relationship without altering the data itself and finds patterns within a multidimensional data set.

Principal component analysis (PCA) based on SSR data for 101 individual isolates from five populations in Ilam province. Individuals within the same population are marked using the same symbols. The first and second principal coordinates account for 40.53% and 20.08% of the variation, respectively. There was no clear separation among individuals from different populations, but isolates in the same populations tended to gather together
Discussion
Present study clearly indicated the variation among 101 FOL isolates, collected from different regions. The present work was carried out to explore the possible utilization of SSR technique for identifying genetic structure in FOL isolates too.
The advantage of microsatellite markers over Random Amplified Polymorphic DNA (RAPD) and Restriction Fragment Length Polymorphism (RFLP) markers are their high specificity, high polymorphism, good reproducibility and unambiguous scorability (Tenzer et al. 1999; Saharan and Naef 2008).
In this research SSR markers grouped FOL isolates into three major groups based on their geographical regions as earlier reported by Belabid et al. (2004). Similar studies on other plant pathogenic fungi have emphasized the importance of molecular approaches to characterize genetic diversity within and between isolates (Sivaramakrishnan et al. 2002a, b; Bentley et al. 1995; Belabid et al. 2004). By SSR markers, different allele sizes were recorded in FOL genome, the smallest allele size was detected as 120 bp and the largest one was 1000 bp as reported by Mohammadi et al. (2004). The polymorphic character of SSRs produces highly discriminating fingerprints that often allow characterization of fungi at a strain level (Migheli et al. 1998; Barres et al. 2006). The variable numbers of alleles per loci is an indication of high level of polymorphism and was reported by Mwang Òmbe et al. (2007) in Kenyan R. solani isolates. Polymorphism has been observed in other fungi as a direct record of genetic evolution (Sanders 2002). The results showed that there was a low level of genetic variation among the isolates of FOL populations in Iran. In this study the relationship detected between isolates within the five populations was probably due to exchange of lentil seeds between sampled regions and geographical closeness as well. In this study, Iranian FOL isolates showed a different level of diversity within and between populations in comparison with the international isolates. Similar results were also reported in F. oxysporum f.sp. phaseoli by Woo et al. (1996) and in F. oxysporum f. sp. ciceris by Jimenez-Gasco et al. (2001). Similar findings have also been reported in the Ethiopian F. oxysporum isolates by AFLP, SSR and ITS sequence analysis by Bogale et al. (2006). In Iranian populations, over 89% of the gene diversity was distributed on a local level within populations. However there was a high degree of genetic similarity among populations separated by low geographical distances. The low level of gene diversity (Gst = 0.126) was detected among all five populations. The low Gst value indicated little genetic differentiation among the five populations and showed little evidence for geographical subdivision among populations (Bayraktar et al. 2010).
The genetic distances were very small, indicates that populations linked by movement of conidia through contaminated seeds, infected plant debris as gene flew because the geographic distances between sampling regions are short with range of 50 to 300 km. Gene flow is one of the evolutionary forces that can have a significant force on the genetic diversity of a population. In the absence of gene flow, genetic drift cause developing different allele frequencies at neutral loci, leading to differentiation in isolate populations (Keller et al. 1997). The high genetic similarity among FOL populations suggests that gene flow occurred across long distances. Infected seed can lead to persistence of genotypes; we consider that infected seed can explain the distribution and diversity of genotypes found at the end of the growing season in natural populations. Genetic drift and selection would limit the number of genotypes present in field populations (Shah et al. 1995). In this study genetic characterization of FOL isolates could be essential for the efficient management of Fusarium wilt through use of resistant cultivars in Lentils growing areas. Low FOL genetic diversity in the west of Iran is a warning for breeder to apply the successful use of resistance genes in local disease management.
However, an understanding of occurrence, distribution, and genetic relatedness of such pathogenic variants is a necessary for developing effective and efficient integrated disease management. Quarantine regulations will need to prevent the introduction of more diverse isolates into these populations and prevent transmission any isolates from these area to other regions of the country. Understanding the genetic structure of pathogen populations in the present study may provide insights into the epidemiology and evolutionary potential of pathogens and could lead to developing integrated strategies for disease management and breeding programs.
References
Abbas A (1995) Variation in some cultural and physiological characters and host/pathogen interaction of Fusarium oxysporum f. sp. lentis, and inheritance of resistance to lentil wilt in Syria. Ph.D. Thesis, Faculty of Agriculture, University of Aleppo, Syria
Abd-Elsalam KA, Omar MR, Migueli Q, Nirenberg HI (2004) Genetic characterization of Fusarium oxysporum f. Sp. vasinfectum isolates by random amplification of polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP). J Plant Dis and Protec 111:534–544
Agrios GN (1997) Plant pathology 4th edition. Academic Press Inc, San Diego
Baayen RP, O'Donnell K, Bonants PJM, Cigelnik E, Kroon LPNM, Roebroeck EJA, Waalwijk C (2000) Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathol 90:891–900
Barnes I, Nakabonge G, Roux J, Wingfield BD, Wingfield MJ (2005) Comparison of populations of the wilt pathogen Ceratocystis albifundus in South Africa and Uganda. Plant Pathol 54:189–195
Barnett HL, Hunter BB (1972) Illustrated genera of imperfect fungi. Burgess, Publication Company, p 241
Barres B, Dutech C, Andrieux A, Caron H, Pinon J, Frey P (2006) Isolation and characterization of 15 microsatellite loci in the poplar rust fungus, Melampsora larici-populina, and cross-amplification in related species. Mol Ecol Not 6:60–64
Barve MP, Haware MP, Sainani MN, Ranjekar PK, Gupta VS (2001) Potential of microsatellites to distinguish four races of Fusarium oxysporum f. sp. ciceri prevalent in India. Theo App Gen 102:138–147
Bayraktar H, Dolar FS (2008) Genetic diversity of wilt and root rot pathogens of chickpea. Turk J Ag 33:1–10
Bayraktar H, Türkkan M, Dolar FS (2010) Characterization of Fusarium oxysporum f. sp. cepae from onion in Turkey based on vegetative compatibility and rDNA RFLP analysis. J Phytopathol 10:1439–0434
Belabid L, Baum M, Fortas Z, Bouzand Z, Eujal I (2004) Pathogenic and genetic characterization of Algerian isolates if Fusarium oxysporum f. sp. lentis by RAPD and AFLP analysis. Af J Biotech 3:25–31
Beniwal SPS, Bayaa B, Weigand S, Makkouk K, Saxena MC (1993) Field guide to lentis diseases and insect pests. International Centre for Agricultural Research in the Dry Areas, Aleppo, p. 106
Bentley S, Pegg KG, Dale JL (1995) Genetic variation among a worldwide collection of isolates of Fusarium oxysporum f. sp. cubense analyzed by RAPD-PCR fingerprinting. Mycol Res 99:1378–1384
Bhalla MK, Nozzolillo C, Schneider E (1992) Observation on the responses of lentil root cells to hypha of Fusarium oxysporum. J Phytopathol 135:335–341
Bogale M, Wingfield BD, Wingfield MJ, Steenkamp ET (2005) Simple sequence repeats markers for species in the Fusarium oxysporum complex. Mol Ecol Notes 5:622–624
Bogale M, Wingfield BD, Wingfield M, Steenkamp ET (2006) Characterization of Fusarium oxysporum isolates from Ethiopia using AFLP, SSR and DNA sequence analyses. Fungal Divers 23:51–66
Britz H, Coutinho TA, Wingfield BD, Wingfield MJ (2002) Sequence characterized amplified polymorphic markers for the pitch canker pathogen Fusarium circinatum. Mol Ecol Notes 3:577–580
Burgess T, Wing M, Wing B (2001) Simple sequence repeat markers distinguish among morphotypes of Sphaeropsis sapinea. Appl Environ Microbiol 67:354–362
Chaudhary RG, Amarjit K (2002) Wilt disease as a cause of shift from lentis cultivation in Sangod tehsil of Kota Rajasthan. Ind J Pulse Res 15:193–194
Clulow SA, Lewis BC, Matthews P (1991) A pathotype classification for Mycosphaerella pinodes. J Phytopathol 131:322–332
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Enjalbert J, Duan X, Leconte M, Hovmøller MS, De Vallavielle-Pope C (2005) Genetic evidence of local adaptation of wheat yellow rust (Puccinia striiformis f. sp. tritici) within France. Mol Ecol 14:2065–2073
Groenewald S, Berga NVD, Marasasb WFO, Viljoen A (2006) The application of high-throughput AFLP’s in assessing genetic diversity in Fusarium oxysporum f. sp. cubense. Mycol Res 110:297–305
Haware MP, Nene YL, Natarajan M (1996) Survival of Fusarium oxysporum f. sp. ciceri. Plant Dis 66:809–810
Hawker LE (1950) Physiology of fungi. University of London Press Ltd, London
Jalali BL, Chand H (1992). Disease of cereals and pulses Chickpea wilt Plant disease of International importance Vol I. In: Singh US, Mukhtopadhyay AN, Kumar J, Chaube HS (eds) Prentice Hall Englewood, Cliff NJ pp 420–444
Jarne P, Lagoda PJL (1996) Microsatellites from molecules to populations and back. Trend Ecol Evol 11:424–429
Jens CF, Thrane V, Mathur SB (1991) An illustrated manual on identification of some seed-borne Aspergilli Fusaria Penicillia and their mycotoxins. Danish government institute of seed pathology for developing countries. Ryvans Alle 78 DK 2900 Hellerue Denmark
Jimenez-Gasco MM, Perez-Artes E, Jimenez-Diaz RM (2001) Identification of pathogenic races 0, 1B/C, 5 and 6 of Fusarium oxysporum f. Sp. ciceri with random amplified polymorphic DNA (RAPD). Eur J Plant Pathol 107:237–248
Keller SM, McDermott JM, Pettway RE, Wolfe MS, McDonald BA (1997) Gene flow and sexual reproduction in the wheat glume blotch pathogen Phaeosphaeria nodorum (anamorph Stagonosporanodorum). Phytopathol 87:353-358
Khare MN (1980) Wilt of lentis. Jawaharlal Nehru Krishi Vishwa Vidyalaya Jabalpur MP India, p 155
Kiprop EK, Baudoin JP, Mwang’ombe AW, Kimani PM, Mergeai G, Maquet A (2002) Characterization of Kenyan isolates of Fusarium udum from pigeon pea [Cajanus cajan (L) Millsp] by cultural characteristics aggressiveness and AFLP analysis. J Phytopathol 150:517–525
Kistler HC (2001) Evolution in host specificity in Fusarium oxysporum. In: Summerell BA, Leslie JF, Backhouse D, Bryden WL, Burgess LW (eds) Fusarium. APS Press, St Paul
Kosman E, Leonard J (2005) Similarity coefficients for molecular markers in studies of genetic relationships between individuals for haploid diploid and polyploid species. Mol Ecol 14:415–424
Leslie JF, Summerell BA, Bullock S (2006) The Fusarium laboratory manual. Wiley-Blackwell, New York, p. 388
McDermott JM, McDonald BA (1993) Gene flow in plant pathosystems. Annu Rev Phytopathol 31:353–373
McDonald BA (1997) The population genetic of fungi: tools and techniques. Phytopathol 87:448–453
Migheli Q, González-Candelas L, Dealessi L, Camponogara A, Ramón-Vidal D (1998) Transformants of T. longibrachiatum overexpressing the β-1 4-endoglucanase gene egl1 show enhanced biocontrol of Pythium ultimum on cucumber. Phytopathol 88:673–677
Miller JJ (1946) Taxonomy in Fusarium with particular reference to section Elegans. Can J Res 24:213–223
Mohammadi M, Aminipour M, Banhashemi Z (2004) Isozyme analysis and soluble mycelial protein pattern in Iranian isolates of several formae speciales of Fusarium oxysporum. J Phytopathol 152:267–276
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Mwang Òmbe AW, Thiong G, Olubayo FM, Kiprop EK (2007) DNA microsatellite analysis of Kenyan isolates of Rhizoctonia solani from common bean (Phaseolus vulgaris L.). J Plant Pathol 6:66–71
Nasir M, Hoppe HH (1991) Studies on pathotype differentiation within Mycosphaerella pinodes (Berk, Bloxan) Vestergren a component of the Ascochyta disease complex of peas (Pisum sativum). Zeit Fur Pflam Kran Pflan 98:619–626
Nei M (1973) Analysis of the genetic diversity in subdivided populations. Proc Natl Acad Sci U S A 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. G E N 89:583–590
Nei M, Li WH (1979) Mathematical model for studying genetic variation in terms of restriction endonuclease. Proc Natl Acad Sci U S A 76:5269–5273
Nelson PE, Toussoun TA, Marasas WFO (1983) Fusarium species: An illustrated manual for identification. The Pennsylvania State University Press University Park, p 193
Nourollahi K, Javannikkhah M, Naghavi MR, Lichtenzveig J, Okhovat SM, Richard P, Oliver RP, Ellwood SR (2011) Genetic diversity and population structure of Ascochyta rabiei from the western Iranian Ilam and Kermanshah provinces using MAT and SSR markers. Mycol Prog 10:1–7
O’Donnell K (2000) Molecular phylogeny of the Nectria haematococca-Fusarium solani species complex. Mycol 92:919–938
Ouellet T, Seifert KA (1993) Genetic characterization of Fusarium graminearum strains using RAPD and PCR amplification. Phytopathol 83:1003–1007
Pasquali M, Acquadro A, Balmas V, Migheli Q, Garibaldi A, Gullino ML (2003) RAPD characterization of Fusarium oxysporum isolates pathogenic on Argyranthemum frutescens L. J Phytopathol 151:30–35
Peakall R, Smouse PE (2006) GenAlEx 6: genetic analysis in excel population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Roldan-Ruiz I, Dendauw JE, Vanbockstaele E, Depicker A, Loose M (2000) AFLP markers reveals high polymorphic rates in ryegrasses (Lolium spp.). Mol Breed 6:125–126
Rolhf FJ (1990) NTSYSPc Numerical taxonomy and multivariant analysis system Version 202 Applied Biostatistics. New York
Saharan MS, Naef A (2008) Detection of genetic variation among Indian wheat head scab pathogens (Fusarium spp. /isolates) with microsatellite markers. Crop Protect 27:1148–1154
Sanders IR (2002) Ecology and evolution of multigenomic arbuscular mycorrhizal fungi. Am Nat 160:8–41
Sarker A, Erskine W (2006) Recent progress in the ancient lentil. J Ag Sci 144:19–29
Shah D, Bergstrom GC, Ueng PP (1995) Initiation of septoria nodorum blotch epidemics in winter wheat by seedborne Stagonospora nodorum. Phytopathol 85:452–457
Sirjusingh C, Kohn LM (2001) Characterization of microsatellite in the fungal plant pathogen S. sclerotiorum. Mol Ecol Notes 1:267–269
Sivaramakrishnan S, Kannan S, Singh SD (2002a) Genetic variability of Fusarium wilt pathogen isolates of chickpea (Cicer arietinum L) assessed by molecular markers. Mycopathol 155:171–178
Sivaramakrishnan S, Kannan S, Singh SD (2002b) Detection of genetic variability in Fusarium udum using DNA markers. Ind J Phytopathol 55:258–263
Slippers B, Burgess T, Wingfield BD, Crous PW, Coutinho TA, Wingfield MJ (2004b) Development of simple sequence repeat markers for Botryosphaeria spp. with Fusicoccum anamorphs. Mol Ecol Notes 4:675–677
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. WH Freeman and Company, San Francisco, p. 573
Snyder WC, Hansen HN (1940) The species concept in Fusarium. Am J Bot 27:64–67
Stewart JE, Kim M, James RL, Dumroese RK, Klopfenstein NB (2006) Molecular characterization of Fusarium oxysporum and Fusarium commune isolates from a conifer nursery. Phythopathol 96:1124–1133
Tenzer I, Ivanissevich SD, Morgante M, Gessler C (1999) Identification of microsatellite markers and their application to population genetics of Venturia inaequalis. Phytopathol 89:748–753
Tosi L, Cappelli C (2001) First report of Fusarium oxysporum f. Sp. lentis of lentis in Italy. Plant Dis 85:562
Varshney RK, Chabane K, Hendre PS, Aggarwal RK, Graner A (2007) Comparative assessment of EST-SSR; EST-SNP and AFLP markers for evaluation of genetic diversity and conservation of genetic resources using wild cultivated and elite barleys. Plant Sci 173:638–649
Welsh J, Petersen C, Mc Clelland M (1991) Polymorphisms generated by arbitrary primed PCR in mouse: application to strain identification and genomic mapping. Nucleic Acids Res 19:303–306
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphism amplified by arbitrary primers as useful genetic markers. Nucleic Acids Res 18:6531–6535
Woo SL, Zoina A, Sorbo G, Lorito MD, Scala NBF, Noviello C (1996) Characterization of Fusarium oxysporum f. sp. phaseoli by pathogenic races VCGs RFLPs and RAPD. Phytopathol 86:966–973
Yadav SS, McNeil DL, Stevenson PS (2007) Lentis an Ancient Crop for Modern Times. PO Box 17 3300 AA Dordrecht the Netherlands, p 443
Yeh F, Yang R, Boyle T (1999) POPGENE version 131 Microsoft Windows-based software for population genetics analysis. University of Alberta and Centre for International Forestry Research Alberta
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nourollahi, K., Madahjalali, M. Analysis of population genetic structure of Iranian Fusarium oxysporum f. sp. lentis isolates using microsatellite markers. Australasian Plant Pathol. 46, 35–42 (2017). https://doi.org/10.1007/s13313-016-0458-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13313-016-0458-8