
Abstract
A rapid and low-cost sample preparation method modifying the so-called QuEChERS (quick, easy, cheap, effective, rugged and safe) method was developed for the determination of H2SO4 stable organic compounds in fish samples. The method involves acetonitrile salting-out extraction and a novel clean-up procedure combining the pH-tuned dispersive liquid–liquid microextraction (DLLME) with conc. H2SO4 digestion. The H2SO4 treatment is carried out by a simple operation in contrast to laborious and time-consuming procedures reported in the literature. The method was developed, optimised and validated using selected polybrominated diphenyl ethers (PBDEs) as representatives of H2SO4 stable compounds. For instrumental analysis of sample extracts, thermal desorption–gas chromatography–triple quadrupole tandem mass spectrometry (TD–GC–QqQ-MS/MS) was used. The proposed sample preparation method provides better co-extractives removal efficiency as compared to the original QuEChERS method and, unlike the QuEChERS dispersive solid-phase extraction (dSPE), allows complete removal of fatty acids. The sample preparation is low-cost due to the employment of small amounts of low-cost chemicals (no need of expensive sorbents), and it does not require special instrumentation. The whole analytical method showed satisfactory performance (linearity, recovery, repeatability, accuracy, limits of the method), and its applicability was confirmed by determination of PBDEs in samples of four fish species with variable lipid content in the range of 0.7–19%. The obtained recoveries for six PBDEs in the NIST standard reference material SRM 1946 were in the range of 81–95%, and relative standard deviations were in the range of 3–13%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
It is well-established that sample preparation for the determination of organic micropollutants is the bottleneck of the whole analytical process. This is particularly the case for samples derived from fish, whereby their high lipid content and complexity require employing efficient extraction, clean-up and preconcentration techniques prior to instrumental analysis. Determining factors in selecting and combining these techniques include the physical–chemical properties of analytes, the available instrumentation, as well as the time, labour and cost consumption, especially if large numbers of samples are to be analysed.
Among the frequently monitored fish contaminants belong polybrominated diphenyl ethers (PBDEs), polychlorinated biphenyls (PCBs) and organochlorine pesticides (OCPs) (Chung and Chen 2011; LeDeoux 2011; Berton et al. 2016; Pietroń and Małagocki 2017), which are lipophilic compounds, mostly stable in conc. H2SO4, and usually determined by GC methods. Efficiently removing lipids (and other co-extractives) from fatty sample extracts without removing analytes is the key to improve chromatographic performance for the best quality data (without interferences and false-positive results) and to reduce demands for GC system maintenance (because of injector and column fouling). Therefore, for high-lipid samples, an effective lipid removal step has to be incorporated into the sample preparation procedure.
Among the existing techniques of lipid removal from biological matrices analysed for acid stable compounds, a significant role is played by the clean-up with H2SO4 providing clean sample extracts (Chung and Chen 2011; Pietroń and Małagocki 2017). Various procedures for H2SO4 treatment are described in the literature, which involve clean-up by direct addition of concentrated acid to the extract or by using H2SO4-modified silica gel columns. For the analysis of OCPs in human milk, Brevik (1978) added 1.5 mL of conc. H2SO4 to 1 mL of hexane extract (prepared by solvent extraction under sonication) and allowed it to stand for 1 h at 20 °C. Then, the sample was centrifuged, frozen and analysed by GC–ECD. In the study of acid stability and recovery of 84 pesticides and 12 PCBs, Bernal et al. (1992) employed H2SO4 clean-up comprising equal 1 mL portions of hexane extracts (with different fat contents) and conc. H2SO4 and sonication for 10 min. The separated acid phase was then washed with 2 mL of hexane, and the combined hexane fractions were washed with 5 mL of ultrapure water. In the sample preparation procedure for the analysis of organochlorine compounds in fish developed by Berdié and Grimalt (1998), the sample clean-up involved vigorous stirring (2 min) of a mixture of 2 mL hexane extract (after Soxhlet extraction) and 2 mL conc. H2SO4. After agitation, the mixture was centrifuged and the H2SO4 layer was discarded. This clean-up step was repeated until a colourless transparent hexane layer (2 mL) was obtained (4–6 times). The final H2SO4 mixture was re-extracted with hexane (2 × 2 mL), and all hexane solutions were combined and evaporated just to dryness and re-dissolved in isooctane for instrumental analysis. For the clean-up of 30 mL extract after microwave-assisted extraction or accelerated solvent extraction of freeze-dried fish samples analysed for PCBs and PBDEs, Tapie et al. (2008) employed shaking five times with H2SO4 (5 × 10 mL). The organic and acid phases were separated, and acid phases were eliminated. Ultrapure water was used to remove the excess acidity from the organic phase, and then, the extract was dried with anhyd. Na2SO4, concentrated and subsequently purified on a silica gel column. In the recently published study by Dosis et al. (2016), 6 mL hexane extract of homogenised mussels analysed for PBDEs was treated with 4 mL of conc. H2SO4 and centrifuged. The overhead hexane phase was transferred into a test tube, and the remaining acid phase was re-extracted with 3 mL of hexane. After centrifugation, the second hexane phase was pooled with the first, concentrated, and subsequently purified using H2SO4-modified silica and no-modified silica columns. The use of H2SO4-modified silica gel columns is another technique of H2SO4 treatment that comprises preparation of columns, column conditioning, loading of extracts, elution of analytes and concentration of collected extracts (Chung and Chen 2011; Pietroń and Małagocki 2017).
As can be seen, the above-presented procedures are laborious and tedious, and for direct acid digestion, the volume (mL) ratios of H2SO4 to solvent extract were from 10/30 to 1.5/1. In this study, we found out that adding an abundant volume of H2SO4 to the treated extract (ratio of 1 to 0.08) after previous clean-up using dispersive liquid–liquid microextraction (DLLME) changed the H2SO4 treatment to a simple operation.
In the refinement of sample preparation method, we used an acetonitrile salting-out extraction, which is the first part of the QuEChERS method originally designed for the analysis of fruits and vegetables (Anastassiades et al. 2003). The second—clean-up—part of this method carried out by dispersive solid-phase extraction (dSPE) is not fully sufficient for the analysis of fatty biological samples and, therefore, has gone through many modifications to increase the removal of co-extractives (mainly lipids) from the final extract (use of freezing, dual dSPE, gel permeation chromatography, silica mini-column, EMR-lipid sorbent) (Norli et al. 2011; Molina-Ruiz et al. 2015; Tölgyessy et al. 2016; Morrison et al. 2016; Cloutier et al. 2017; Han et al. 2016). Another drawback of using the original QuEChERS method is that it provides no additional concentration factor making it difficult to achieve stricter limits of quantification (LOQ).
To increase the concentration factor, it is advantageous to combine QuEChERS extraction with DLLME as has been done in multiple studies dealing with the analysis of food samples (Andraščíková et al. 2015; Andraščíková and Hrouzková 2016; Faraji et al. 2017; Wang et al. 2017; Lawal et al. 2016). DLLME is another fast, simple and inexpensive sample preparation method, which was developed by Rezaee et al. (2006) for extraction and preconcentration of organic compounds from aqueous samples. In this method, which is based on a ternary component solvent extraction system, an appropriate mixture of extraction solvent (water immiscible solvent) and dispersive solvent (water miscible solvent) is rapidly injected into the aqueous sample to form a cloudy solution. The hydrophobic solutes are quickly enriched in the fine droplets of extraction solvent which is dispersed entirely into the aqueous phase. Here, an advantage of the possibility to tune the pH of the extraction system can be emphasised (Campone et al. 2012). After centrifugation, the separated organic phase is taken with a microsyringe for its later instrumental analysis. In the case of coupling DLLME with the QuEChERS extraction, the dispersive solvent is obtained from the QuEChERS procedure. When analysing high-fat samples, such as fish, the clean-up effect of DLLME on the raw QuEChERS extract can be further enhanced by H2SO4 treatment to remove the residual lipids.
In this paper, as to our knowledge, we are for the first time presenting sample extract clean-up by combining the pH-tuned DLLME with H2SO4 digestion, which enables obtaining high lipid-removal efficiency. The method was developed and optimised using selected PBDEs as representatives of H2SO4 stable compounds. The final sample extracts were analysed by thermal desorption–gas chromatography–triple quadrupole tandem mass spectrometry (TD–GC–QqQ-MS/MS).
Experimental
Standards and Reagents
Individual PBDE standards BDE-28, BDE-47, BDE-77, BDE-99, BDE-100, BDE-153 and BDE-154, each at 50.0 μg mL−1 in nonane (≥ 98% pure), were from Cambridge Isotope Laboratories (CIL, Andover, MA, USA).
Anhydrous magnesium sulphate, chloroform, toluene and sulphuric acid, all Emsure grade, and n-hexane, UniSolv grade, were purchased from Merck (Darmstadt, Germany). Anhydrous sodium acetate, sodium chloride (both ReagentPlus) and acetonitrile (Chromasolv) were obtained from Sigma–Aldrich (Steinheim, Germany). Milli-Q water was produced by a Direct-Q 3 water purification system (Millipore, Molsheim, France).
Sodium acetate solutions of 0.1, 0.3, 0.5, 0.7 and 1.0 M were prepared by dissolution of corresponding amounts of CH3COONa in Milli-Q water placed in 250-mL volumetric flasks. Working standard mixture solutions of six PBDEs (BDE-28, 47, 99, 100, 153 and 154) at concentrations of 5 and 0.5 μg mL−1 were obtained by dilution of the individual standard solutions with toluene. An internal standard (IS) solution of BDE-77 at 5 μg mL−1 was prepared from the stock standard solution also by dilution with toluene.
Fish Samples
Chub (Squalius cephalus) was chosen as a source of testing matrix for the method development. They were obtained during a fish survey performed in Slovak water bodies in 2015 within the project: Monitoring and assessment of water body status (see the “Acknowledgement” section). The fish were collected by electrofishing and were deep frozen after catch until sample preparation. After thawing, composite samples were prepared from several pieces (from 2 to 6) of the whole fish from each sampling site to give a wet weight mostly of about 600 g. The samples were homogenised using an electric meat grinder TC-22 Elegant Plus (Tre-Spade, Turin, Italy) and kitchen mixer Eta Mira 011 (ETA, Hlinsko, Czech Republic). The fish homogenates were stored in a freezer at − 20 °C until extraction and analysis.
For validation of the proposed method, the standard reference material SRM 1946 (Lake Superior Fish Tissue) was obtained from the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA). This SRM was a frozen fish tissue homogenate prepared from lake trout (Salvelinus namaycush), with 10.2% of extractable fat and 71.4% of water.
For the method applicability demonstration, frozen skinless fillets of Atlantic salmon (Salmo salar) and Alaska pollock (Theragra chalcogramma) were purchased in a local supermarket. The fish fillets were homogenised in the same way as the chub samples.
The linear range, recovery, repeatability and limits of the method were tested by analysing chub composite samples (with 5.6% of total lipids and 76% of water) spiked with the studied analytes. The employed spiking concentrations were 0.0, 0.1, 0.5, 1, 5, 10, 25, 30 and 50 μg kg−1, respectively, and for IS constant concentration of 20 μg kg−1.
Lipid and Moisture Determination
The lipid and moisture determination was accomplished according to our previous work (Tölgyessy and Miháliková 2016). For the lipid content determination, 5 g of fish tissue homogenate in a centrifuge tube was extracted with 5 mL of acetone/ethyl acetate mixture (6:4, v/v) using vigorous shaking for 3 min, and after addition of inorganic salts (2 g MgSO4 and 0.5 g NaCl) and agitation (3 min), the organic phase was separated by centrifugation. An aliquot of the organic phase was dried to constant weight at 103 °C, and the percent lipid content was calculated from the mass of the final residue. The moisture content was also determined gravimetrically from the mass difference of 2–3 g portions of fish homogenate before and after a 24 h drying at 60 °C. For all fish sample homogenates (except SRM 1946), the lipid content was determined in triplicates (results in the range 0.7–19%) and the moisture content in duplicates (59–83%).
Sample Preparation Method
The initial step of sample preparation—the acetonitrile salting-out extraction—was based on the first part of the unbuffered QuEChERS method (Anastassiades et al. 2003; Sapozhnikova and Lehotay 2013). Five grammes of fish homogenate weighed in a 50-mL polypropylene centrifuge tube (Corning CentriStar, Sigma–Aldrich) was spiked with IS solution and mixture of analytes (in case of standard addition). After 15 min, 5 mL of acetonitrile was added, the tube was closed with a stopper and shaken using a vortex mixer (Stuart SA8, UK) at 800 rpm for 1 min. Then, 2 g of anhyd. MgSO4 and 0.5 g of NaCl was added, and again, the tube was shaken vigorously for 1 min. Next, it was centrifuged (centrifuge Rotina 380, Hettich, Tuttlingen, Germany) at 5000 rpm (4863 rcf) for 5 min.
In the DLLME step, 1 mL of supernatant was transferred to a 15-mL centrifuge tube containing 4 mL of 0.5 M CH3COONa solution. Then, 50 μL of chloroform was added; the tube was stoppered, shortly shaken and centrifuged for 5 min at 5000 rpm.
For the final H2SO4 clean-up step, the whole sedimented CHCl3 phase was transferred to a 1.5-mL micro test tube (Eppendorf, Hamburg, Germany) and 1 mL of conc. H2SO4 was added slowly along the sides of the tube. The tube was stoppered and shortly shaken by hand and then 80 μL of hexane was added to the top of the solution. Next, the tube was shaken and centrifuged in a microcentrifuge (Millifuge—Millipore, Hitachi Koki, Japan) for 3 min at 10,000 rpm. The upper phase was the final organic extract taken for analysis.
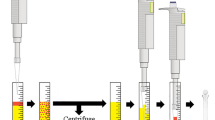
The sample preparation workflow chart is presented in Fig. 1.

Sample preparation workflow
Instrumental Analysis
Analyses were carried out on an Agilent Technologies 7890B GC coupled to a 7000C QqQ-MS/MS system (Wilmington, DE, USA). The GC was equipped with a thermal desorption unit (TDU) assembled to a cooled injection system (CIS4 PTV) (both Gerstel, Mülheim a/d Ruhr, Germany). A Gerstel multipurpose sampler MPS equipped with a TDU tray allowing automated desorption of 98 thermal desorption (TD) glass tubes was installed on top of the GC. The analytes were separated on two identical Agilent J&W HP-5 ms UI capillary columns (15 m × 0.25 mm I.D., 0.25 μm film thickness) connected in series by an Agilent Purged Ultimate Union. To protect the analytical column from contamination, a deactivated silica retention gap (1 m × 0.32 mm) was installed in front of the first column. Helium was used as the carrier gas at constant flow rates of 1.1 and 1.3 mL min−1 for the first and the second column, respectively.
The sample extracts were injected (injection volume of 5 μL) into short homemade glass inserts (with length of 10 mm and I.D. of 3.2 mm) placed inside TD tubes. The loaded TD tubes were closed with a locking cone and placed in the TDU tray for TD–GC analysis.
TD of the loaded TD tubes was performed by ramping TDU from 50 to 275 °C (6 min) at a rate of 720 °C min−1 in the splitless mode under helium flow of 80 mL min−1 (vent pressure 47 kPa), while the CIS was programmed in the solvent venting mode. Following TD, analytes were cryofocussed in a glass baffled liner at the CIS4 PTV at 20 °C using liquid CO2. After desorption, the analytes were transferred to the analytical column (splitless period 2 min) by heating the CIS4 PTV to 275 °C (10 min) at a rate of 12 °C s−1. Simultaneously, temperature programming was started for a GC oven. The oven temperature was programmed from 105 to 305 °C at a rate of 40 °C min−1 with initial and final hold times of 1 and 5 min, respectively. After each analysis, a 2 min post-run column clean-up was performed employing a mid-column backflush. The backflush was conducted at 305 °C, by applying helium to Purged Ultimate Union at 305 kPa.
The MSD was operated using electron ionisation (70 eV) in the multiple reaction monitoring (MRM) mode. The MRM conditions and retention times (tR) for the selected analytes are presented in Table 1. Dwell times were in all cases set at 10 ms. The MSD transfer line was at 280 °C, ion source at 300 °C and quadrupoles at 150 °C. Helium was used as the quench gas (at 2.25 mL min−1) and nitrogen as the collision gas (at 1.5 mL min−1). Agilent MassHunter software was used for instrument control and data analysis.
The quantification process was performed using single-point standard addition method. For this purpose, the concentration of each added PBDE was 10 μg kg−1 and the concentration of the IS was 20 μg kg−1, which was appropriate for the studied range and in agreement with the study of Frenich et al. (2009).
Results and Discussion
TD Conditions
In the GC analysis, sample loading was carried out by thermal desorption of fish extract from the insert placed in the TD tube. The advantage of this sample introduction is in possibility of loading of larger extract volumes (5 μL) and in protection of the injector and GC column, because the non-volatilised co-extractives remain in the insert and do not enter the chromatographic system.
For desorption of analytes from the loaded insert (5 μL CHCl3 extract with PBDEs at 0.1 μg mL−1) placed in the TD tube, flow rates of helium in the range of 50–100 mL min−1 were tested and 80 mL min−1 was found as optimal flow, when largest chromatographic peak areas for the studied PBDEs were obtained. For optimization of duration of analyte desorption, desorption times from 5 to 11 min (at desorption flow rate of 80 mL min−1) were tested. Again, the desorption efficiency was evaluated through the PBDE peak areas. After desorption time of 7 min, the peak areas were fairly constant, and therefore, the time of the whole desorption process was set at 7 min.
DLLME Procedure Optimisation
In the DLLME procedure, acetonitrile extract obtained using conventional QuEChERS extraction (with MgSO4 and NaCl, see the “Sample Preparation Method” section) served as a dispersive solvent. Chloroform was chosen as an extraction solvent on the basis of published works (Andraščíková and Hrouzková 2016; Kaart and Kokk 1987) and our preliminary study. Generally (unless otherwise stated), the experiments were carried out with 1-mL aliquots of raw acetonitrile extract obtained by QuEChERS extraction of chub fish homogenate (lipid content of 5.6%, moisture content of 75. 8%), which was transferred into a 15-mL centrifuge tube and spiked with a mixture of PBDEs at 0.005 μg mL−1. In the initial experiment, 3 mL of Milli-Q water, as third component of the ternary solvent system for the DLLME, was added to the tube. Then, 50 μL of chloroform was added; the tube was stoppered, shortly shaken and centrifuged for 5 min at 5000 rpm. After centrifugation, a few millimetre-thick lipid layer was formed between the aqueous and organic phases. Therefore, 0.1 M CH3COONa solution was used instead of pure water, which improved the separation of sedimented CHCl3 phase without formation of the lipid layer. The basic pH causes that organic acids are in dissociated form, leading to partitioning into aqueous phase.
Effect of CH3COONa Solution Molarity
Subsequently, the effect of CH3COONa solution molarity on the extraction efficiency of selected PBDEs was tested using spiked QuEChERS extracts. For the experiment, 3 mL of pure Milli-Q water and of CH3COONa solutions at 0.1, 0.3, 0.5, 0.7 and 1.0 M were taken. DLLME was carried out with 50 μL of CHCl3. After centrifugation, the volume of the sedimented CHCl3 phase was measured with a 250-μL Hamilton microsyringe and 5 μL of the extract was analysed by GC–MS/MS. For visualisation of the results, responses are shown as average (n = 2) chromatographic peak areas normalised to the highest peak area obtained for individual CH3COONa solutions with different molarity and for each compound (Fig. 2). As can be seen in Fig. 2, the best recovery of the studied PBDEs was obtained by using 0.5 M CH3COONa solution. The recovery increased with increasing molarity of CH3COONa solution despite the fact that increasing the molarity enhanced the volume of the analysed sedimented phase from ca. 60 μL for 0.1 M to ca. 100 μL for 0.5 M solution. The higher molarities of CH3COONa (0.7 M, 1.0 M) caused problems with extraction solvent phase sedimentation (and separation) because instead of settling at the bottom of the centrifuge tube, it moved to the upper layer. Therefore, further experiments were performed with 0.5 M CH3COONa solution.

Effect of CH3COONa solution molarity on the extraction efficiency of selected PBDEs
Effect of CH3COONa Solution Volume
For optimisation of the volume of CH3COONa solution in the DLLME procedure, the spiked QuEChERS extract was mixed with 3, 4 or 5 mL of 0.5 M CH3COONa and 50 μL of CHCl3. The resulting volumes of the sedimented CHCl3 phase decreased from 102 μL for 3 mL, to 78 μL for 4 mL and to 59 μL for 5 mL of CH3COONa solution. The decrease of the volume of the sedimented phase was accompanied by increase of the concentration of co-extractives (mainly lipids) in the obtained CHCl3 extract, which caused problems in the TD–GC analysis (higher background, decrease of PBDE peak’s areas). During development of the procedure, it was also observed that in some cases, when using 3 mL of CH3COONa solution, the CHCl3 phase moved to the upper layer. So, the volume of 3 mL showed to be critical. Therefore, 4 mL of 0.5 M CH3COONa solution was employed in the DLLME procedure.
Volume of Extraction Solvent and of Sedimented Phase
To evaluate the effect of extraction solvent volume, the spiked QuEChERS extract was subjected to DLLME procedure by using 4 mL of 0.5 M CH3COONa solution and 30, 50, 70 or 100 μL volumes of CHCl3. After centrifugation, the lowest studied volume (30 μL) of CHCl3 provided ca. 32 μL of sedimented phase, but the separation was problematic because the part of the CHCl3 remained dispersed in the aqueous phase. The volumes of sedimented phase were approximately 80, 130 and 180 μL when the volumes of extraction solvent were 50, 70 and 100 μL, respectively. The effect of extraction solvent volume on the chromatographic response for the selected PBDEs is presented in Fig. 3 (n = 2). It can be seen that there is a great difference (36–54%) in normalised responses between using 50 and 70 or 100 μL of extraction solvent volumes, in favour of 50 μL volume. Thus, 50 μL of extraction solvent was selected to obtain better sensitivity of the method.

Effect of extraction solvent volume on the chromatographic response of selected PBDEs
For the volume of sedimented CHCl3 phase, dependence on the fish homogenate lipid content was also studied. In the experiment, 1-mL aliquots of QuEChERS extracts obtained from homogenates of chub fish with a lipid content of 1.2, 5.6 and 12.0%, respectively, were subjected to DLLME by using 4 mL of 0.5 M CH3COONa solution and 50 μL of CHCl3. The resulting volumes of sedimented CHCl3 phase were for all tested samples constant (78 ± 1.6 μL), and thus independent of the lipid content. On the other side, the lipid content of the CHCl3 extract was increasing with the increasing lipid content of fish homogenate (determined gravimetrically after drying at 103 °C). The increasing lipid content of the CHCl3 extract had a negative impact on the TD–GC analysis, causing decreasing of the PBDE peak’s areas.
Clean-Up with H2SO4
Because of the remaining lipids in the extract after DLLME, the amounts of which depend on the lipid content of the fish homogenate, a further clean-up step using chemical digestion with conc. H2SO4 was incorporated into the sample preparation method. In the clean-up procedure, the whole volume (ca. 80 μL) of the CHCl3 extract obtained by DLLME was transferred into a 1.5-mL micro test tube, 1 mL of conc. H2SO4 was added, and the mixture was shortly shaken. Then, to improve the extraction and separation between organic and acid phases (and also facilitate the collection of sample aliquot), 80 μL of hexane was added to the top of the solution. Next, the tube was stoppered, shortly shaken and centrifuged again for 3 min at 10,000 rpm. The upper phase was the final organic extract taken for analysis.
To evaluate the proposed procedure, this experiment focused on the gravimetric determination of co-extractive residues and co-extractive removal efficiency for different extract clean-up stages was carried out using chub fish homogenates with various lipid contents (1.2, 5.6 and 12.0%). The results show that co-extractive removal efficiency after DLLME was only about 25% in terms of content in the original QuEChERS extract (Table 2). The much higher removal efficiency—of about 90%—was reached after the extract clean-up with conc. H2SO4. For comparison, Sapozhnikova and Lehotay (2013) obtained 80% co-extractive removal efficiency when using unbuffered QuEChERS method with dSPE treatment with a mixture of a primary secondary amine (PSA) and C18 sorbents for the sample preparation of catfish homogenate with lipid content of 6%. When using Z-sep and Z-sep + sorbents, the co-extractive removal efficiencies were 66% and 56%, respectively.
An important observation (see Table 2) from the H2SO4 treatment of the CHCl3 extract after DLLME was that the remaining concentration of co-extractives in the final extract was at the same level for all tested samples independent on the original lipid content of fish homogenate.
In order to compare the clean-up efficiency of the proposed method with the original QuEChERS method involving dSPE clean-up using PSA and C18 sorbents, the extracts obtained after processing the chub homogenate sample by both methods were analysed by GC–MS. In Fig. 4, it can be seen that the proposed clean-up method, unlike the QuEChERS dSPE method, has completely removed fatty acids.

Total ion chromatograms from the GC–MS analysis of co-extractives residues obtained after clean-up of raw fish acetonitrile extract with (a) dSPE [(150 mg PSA + 150 mg C18 + 300 mg MgSO4) per 1 mL of MeCN extract] and (b) employing DLLME and H2SO4. The identified compounds: 1 lauric acid; 2 myristic acid; 3 hexadecenoic acid, Z-11-; 4 palmitic acid; 5 linoleic acid; 6 timnodonic acid; 7 cholesta-3,5-diene. GC–MS analysis conditions as in Tölgyessy et al. (2016)
Recovery of PBDEs After DLLME and H2SO4 Clean-Up and Matrix Effect
To evaluate the recovery of PBDEs after DLLME and H2SO4 clean-up, the blank raw QuEChERS extract obtained from the chub fish homogenate was spiked at concentration of 0.005 μg mL−1 and subsequently processed by the optimised procedure. The recovery of the analytes was evaluated by comparing their peak areas from the analysis of the final purified extract to the peak areas from the analysis of the corresponding standard solution prepared in the mixture of CHCl3 and hexane (1:1). Also, for investigation of the matrix effect, an acetonitrile solution of PBDEs at 0.005 μg mL−1 was processed by the same procedure. The mean PBDE recoveries (n = 3) for the fortified QuEChERS extract and acetonitrile solution after DLLME and H2SO4 clean-up are presented in Fig. 5. The obtained PBDE recoveries were in the range of 83–90% for the QuEChERS extract and 96–105% for the acetonitrile solution, and the matrix suppression of the chromatographic signal was in the range from 6 to 18%.

Recoveries of PBDEs after DLLME and H2SO4 clean-up for the fortified QuEChERS extract and acetonitrile (MeCN) solution. The error bars denote standard deviations from three replicates (SDs 5–15%)
Whole Method Performance
Once the sample preparation procedure (see Fig. 1) and TD–GC–MS/MS conditions have been optimised, the performance characteristics of the whole analytical method were determined.
Response linearity for the studied analytes was evaluated by analysing matrix-matched standards at seven concentration levels in the range of 0.1–50 μg kg−1 and applying a blank correction. To evaluate the linearity of the response, correlation coefficients (r) of the calibration lines as well as relative standard deviations (RSD) of the relative response factors (RRF) were investigated. The RRFs were calculated for the analytes relative to the internal standard at each concentration level. For all the analytes, the obtained calibration functions were linear with r values in the range of 0.9989–0.9999 and RSDs of the RRFs below the value of 18% (see Table 3).
The limits of the method were determined from the analyses of seven replicates of the blank chub composite sample spiked at 0.1 μg kg−1. The limits of detection (LOD) and quantification (LOQ) were calculated as three and ten times the standard deviation (SD) of the measurement, respectively. As can be seen in Table 3, the LOQs for the studied compounds are in the range 0.10–0.19 μg kg−1. Due to the fact that we did not have suitable PBDE-free chub matrix necessary for matrix-matched calibration at low concentration levels, we decided to use a single-point standard addition method for quantification and to compensate for matrix effects. The single-point standard addition method was also applied for determination of recovery and repeatability (RSD) of the method. Results from the evaluation of five repeated analyses of the selected chub composite sample spiked at 5 and 25 μg kg−1 levels are presented in Table 3. The recovery (96–111%) and repeatability (5–15%) for all the studied analytes fall within the acceptable recovery and repeatability range of 70–120% and RSD ≤ 20%, respectively (European Commission 2013).
To show the accuracy of measurements of the developed method, a NIST SRM 1946 standard fish tissue reference material (prepared from lake trout) with the certified values for the selected PBDEs was analysed. Figure 6 presents a total ion MRM chromatogram from the TD–GC–MS/MS analysis of sample extract prepared by the developed sample preparation procedure. The certified values and the obtained results are presented in Table 4. Good agreement with the certified values was found for all the studied PBDEs with the accuracy in the range of 81–95%. The precision of four replicate results expressed as RSD was in the range of 3–13%.

Total ion MRM chromatogram from the analysis of standard reference material SRM 1946. Analytes: 1 BDE-28, 2 BDE-47, 3 BDE-100, 4 BDE-99, 5 BDE-154, 6 BDE-153
The presented characteristics show a satisfactory performance of the proposed method.
Method’s Applicability
Besides the analysis of standard reference sample (lake trout), the method’s applicability was demonstrated by the analysis of homogenate samples prepared from three fish species (pollock, chub and salmon, see the “Fish Samples” section) with different lipid and moisture contents. Firstly, recovery test with homogenates of pollock (lipids 0.68%, moisture 82%), chub (lipids 5.6%, moisture 76%) and salmon (lipids 8.6%, moisture 68%) spiked with studied PBDEs at 5 μg kg−1 was performed. The obtained results illustrated in Fig. 7 show that the recoveries of PBDEs were for all the three fish species similar with values in the range of 90–111% and with SDs in the range of 2–18%. Secondly, ten homogenates of different fish samples were analysed in triplicate by the developed method. Results from determination of PBDEs and also content of total lipids and moisture are presented in Table 5. It can be seen that the sum concentrations of BDE-28, 47, 99, 100, 153 and 154 are in the range from 0.26 to 3.84 μg kg,−1, which are similar contamination levels as were determined in the study of Labadie et al. (2010). Also, it is visible that PBDE concentrations, generally, positively correlate with lipid content of fish.

Recoveries of PBDEs for samples of three fish species fortified at 5 μg kg−1. The error bars denote standard deviations from five replicates (SDs 2–18%)
Conclusions
A rapid and low-cost sample preparation method modifying the clean-up part of the original unbuffered QuEChERS method was developed for the determination of H2SO4 stable organic compounds in fish samples. As to our knowledge, the fatty extract clean-up by pH-tuned DLLME and subsequent digestion with conc. H2SO4 is presented for the first time. The H2SO4 treatment is carried out by a simple operation in contrast to laborious and time-consuming procedures reported in the literature. The proposed sample preparation method provides better co-extractive removal efficiency as compared to the original QuEChERS method and, unlike the QuEChERS dSPE, allows complete removal of fatty acids. The method is low-cost due to the employment of small amounts of low-cost chemicals (no need of expensive sorbents) and does not require special instrumentation. Sample preparation of ten samples can be accomplished within 3 h, including all weighing operations.
The method was developed, optimised and validated using selected PBDEs as representatives of H2SO4 stable compounds and showed satisfactory performance. Applicability of the method was confirmed by determination of PBDEs in samples of four fish species with variable lipid content in the range of 0.7–19%.
References
Agilent Technologies (2014) Pesticides and environmental pollutants MRM database.G9250AA
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Andraščíková M, Hrouzková S (2016) Fast preconcentration of pesticide residues in oilseeds by combination of QuEChERS with dispersive liquid–liquid microextraction followed by gas chromatography-mass spectrometry. Food Anal Methods 9:2182–2193
Andraščíková M, Matisová E, Hrouzková S (2015) Liquid phase microextraction techniques as a sample preparation step for analysis of pesticide residues in food. Sep Purif Rev 44:1–18
Berdié L, Grimalt JO (1998) Assessment of the sample handling procedures in a labor-saving method for the analysis of organochlorine compounds in a large number of fish samples. J Chromatogr A 823:373–380
Bernal JL, Del Nozal MJ, Jiménez JJ (1992) Some observations on clean-up procedures using sulphuric acid and Florisil. J Chromatogr A 607:303–309
Berton P, Lana NB, Ríos JM, García-Reyes JF, Altamirano JC (2016) State of the art of environmentally friendly sample preparation approaches for determination of PBDEs and metabolites in environmental and biological samples: a critical review. Anal Chim Acta 905:24–41
Brevik EM (1978) Gas chromatographic method for the determination of organochlorine pesticides in human milk. Bull Environ Contam Toxicol 19:281–286
Campone L, Piccinelli AL, Celano R, Rastrelli L (2012) pH-controlled dispersive liquid-liquid microextraction for the analysis of ionisable compounds in complex matrices: case study of ochratoxin A in cereals. Anal Chim Acta 754:61–66
Chung SWC, Chen BLS (2011) Determination of organochlorine pesticide residues in fatty foods: a critical review on the analytical methods and their testing capabilities. J Chromatogr A 1218:5555–5567
Cloutier PL, Fortin F, Groleau PE, Brousseau P, Fournier M, Desrosiers M (2017) QuEChERS extraction for multi-residue analysis of PCBs, PAHs, PBDEs and PCDD/Fs in biological samples. Talanta 165:332–338
Dosis I, Athanassiadis I, Karamanlis X (2016) Polybrominated diphenyl ethers (PBDEs) in mussels from cultures and natural population. Mar Pollut Bull 107:92–101
European Commission (2013) Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. Document no. SANCO/12571/2013, Brussels
Faraji M, Noorani M, Sahneh BN (2017) Quick, easy, cheap, effective, rugged, and safe method followed by ionic liquid-dispersive liquid–liquid microextraction for the determination of trace amount of bisphenol A in canned foods. Food Anal Methods 10:764–772
Frenich AG, Martínez Vidal JL, Fernández Moreno JL, Romero-González R (2009) Compensation for matrix effects in gas chromatography–tandem mass spectrometry using a single point standard addition. J Chromatogr A 1216:4798–4808
Han L, Matarrita J, Sapozhnikova Y, Lehotay SJ (2016) Evaluation of a recent product to remove lipids and other matrix co-extractives in the analysis of pesticide residues and environmental contaminants in foods. J Chromatogr A 1449:17–29
Kaart KS, Kokk KY (1987) Spectrophotometric determination of decabromodiphenyl oxide in industrial sewage. Ind Lab (USSR) 53:289–290
Labadie P, Alliot F, Bourges C, Desportes A, Chevreuil M (2010) Determination of polybrominated diphenyl ethers in fish tissues by matrix solid-phase dispersion and gas chromatography coupled to triple quadrupole mass spectrometry: case study on European eel (Anguilla anguilla) from Mediterranean coastal lagoons. Anal Chim Acta 675:97–105
Lawal A, Tan GH, Alsharif AMA (2016) Recent advances in analysis of pesticides in food and drink samples using LPME techniques coupled to GC-MS and LC-MS: a review. J AOAC Int 99:1383–1394
LeDeoux M (2011) Analytical methods applied to the determination of pesticide residues in foods of animal origin. A review of the past two decades. J Chromatogr A 1218:1021–1036
Molina-Ruiz JM, Cieslik E, Cieslik I, Walkowska I (2015) Determination of pesticide residues in fish tissues by modified QuEChERS method and dual-d-SPE clean-up coupled to gas chromatography-mass spectrometry. Environ Sci Pollut Res Int 22:369–378
Morrison SA, Sieve KK, Ratajczak RE, Bringolf RB, Belden JB (2016) Simultaneous extraction and cleanup of high-lipid organs from white sturgeon (Acipenser transmontanus) for multiple legacy and emerging organic contaminants using QuEChERS sample preparation. Talanta 146:16–22
Norli HR, Christiansen A, Deribe E (2011) Application of QuEChERS method for extraction of selected persistent organic pollutants in fish tissue and analysis by gas chromatography mass spectrometry. J Chromatogr A 1218:7234–7241
Pietroń WJ, Małagocki P (2017) Quantification of polybrominated diphenyl ethers (PBDEs) in food. A review. Talanta 167:411–427
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
Sapozhnikova Y, Lehotay SJ (2013) Multi-class, multi-residue analysis of pesticides, polychlorinated biphenyls, polycyclic aromatic hydrocarbons, polybrominated diphenyl ethers and novel flame retardants in fish using fast, low-pressure gas chromatography–tandem mass spectrometry. Anal Chim Acta 758:80–92
Tapie N, Budzinski H, Le Ménach K (2008) Fast and efficient extraction methods for the analysis of polychlorinated biphenyls and polybrominated diphenyl ethers in biological matrices. Anal Bioanal Chem 391:2169–2177
Tölgyessy P, Miháliková Z (2016) Rapid determination of total lipids in fish samples employing extraction/partitioning with acetone/ethyl acetate solvent mixture and gravimetric quantification. Food Control 60:44–49
Tölgyessy P, Miháliková Z, Matulová M (2016) Determination of selected chlorinated priority substances in fish using QuEChERS method with dual dSPE clean-up and gas chromatography. Chromatographia 79:1561–1568
Wang XC, Shu B, Li S, Yang ZG, Qiu B (2017) QuEChERS followed by dispersive liquid–liquid microextraction based on solidification of floating organic droplet method for organochlorine pesticides analysis in fish. Talanta 162:90–97
Acknowledgements
Financial support from the EU Cohesion Funds within the project Monitoring and Assessment of Water Body Status (No. 310011A366 Phase III) is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
P. Tölgyessy declares that he has no conflict of interest. S. Nagyová declares that he has no conflict of interest.
Ethical Approval
All applicable international, national and institutional guidelines for the care and use of animals were followed.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Tölgyessy, P., Nagyová, S. Rapid Sample Preparation Method with High Lipid Removal Efficiency for Determination of Sulphuric Acid Stable Organic Compounds in Fish Samples. Food Anal. Methods 11, 2485–2496 (2018). https://doi.org/10.1007/s12161-018-1241-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1241-y