
Abstract
This paper describes fast and simple extraction methods for the determination of polychlorinated biphenyls and polybrominated diphenyl ethers in biological matrices. Four extraction protocols were tested. The first protocol used microwave-assisted extraction combined with two purification steps. The second one was similar, except that microwave-assisted extraction was replaced by accelerated solvent extraction. The third one combined extraction/purification by accelerated solvent extraction with final purification on a silica gel column. The last one combined microwave-assisted extraction with purification on an acidic silica gel column. The protocols were tested on various matrices: a spiked matrix, two certified matrices (SRM 2977, WMF 01), and natural matrices (mysids and fish). All of the protocols produced good performance in terms of recovery and reproducibility. The two last protocols showed promising results in terms of applicability to natural matrices, as they required a minimum of sample handling and minimal amounts of solvent and time. These methods allowed at least 24 samples to be handled per day, and could easily be used for routine analysis.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Persistent organic pollutants (POPs) such as polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs) are known to be ubiquitous environmental contaminants. Great efforts have been made to find the major sources and pathways by which these compounds are introduced into the environment, and to determine their levels in biotic and abiotic compartments. Due to matrix complexity, several pre-treatment steps are absolutely essential for natural samples: extraction, purification and identification/quantification.
Several techniques have been used to extract these compounds. Soxhlet extraction was the method traditionally used to extract POPs [1], but this method usually requires large amounts of solvent and is time-consuming. A necessary step for Soxhlet extraction is automation. In this respect the commercial equipment called Soxtec™ is the most commonly used device in the field of automated Soxhlet systems, as it performs extraction with similar precision to a conventional Soxhlet, but with a significant saving in time. The Soxtec™ method was invented in the early 1970s and commercialized in 1982 [2]. An alternative to Soxhlet is ultrasound extraction (also called “sonication”), and this has been applied to the extraction of organic compounds from various matrices [2–4]. Soxhlet extraction and sonication are not particularly application-specific, and consequently purification steps are required before analysis [5].
Since then, modern extraction techniques have been developed; these are less time-consuming and require less solvent. The first of these new techniques appeared almost 20 years ago in the form of supercritical fluid extraction [6–7]. More recently, another two techniques have been introduced: microwave-assisted extraction, and accelerated solvent extraction.
Microwaves act directly on materials that rapidly reach high temperatures [8]. Current applications of microwave extraction for pollutants started in the mid-1990s [9]. Microwave-assisted extraction speeds up extraction but it is still however necessary to purify the extract before analysis [10].
In contrast, accelerated solvent extraction—also known as pressurized fluid extraction or pressurized liquid extraction—is the most recent solid-sample extraction method [11–14]. Accelerated solvent extraction speeds up the extraction process by using solvent at high temperature. Pressure is applied to the sample-extraction cell, in order to maintain the heated solvent in a liquid state during extraction. After heating, the extract is flushed from the sample cell into a vial for further steps in the analysis. The major advantage of this technique is the ability to combine extraction with clean-up [15–19].
Both accelerated solvent extraction and microwave-assisted extraction have become widely used extraction techniques for environmental solid samples [5, 20].
The present study compares conventional approaches combining microwave-assisted extraction with post-purification (using sulfuric acid and liquid chromatography on silica) [10, 21] with a procedure that combines pre-purification with accelerated solvent extraction.
Analyses have been performed for two types of contaminants—PCBs and PBDEs—from various matrices: a spiked sample of muscle from trout (Oncorhynchus mykiss), two certified matrices [SRM 2977: tissues from mussel Perna perna taken from Guanabara Bay, Brazil; provided by the National Institute of Standards and Technology (NIST; Gaithersburg, MD, USA), and WMF 1 (fish tissue) provided by Wellington Laboratories (Irigny, France)], and two pooled natural samples: one sample made from small crustaceans (Neomysis integer and Mesopodopsis slabberi) and another from eel muscle (Anguilla anguilla).
Experimental
Chemicals
Pestinorm® dichloromethane was purchased from Acros Organics (Noisy-le-Grand, France); pentane from Atlantic Labo (Bruges, France); and Sharlau’s sulfuric acid (95–98% extra pure) and Sharlau’s iso-octane from ICS (Gradignan, France). The silica gel (0.063–0.2 mm, Merck) was purchased from VWR International (Fontenay sous bois, France). It was cleaned with dichloromethane to prepare acidic silica by mixing 600 g and 400 g of sulfuric acid and leaving this overnight at 200 °C [16]. Sodium sulfate (purity 99%) was obtained from Sigma–Aldrich (Saint-Quentin-Fallavier, France). Ultrapure water was further purified by elution through an Elix®/ Rios™-system cartridge, from Millipore (Saint-Quentin-en-Yvelines, France).
The standard reference material SRM 2262 (chlorinated biphenyl congeners in isooctane) was provided by the NIST. Solutions of individual PBDEs with a purity of 99% (BDE 47, BDE 99, BDE 119 and BDE 153) were purchased from Cambridge Isotope Laboratories (Andover, MA, USA). SRM 2262 and solutions of individual PBDEs were used for calibration. CB 30, CB 103, CB 155 and CB 198 were used as internal standards. The CB 30, CB103, CB 155 and CB 198, with a purity of 99%, were supplied as neat crystals by Promochem (Molsheim, France). Octachloronaphthalene (OCN), with a purity of 95%, was purchased from Ultra Scientific (North Kingstown, RI, USA) and used as a syringe standard.
Sample material
To study the matrix effect, three types of matrix were analyzed: (1) a natural matrix (trout muscle) spiked with standard solutions; (2) two certified matrices, SRM 2977 provided by the NIST and WMF 1 provided by Wellington Laboratories; (3) two natural matrices, muscle from eels and pooled mysids. All of the matrices were freeze-dried and ground up before carrying out extraction. Seven marker PCBs (CB 28, CB 52, CB 101, CB 118, CB 138, CB 153 and CB 180) and five PBDEs (BDE 47, BDE 99, BDE 119, BDE 153 and BDE 190) were studied.
The spiked matrix (muscle from trout) was spiked with a mixture of PCB congeners (SRM 2262) and with solutions of individual PBDEs at concentrations of around 700 ng g−1 in isooctane. A 0.5 g sample of homogenized freeze-dried matrix was spiked with 50 μL of the solution of SRM 2262 diluted ten times as well as with 20 μL of each PBDE solution, in order to obtain nominal quantities in the range of 10 ng of compounds in 0.5 g of matrix. All operations were controlled by gravimetry in order to obtain experimental concentrations and not nominal ones. The final exact quantities of compounds in the spiked matrices are given Table 1.
In order to validate the extraction procedures, two certified matrices were tested. The SRM 2977 (tissues from mussel, Perna perna) is certified for PCB concentrations but not for PBDEs. Reference values for BDEs were obtained from Zhu and Hites [22]. WMF 01 (fish tissue) is certified for PBDE concentrations, but not for the seven studied marker PCBs.
Finally, two natural matrices were used in order to test the extraction procedures with natural samples containing various amounts of lipids. Analyses were undertaken from (1) a pooled mysid sample (with a lipid content of about 7% dry weight), caught in the Gironde estuary (S-W France), and (2) from eel muscle (Anguilla anguilla), also caught in the Gironde Estuary; this latter matrix had a high fat content (lipids accounted for more than 40% dry weight).
GC analyses
Analyses were performed on an HP 5890 series II gas chromatograph from Hewlett-Packard (Avondale, CA, USA) coupled to a 63Ni electron-capture detector (ECD). An HP5-MS capillary column from Agilent Technologies (Massy, France) was used (60 m × 0.25 mm × 0.25 μm) for PCB and PBDE analyses. Helium (He, 5.6 quality) provided by Linde Gas (Toulouse, France) was used as carrier gas at a flow rate of 1 mL min −1, and nitrogen (N2, 5.0 quality; Linde Gas) was used as make-up gas at 60 mL min −1. The temperature program was as follows: 80 °C for 2 min; 10 °C min−1 to 200 °C; 200 °C for 2 min; 2 °C min−1 to 320 °C; and 320 °C for 20 min.
PCBs (CBs 28, 52, 101, 118, 138, 153 and 180) and PBDEs (BDEs 47, 99, 119, 153 and 190) were quantified relative to internal standards (CBs 30, 103, 155 and 198). Octachloronaphthalene was used as a syringe standard. The use of a syringe standard allows the internal standards (CBs 30, 103, 155 and 198) used for the quantification of PCBS and PBDEs to be quantified, in order to estimate the recovery of each extraction protocol. The response factors of the various compounds were measured by injecting a mixture of Standard Reference Material (SRM 2262) solutions of individual PBDEs, internal standards and syringe solutions.
Extraction procedures tested
Four extraction procedures were tested in triplicate for each matrix. Blank experiments (complete procedure but without matrix) were performed. The concentrations determined by analysis were corrected by taking into account the blank values. The surrogate internal standards (CBs 30, 103, 155 and 198) were added gravimetrically to the matrix prior to extraction (using 100 μL of isooctane solution). They were used to quantify the PBBs and PBDEs of interest. A syringe standard (octachloronaphthalene) was added gravimetrically at the end of the extraction and purification steps (using 30 μL of isooctane solution), followed by analysis in order to calculate the percentage recovery of internal standards.
Extraction conditions for ASE were not optimized individually for each method, but were selected from an extensive reference list which includes some of the most important articles published on POP extraction [11, 12, 16, 23]. The conditions used for MAE were derived from methods already published by our laboratory [10, 21].
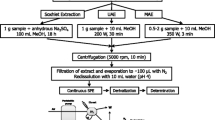
Microwave-assisted extraction and liquid–liquid purification with sulfuric acid (MAE)
The first protocol (MAE) related to microwave-assisted extraction using dichloromethane, coupled with liquid–liquid purification using sulfuric acid, and a final step of purification on a silica gel column [20].
Internal standards were added gravimetrically to 0.5 g of dry material. Microwave-assisted extraction was performed using a Maxidigest MX 4350 from VWR Prolabo (Paris, France) at 30 W for 10 min, using dichloromethane (30 mL). Dichloromethane was chosen due to its ability to absorb microwaves and to extract persistent organic pollutants; it can also be easily removed during reconcentration [24].
Finally, the organic extract was filtered and then purified by shaking five times with sulfuric acid (5 × 10 mL). The organic and acid phases were separated and acid phases were eliminated. Ultrapure water was used to remove the excess acidity from the organic phase and then the extract was dried with anhydrous sodium sulfate. It was concentrated under a gentle flow of nitrogen before a second purification on silica gel (particle size 0.063–0.2 mm) [10].
The PCBs and PBDEs were eluted with 3 × 5 mL of a pentane–dichloromethane mixture (90:10 v/v). The extract was concentrated and transferred to isooctane. The syringe standard was then added gravimetrically, followed by analysis using GC/ECD. This protocol allows six determinations (five samples and a blank) to be carried out per day, per operator.
Accelerated solvent extraction and liquid–liquid purification with sulfuric acid (ASE)
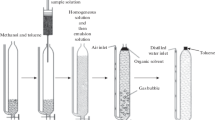
The second protocol (ASE) was a single extraction performed using an ASE 200 System from Dionex (Voisins-le-Bretonneux, France). This protocol followed exactly the same pattern as described above, except that microwave-assisted extraction was replaced by accelerated solvent extraction. The procedure used to pack the extraction cell (11 mL) can be seen in Fig. 1. The matrix (0.5 g) was weighed and mixed with glass pearls in the cell. Internal standards were added directly to the collection vials in order to avoid contamination and memory effects. The cell was loaded into the oven and filled with dichloromethane. The cell was first heated for 5 min at 100 °C, and then extracted at 100 °C under 130 bars of pressure for 8 min. The cell was then flushed with fresh dichloromethane (60% of the total cell volume). Finally the solvent was purged from the cell by nitrogen for 60 s. Organic extracts were collected in the vials and treated using the same purification steps as before (use of sulfuric acid, ultrapure water and silica gel: see previous section).This protocol allows eight determinations to be carried out (seven samples and a blank) per day, per operator.

Packing procedure for cells in (a) ASE and (b) acidic ASE protocols
Microwave-assisted extraction with clean-up on an acidic silica gel column (acidic MAE)
The third protocol combined microwave extraction with purification on an acidic silica gel column (acidic MAE). First, 0.5 g of material were weighed. Internal standards were added gravimetrically. Microwave-assisted extraction was then carried out at 30 W for 10 min, using dichloromethane (30 mL). The organic extract was filtered, reconcentrated to a few μL in isooctane (300 μL), and purified on an acidic silica gel column (about 2 g). Analytes were eluted with 3 × 5 mL of a pentane–dichloromethane mixture (90:10 v/v). The extract was concentrated and transferred to isooctane. The syringe standard was added and then analysis by GC/ECD was carried out. This protocol allows 24 determinations (23 samples and a blank) to be carried out per day, per operator. A skilled operator can manage 32 determinations (one blank and 31 samples).
Accelerated solvent extraction with on-line acid purification and clean-up on an acidic silica gel column (acidic ASE)
The fourth protocol involves extraction–purification on-line, performed by the ASE 200 System (Dionex). The parameters used for the extraction procedure were the same as used previously (see “Accelerated solvent extraction and liquid–liquid purification with sulfuric acid (ASE)”). The procedure used to pack the extraction cell (22 mL) can be seen in Fig. 1. First acidic silica gel (5 g) was added to the extraction cell, and then the matrix (0.5 g) was introduced, with mixing carried out using glass pearls. After extraction, the sample was collected and reconcentrated into 300 μL of isooctane, using a RapidVap vacuum evaporation system from Labconco (Kansas City, MO, USA). A second purification of the extract was performed. The extract was then put on an acidic silica gel column. The PCBs and PBDEs were eluted with 3 × 5 mL of a pentane–dichloromethane mixture (90:10 v/v). The extract was concentrated and transferred to isooctane; the syringe standard was then added and GC/ECD analysis carried out. This protocol allows 24 determinations (one blank and 23 samples) to be undertaken per day, per operator.
Statistics
Statistical analyses were conducted using STATISTICA 7.1 from StatSoft (Maison Alfort, France). The Shapiro–Wilk test and the Kolmogorov–Smirnov test were used to test the normality. In order to compare the efficiency of the four tested protocols, two types of nonparametric tests were used: the Kruskal–Wallis test and the Mann–Whitney U-test.
Results and discussion
Extraction of spiked matrix
The PCB and PBDE quantities obtained as described above were compared to reference values. The reference values were calculated from the nominal theoretical quantity (introduced by spiking 0.5 g of trout muscle). Standard deviations were obtained from the mean values of compounds added to trout muscle (gravimetric data). The spiked levels were about 10 ng for each compound. The quantities of individual PCBs and PBDEs recovered, calculated from the spiked values, are presented in Table 1 (please see Table S1 and Figure S1 of the “Electronic Supplementary Material” for limits of detection and for chromatograms). Each result is the average value calculated from three replicates. Typical associated standard deviations are shown in Table 1. No significant differences can be observed between the quantities obtained by the four tested protocols, except for the BDE 190. Indeed, the BDE 190 quantity obtained by the ASE protocol is significantly lower than those obtained by the other protocols (Mann–Whitney, p < 0.05).
Recovery percentages for the tested PCBs and PBDEs were higher than 70% (please see Fig. S2 of the “Electronic Supplementary Material”). Carrying out each determination three times clearly illustrates the variability. For the majority of compounds this variability was about 15%, but it was as high as 30% for CB 118 and CB 52. Moreover, high recovery values (about 120%) were observed for CB 28, probably due to chromatographic interference.
The last two protocols tested (acidic ASE and acidic MAE) showed the best recovery percentages, over 80%. The detection limits (S/N = 3) for the enhanced protocols ranged from 0.8 to 1.2 ng g−1 for PCBs and from 1.1 to 2.6 ng g−1 for PBDEs. The quantification limits (S/N = 10) ranged from 2.4 ng g−1 to 4.1 for PCBs and from 3.6 to 8.6 ng g−1 for PBDEs. It should also be stressed that with both protocols—acidic MAE and acidic ASE—it was possible to manage more samples than with the earlier protocols. At this stage, both protocols could be considered reliable and usable for environmental monitoring in the laboratory.
Extraction of certified reference material
SRM 2977 from NIST
In order to confirm that the last two protocols can be considered reliable and usable in a laboratory, SRM 2977 was extracted using both of the new protocols—acidic ASE and acidic MAE—and also by our reference protocol, MAE. Extraction was not carried out by the ASE procedure in this case. Extraction by the ASE procedure without on-line purification takes longer than extraction by MAE and is not more efficient.
Quantitative results obtained for individual PCBs and PBDEs in SRM 2977 are shown in Table 2. No significant differences can be observed between results obtained for the three tested protocols (Kruskal–Wallis, p ≥ 0.05). The recovery percentages, calculated from the certified values and for all of the protocols tested, are presented in Table 2. Recoveries were higher than 80% for all of the compounds, with all of the protocols (please see Fig. S3 of the “Electronic Supplementary Material”). The analyses were carried out in triplicate in order to demonstrate the variability of results, and this was fairly uniform, at around 10%. Nevertheless, an overestimation for CB 28 is still noticeable, due to coelution with CB 50. No indicative concentration value is given for CB 50 in SRM 2977 in order to correct the reference value for CB 28. The results obtained for BDEs 47 and 99 are, respectively, of the same order and slightly higher than the values reported by Zhu and Hites [22]. No traces were found of BDEs 119, 153 or 190.
WMF 01 from Wellington Laboratories
In order to confirm the robustness of these protocols for PBDEs, another matrix certified for PBDEs, WMF 01 from Wellington Laboratories, was analyzed. WMF 01 was extracted using the new protocols (acidic ASE and acidic MAE procedures) and our reference MAE protocol. The quantitative results obtained for individual PBDEs in WMF 01 are shown in Table 3. The recovery percentages were satisfactory in all cases, and about 100% for BDEs 47 and 99 (Table 3; please see also Fig. S4 of the “Electronic Supplementary Material”). For BDE 153, the recovery percentage was about 80%. The variability of results was fairly uniform: around 10% for all the compounds and for all of the protocols. No significant differences can be observed between results obtained by the tested protocols (Kruskal–Wallis, p ≥ 0.05).
Extraction of natural matrices
In order to test the applicability of these protocols to environmental monitoring, they were tested on natural matrices.
Mysids
A pooled sample of mysids (small crustaceans) was chosen as a matrix with a low fat content, with lipids accounting for less than 10% of the dry weight. In this case, all four protocols were applied in order to confirm their potential. The quantitative results obtained are shown in Table 4. All of the PCBs investigated were found to have PCBs conforming to a pattern that is typical in biota, with CB 153 and CB 138 dominating. All of the protocols produced similar results. To assess the new ASE protocols, acidic ASE and acidic MAE, PCB and PBDE concentrations were compared with the MAE procedure. The recovery values generally varied between 90% and 120% (please see also Fig. S5 of the “Electronic Supplementary Material”). Test variability, calculated from analyses carried out in triplicate for each protocol, ranged between 5% and 20%, and averaged around 10%. All of the protocols tested produced satisfactory results. No significant differences can be observed between the results (Kruskal–Wallis, p ≥ 0.05). Before injection, the organic extracts were (visually) clear,and they produced chromatograms free of interference, enabling us to conclude that the two tested new protocols are reliable and can be used for matrices with a low fat content, such as crustaceans.
Eels
Eel muscle was chosen as a high-fat content matrix, since it has more than 40% lipids by dry weight. This matrix was chosen to test the efficiency of the purification steps in each protocol. Lipids typically pose a problem during purification. This testing was carried out with all of the protocols, in order to check their performances. The quantitative results obtained are shown in Table 5. No significant differences can be observed between results obtained with the three protocols (Kruskal–Wallis, p ≥ 0.05). The purification step in the acidic MAE protocol is not effective with this type of matrix. The final organic extract was found to be insufficiently clear (visually speaking) for injection into the chromatograph. During the last concentration step before injecting the material, the organic extract became viscous, and this can probably be attributed to lipids not being completely eliminated during the purification procedure. This problem means that no results could be obtained for the acidic MAE protocol; further purification steps are needed for the extract to be injected into the chromatograph. The acidic MAE protocol cannot be used for a matrix with a high fat content. Using the other protocols, idiosyncratic patterns of PCBs were found in the biota in all cases, with CB 153 and CB 138 dominating.
In order to assess the new ASE and acidic ASE protocols, they were compared with our reference protocol, MAE, to determine the PCB and PBDE concentrations. The recovery percentages range from 90 to 120%) (please see also Fig. S5 of the “Electronic Supplementary Material”). The variability of the results from analyses carried out in triplicate using each protocol ranged between 5% and 20%, and averaged around 10%.
The acidic ASE protocol shows promising results for PCB and PBDE extraction from matrices with a high fat content. The organic extracts obtained after purification and final reconcentration were visually clear, allowing injection into the gas chromatograph.
Control cards
To test the reliability of the acidic MAE and acidic ASE protocols, both of the natural matrices were used as internal reference matrices during each series of sample laboratory analyses for environmental monitoring. The reference materials used were mysids in the case of the acidic MAE protocol and eel muscle in the case of the acidic ASE protocol. The results in terms of recovery (shown as the mean value of concentration determined) from each matrix (mysids and eels) are presented in Fig. 2.

Control cards for PCBs and PBDEs investigated in mysids and eels (gray zone represents ±20% variation)
Acidic MAE carried out over ten months shows a variability of about 15% in the PCB and PBDE concentrations in mysids. Acidic MAE is reliable for investigating the PCBs and PBDEs in matrices with a low fat content, such as crustaceans.
Acidic ASE carried out during the same period showed a variability of about 15% for PCBs in eels. The variability in this case for BDEs was about 20%. Acidic ASE was found to be reliable for investigating PCBs and PBDEs in matrices with a high fat content, such as eel.
Conclusion
The four tested protocols were found to be reliable and usable for PCB and PBDE analyses of biological matrices. The acidic MAE protocol and the acidic ASE protocol are promising in terms of applicability to natural matrices as well as savings in time and solvent. The use of acidic MAE protocol (combining microwave-assisted extraction with purification on an acidic silica gel column) makes it possible to handle 24–32 samples per day when analyzing the extracts by injection onto a GC coupled to ECD, but it should be reserved for low-fat content matrices.
The second promising protocol is acidic ASE. This procedure combines two purification steps, an on-line step and one using an acidic silica gel column. This protocol shows good performance in terms of recovery and reproducibility when using a matrix with a high fat content (over 40% lipids by dry weight). Extraction and purification by ASE allows the elimination of interfering compounds (such as lipids, proteins, pigments, etc.) that are hydrolyzed and/or oxidized by sulfuric acid. The second stage of purification (on a chromatographic column) is more efficient in this case, with the chromatographic separation being combined with the use of acidic silica gel. This procedure allows 24 samples a day to be handled.
Moreover, it could be useful to continue developing this type of extraction technique with the use of on-line purification in order to avoid the need for a second purification. Purification of the samples is preferable for obtaining the best results in terms of detection limits for environmental POPs that are being investigated.
References
De Boer J (1988) Chemosphere 17:1803–1810
Luque de Castro MD, Garcia-Ayuso L (1998) Anal Chim Acta 369:1–10
Sun F, Littlejohn D, Gibson MD (1998) Anal Chim Acta 364:1–11
Evans J, Kaake RH, Orr MH, Watwood H (1998) J Soil Contam 7:589–597
De Boer J, Law RJ (2003) J Chromatogr A 1000:223–251
Dean JR (2000) Trends Anal Chem 9:553–564
Zougagh M, Valcarcel M, Rios A (2004) Trends Anal Chem 23:399–405
Abu-Samra A, Morris SJ, Koirtyohann SR (1975) Anal Chem 47:1475–1477
Eskilsson CS, Björklund E (2000) J Chromatogr A 902:227–250
Thompson S, Budzinski H (2000) Int J Environ Anal Chem 76:49–60
Suchan P, Pulkrabová J, Hajlová J, Kocourek V (2004) Anal Chim Acta 520:193–200
Bjorklund E, Nilsson T, Bøwadt S (2000) Trends Anal Chem 19:434–445
Abrha Y, Raghavan D (2000) J Hazard Mater 80:147–157
Wenzel KD, Hubert A, Manz M, Weissflog L, Engewald W, Schuurmann G (1998) Anal Chem 70:4827–4835
Draisci R, Marchiafava C, Ferretti E, Palleschi L, Catellani G, Anastasio A (1998) J Chromatogr A 814:187–197
Müller A, Björklund E, Von Holst C (2001) J Chromatogr A 925:197–205
Gomez-Ariza JL, Bujalance M, Giraldez I, Velasco A, Morales E (2002) J Chromatogr A 946:209–219
Sporring S, Bjorklund E (2004) J Chromatogr A 1040:155–161
Bjorklund E, Sporring S, Wiberg K, Haglund P, Von Holst C (2006) Trends Anal Chem 25:318–325
Thompson S, Budzinski H, Le Menach K, Letellier M, Garrigues P (2002) Anal Chim Acta 372:196–204
Budzinski H, Letellier M, Thompson S, Le Menach K, Garrigues P (2000) Fresenius J Anal Chem 367:165–171
Zhu LY, Hites RA (2003) Anal Chem 75:6696–6700
De Boer J, Cofino WP (2002) Chemosphere 46:625–633
Letellier M, Budzinski H, Bellocq J, Connan J (1999) Org Geochem 30:1353–1365
Acknowledgements
This work was supported by CEFIC’s Long-Range Research Initiative (LRI). We would also like to thank Gis Ecobag, SMIDDEST, ORQUE and Région Aquitaine for their financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material
Rights and permissions
About this article
Cite this article
Tapie, N., Budzinski, H. & Le Ménach, K. Fast and efficient extraction methods for the analysis of polychlorinated biphenyls and polybrominated diphenyl ethers in biological matrices. Anal Bioanal Chem 391, 2169–2177 (2008). https://doi.org/10.1007/s00216-008-2148-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2148-z