
Abstract
A fast, efficient, and simple method for determination of pesticide residues in pumpkin seeds has been developed combining QuEChERS and dispersive liquid–liquid microextraction (DLLME) followed by gas chromatography and mass spectrometry (GC-MS). Parameters affecting the DLLME performance such as solvent selection and volume of extractive and dispersive solvent, salt effect, and extraction time were studied. Under the selected conditions (50 μL extractive solvent chloroform, 1 mL QuEChERS extract, and 3 mL water), the developed method was validated. Linearity was evaluated at nine concentrations in the broad range of 0.1–500 μg/kg with correlation coefficients from 0.9842 to 0.9972. The relative standard deviations at lowest calibration level varied from 0.3 to 22 %. Under the optimum conditions, an enrichment factor was 6–17-fold and detection limits 0.01–12.17 μg/kg were achieved. Finally, the developed and validated method was successfully applied for the extraction and determination of pesticide residues in 16 real samples with 2 positive findings below maximum residue limits (MRL). Limits of detection (LODs) of the proposed method are below the MRLs established by the European Union.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticides differ from other chemical substances because they are toxic chemicals deliberately spread into the environment with the aim of controlling undesired living species. Since their toxicity may not be completely specific for the target organisms, their use may pose a risk to human health (Colosio et al. 2003). In recent years, potential health risks from pesticides and other organic pollutant residues in food stuffs have become a main public concern (Matsadiq et al. 2011).
The social concern about the levels of pesticides in food and the constant trend observed in the current legislations to reduce the maximum residue levels (MRLs) allowed in a variety of fruits and vegetables is increasing the number of samples to be analyzed as well as the need for their accurate determination reducing the analysis’ costs (Nasreddine and Parent-Massin 2002).
Sample preparation is normally required to isolate and concentrate compounds of interest from the sample matrix prior to chromatographic analysis. Liquid–liquid extraction (LLE) and solid-phase extraction (SPE) are still the most common methods to extract pesticides from environmental samples. LLE was used for the extraction of fungicides in white grape juices (Pose-Juan et al. 2006). However, LLE is time consuming and requires high volumes of organic toxic solvents. SPE uses much less solvent than LLE, but the column needs pretreatment and can be relatively expensive (Fu et al. 2009). Therefore, a lot of research efforts in separation science and related fields have been focused on the development of new sample preparation techniques, which are less time consuming, more effective, and require smaller volumes of organic solvents (Zang et al. 2009). Recently, a new microextraction method, dispersive liquid–liquid microextraction (DLLME), has been developed by Rezaee and co-workers in 2006 as an efficient sample preparation and preconcentration method. The extraction by DLLME is based on the ternary component solvent system (aqueous sample, dispersive solvent, and extractive solvent). In this method, an appropriate mixture of extractive and dispersive solvents is injected rapidly into an aqueous sample with a syringe, resulting in the formation of a cloudy solution. A cloudy solution containing fine droplets of the extractive solvent, which have been dispersed in the sample solution, is formed in a test tube. The extractive phase is then separated by centrifugation, and the enriched analytes in the sedimented phase (with or without further treatment) are determined by analytical methods (Rezaee et al. 2006). DLLME is a simple and fast microextraction technique, the major benefits of which are mainly (1) the negligible volumes (a few microliters only) of extractive solvents used; (2) the very large surface area between the fine droplets of the extractive solvent and the aqueous sample, and the accordingly fast extraction kinetics that result in the rapid achieving of a state of equilibrium; and (3) the high enrichment factor usually obtained (Kocúrová et al. 2012). The additional advantages of DLLME method are low cost and high recovery (Yang et al. 2011). Dispersive liquid–liquid microextraction is more suitable for the treatment of the target compounds with simple matrix, resulting in its wide application in the analysis of water samples. In accordance with that, many methods based on DLLME have been developed in the last few years for the determination of pesticide residues mainly in liquid samples including water (Fu et al. 2009; Yang et al. 2011; Shen et al. 2013), tea (Xiong and Hu 2008; Farajzadeh et al. 2011), and juice (Fu et al. 2009; Matsadiq et al. 2011; Boonchiangma et al. 2012; Zhang et al. 2014). DLLME has been applied to pesticide isolation and preconcentration in various food matrices, mainly from fruit (Zang et al. 2008; Huo et al. 2011) and vegetable such as tomato (Qiao et al. 2010; Melo et al. 2012, 2013) and cucumber (Zhao et al. 2007; Zhang et al. 2012).
By now, the pretreatment of samples with complex matrix by DLLME is still at the beginning stage of exploration (Zang et al. 2009). DLLME technique in food analysis of solid or semisolid samples requires a previous extraction step (Asensio-Ramos et al. 2011). Additionally, the matrix presents a very complex composition and potential interferences can be coextracted (Melo et al. 2012).
For solid samples, DLLME has been used in combination with other extraction methods such as dispersive solid-phase extraction for determination of neonicotinoid insecticides in grain (Wang et al. 2012). Also, the combination of QuEChERS method with DLLME for determination of pesticide residues became very popular for fruit (Zhao et al. 2007; Andraščíková et al. 2013) and vegetable samples (Zhao et al. 2007; Cunha and Fernandes 2011; Melo et al. 2013). However, different sample-pretreatment procedures have been proposed due to the wide variety of pesticides used and the inherent complexity of the matrices. In particular, the high lipid content of some samples may interfere with analysis (González-Curbelo et al. 2012). Methods for fatty food analysis are usually laborious and not fully effective in cleaning up the sample if the compounds happen to be evaluated below a few milligrams per kilogram of lipid weight (Farajzadeh et al. 2014). In fact, it is very difficult to avoid the coextraction of fatty material, even more, taking into account that some of the pesticides which are usually targeted are fat-soluble non-polar compounds, and tend to concentrate and remain in the fat. Since high recoveries of most multi-class pesticides must be obtained in an ideally fat-free extract, an additional clean-up step is usually included in the analytical process (Gilbert-López et al. 2009). Several extraction methods were evaluated for the extraction of pesticides in high oil vegetal commodities by Rajski et al. (2013), QuEChERS with various d-SPE clean-ups (Z-Sep, Z-Sep+, PSA + C18, and silica), miniLuke, and ethyl acetate method. To overcome difficulties with extraction of pesticides from avocado and almonds, Rajski et al. (2013) applied sorbent containing ZrO2, which improves fat removal from the extracts. The method provided the highest number of pesticides with satisfactory recoveries and the lowest amount of coextracted matrix compounds. Lacina et al. (2012) evaluated extraction methods for determination of pesticide residues in various food matrices including oil seeds. The most efficient extraction method was based on QuEChERS in terms of low quantification limits, repeatability, and recoveries.
Sobhanzadeh et al. (2011) developed a multi-residue method based on precipitation at low temperature followed by matrix solid-phase dispersion-sonication for the determination of multiclass pesticides in palm oil. Dispersive solid-phase extraction clean-up method for determination of pesticide residues in edible oils was improved by evaporation of acetonitrile fraction to dryness and reconstitution in 400 μL of ethyl acetate (Deme et al. 2014).
Determination of pesticide residues is generally performed by gas chromatography (GC) (González-Rodríguez et al. 2008; Andraščíková et al. 2013) or high performance liquid chromatography (HPLC) (López-Fernández et al. 2012).
The main objective of this study was to develop a sample preparation method that combines QuEChERS method with DLLME for fatty food matrices. In this study, acetonitrile was used as an extractive solvent for QuEChERS, and consequently, in a purification step of the procedure, it was used as a dispersive solvent for the DLLME. Special attention was given to the optimization of DLLME parameters. To the best of our knowledge, such combination of sample preparation method was not applied to high lipid content samples such as oil seed samples.
Experimental
Chemicals
Pesticide standards were obtained from different sources with purity >95 % (Dr. Ehrensdorfer, Augsburg, Germany; Bayer, Leverkusen, Germany; Cheminova, Harboore, Denmark; Agrovita, Ivanka pri Dunaji, Slovak Republic). Individual standard stock solution of pesticides was prepared at 1 mg/mL in toluene (Merck KGaA, Darmstadt, Germany), and consequently composite stock standard solution at a concentration of 0.020 mg/mL in acetonitrile was prepared. Additional dilutions needed for preparation of calibration standards were done in acetonitrile (MeCN) (Merck KGaA, Darmstadt, Germany). All standards and stock solutions were stored at −18 °C and working solutions at +4 °C. Sartorius Analytic MC1 scales (Sartorius, Göttingen, Germany) were used for standards weighing. Magnesium sulfate (MgSO4)—clean, anhydrous, and sodium chloride (NaCl)—per analysis were from Lachema (Lachema a.s., Brno, Czech Republic). MgSO4 was baked at 500 °C (5 h) and NaCl at 600 °C (6 h). Tetrachloroethane and carbon tetrachloride purity reagent grade, chloroform, and water for residual analysis were obtained from Sigma-Aldrich, GmbH (Steinheim, Germany).
Sample Preparation
The pumpkin seeds with water (1:1, w/v) were homogenized with a Braun MX 2050 blender (Kronberg, Germany) and the pumpkin seeds pyre was stored at −16 °C until further sample treatment. For sample preparation, QuEChERS methods (Anastassiades et al. 2003) based on acetonitrile extraction and DLLME were used.
Ten grams of homogenized sample was accurately weighed into a 50-mL centrifuge tube (polypropylene; Bio-Chrom s.r.o., Bratislava, Slovakia) and extracted with 10 mL of MeCN. Following liquid–liquid partitioning, 1 g NaCl and 4 g MgSO4 were added and the mixture was shaken by hand for 1 min. Subsequently, the mixture was centrifuged (ROTOFIX 32; Hettich centrifugen, Tuttlingen, Germany) at 4000 rpm for 5 min. After centrifugation, a 1-mL aliquot of acetonitrile extract, also serving as dispersive solvent, was transferred into the 15-mL centrifuge tube for DLLME procedure. Into the MeCN extract (spiked at the appropriate concentration by working standard solution in MeCN), 50 μL extractive solvent chloroform (alternatively tetrachloroethane, or carbon tetrachloride for optimization studies) and 3 mL water were added. The mixture was shaken by hand and a cloudy solution that consisted of very fine droplets of extractant dispersed into aqueous sample was formed, and the analytes were extracted into the fine droplets. After centrifugation at 4000 rpm for 2 min, the organic phase was sedimented at the bottom of the centrifuge tube.
Validation Experiments
Matrix-Matched Standards
Pumpkin seeds without pesticides (checked before utilization) were used as a blank matrix in recovery study. The matrix-matched standard solutions were prepared by adding working pesticide solutions of respective concentration into chloroform extract obtained by DLLME after liquid–liquid extraction using QuEChERS method.
Recovery Studies
The recovery studies were performed at four concentrations (10, 50, 100, and 250 μg/kg) using 950 μL of QuEChERS extract spiked with a 25-μL mixture of working standard solutions at appropriate concentrations (0.4, 2, 4, and 10 ng/μL) and triphenylphosphate (TPP) (6 ng/μL), followed by DLLME. Preparation of matrix-matched standard solutions needed for recovery evaluation was as follows: 950 μL of chloroform extract after QuEChERS and DLLME + 25 μL TPP (6 ng/μL) + 25 μL of appropriate working pesticide solution in MeCN (0.4, 2, 4, and 10 ng/μL). Recovery studies at each concentration were performed five times.
Linearity and Precision
Linearity was evaluated at nine concentrations of 0.1, 0.5, 1, 5, 10, 50, 100, 250, and 500 μg/kg. Calibration solutions were prepared from 950 μL MeCN extract spiked with 25 μL mixture of working standard solutions at required concentration (0.004, 0.02, 0.04, 0.2, 0.4, 2, 4, 10, and 20 ng/μL) in MeCN + 25 μL TPP (6 ng/μL) in MeCN followed by DLLME. Each extract was analyzed in triplicate.
GC-MS Equipment
The gas chromatograph Agilent 6890 (Agilent, Little Falls, DE, USA) coupled to mass selective detector Agilent 5975 equipped with programmable temperature vaporization (PTV) injector and an Agilent 7683B autosampler was used in this study. The capillary column 15 m × 0.15 mm I.D. × 0.15 μm film thickness with 5 % diphenyl 95 % dimethylsiloxane stationary phase (CP-Sil 8 CB; Agilent Technologies, Middelburg, The Netherlands) connected to a non-polar deactivated precolumn (1 m × 0.32 mm I.D.) was used for separation under the following temperature programmed conditions optimized in our previous study (Purdešová et al. 2013): 40 °C, held for 1.75 min, increased at 60 °C/min to 150 °C, then increased at 23.8 °C/min to 300 °C and held for 3.20 min. Total chromatographic run was 13.09 min. Helium was used as the carrier gas at constant flow of 1.2 mL/min. The mass spectrometer was operated in electron ionization mode (70 eV). The ion source temperature in EI mode was maintained at 250 °C and a solvent delay of 3 min was selected. In selected ion monitoring (SIM) mode, ions were sorted into groups; the dwell time used was 10 ms.
Results and Discussion
Optimization of GC Conditions
Twenty-four pesticides possessing a wide range of physical-chemical properties such as volatility, polarity, and belonging to different chemical classes (phenol, dinitroaniline, amine, organochlorine, organophosphate, carbamate, chloroacetamide, dicarboximide, triazole, pyrethroid, azole, strobilurin) were the subject of this research. For preliminary experiments, standards prepared in neat solvents (chloroform, carbon tetrachloride, tetrachloroethane) at the concentration corresponding to 100 μg/kg in a pumpkin seed sample were used. Fast GC-MS conditions were optimized taking into consideration different boiling points of solvents used in this study (chloroform, carbon tetrachloride, tetrachloroethane). These GC-MS analyses were performed in full scan acquisition mode. Different temperatures of injector and initial oven temperatures were set up for each studied extractive solvent. For example, chloroform requires the use of relatively low injector and initial oven temperatures due to its low boiling point (61.1 °C). In this study, different initial oven temperatures ranging from 30 to 60 °C and initial temperatures of injector ranging from 15 to 40 °C were assayed for chloroform. Based on these results (data are not shown), 20 °C was adopted for injector and 40 °C for oven. At lower initial temperature of injector, problems with cooling of PTV injector were observed. Based on the results obtained for tetrachloroethane, 80 °C was adopted as the initial injector temperature and an initial oven temperature of 100 °C was selected. For carbon tetrachloride, initial temperature of injector was set at 40 °C and initial oven temperature at 60 °C. The temperature program was modified to achieve the separation of pesticide mixture.
DLLME
Pesticide-free pumpkin seeds (previously checked for the presence of the target pesticide residues) were used in this study. For the preliminary experiments, acetonitrile extract obtained by QuEChERS method (described in “Sample Preparation” section) was spiked with a mixture of pesticides at the concentration corresponding to 100 μg/kg in the sample for GC-MS measurements in full scan mode. In order to obtain the highest extraction efficiency of DLLME, several parameters including selection of extractive and dispersive solvent and volume, salt addition, and extraction time were modified. The peak areas of pesticides or extraction recovery were used to evaluate extraction efficiency under various extraction conditions.
Selection of the Extractive Solvent and its Volume
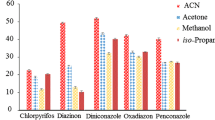
The choice of extractive solvent is of the highest priority to a successful DLLME method and its chemical nature critically determines the ability to extract the target pesticides. The proper extractive solvent should meet some requirements, such as higher density than water, extraction capability of the compounds of interest, good chromatographic behavior, and low solubility in water (Zang et al. 2008). Among the solvents with density higher than water, chloroform, carbon tetrachloride, and tetrachloroethane (densities 1.48, 1.59, and 1.59 g/m3) were investigated for the extraction. The effect of the solvent type was studied by using 1 mL of acetonitrile extract after QuEChERS spiked at the level corresponding to 100 μg/kg in sample as dispersive solvent, 3 mL water, and 50 μL each of the abovementioned extractive solvents. The separations were performed in triplicate and the relative standard deviation (RSD) of pesticide peak areas for different extractive solvents varied in the range of 1–17 %. As can be seen in Fig. 1, for the majority of the analytes, the increase of analytical responses followed the order tetrachloroethane < carbon tetrachloride < chloroform. Therefore, chloroform was selected as the extractive solvent in the subsequent study.

Selection of extractive solvent for the extraction of pesticide residues from pumpkin seeds. Extraction conditions: 1 mL of MeCN extract spiked at 100 μg/kg, 50 μL extractive solvent, and 3 mL water (n = 3)
In order to evaluate the effect of extractive solvent volume on the extraction efficiency, the experiments were performed by using different volumes of chloroform (25, 50, 75, 100 μL) with a constant volume of the dispersive solvent (1 mL) and water (3 mL). The lowest studied volume of the extractive solvent (25 μL) provided insufficient volume of the settled sediment phase for the repetitive injections into the GC by autosampler from the insert vial (after centrifugation, a considerable part of chloroform was not sedimented but dispersed in solution). The volume of sedimented phase was approximately 65, 150, and 200 μL when the volume of extractive solvent was 50, 75, and 100 μL, respectively. Slight solubility of acetonitrile extract in chloroform can cause the increase in the volume of sediment phase. Figure 2 depicts the comparison of enrichment factors (EF) of the pesticides using different volumes of extractive solvent. Enrichment factors were decreased with increasing extractive solvent volume. Therefore, 50 μL was selected as the optimum volume of extractive solvent in order to obtain high enrichment factors and lowest detection limits values. This volume also minimizes the environmental impact and also provides enough sediment phase volume for replicate chromatographic analysis, if required. All the experiments were performed in triplicate, and the average values were used for the creation of graphs.

Comparison of enrichment factors (EF) using different volumes of the extractive solvent chloroform for DLLME for pumpkin seeds at a concentration corresponding to 100 μg/kg in the sample. Extraction conditions: 1 mL of MeCN extract, different volumes of extractive solvent chloroform, and 3 mL water (n = 3)
Effect of Salt
DLLME is an equilibrium-based extraction approach, and the increase of the ionic strength of the aqueous solution could affect the analyte transfer. Therefore, the effect of salt addition on the analytical response was investigated at the different mass concentration of salt in the range of 0–30 % (w/v). Figure 3 illustrates the effect of the salt addition on the peak areas with the use of acetonitrile extract as dispersive solvent. The peak areas decreased with the increasing concentration of NaCl. Although visually cleaner extracts were achieved using NaCl, addition of NaCl caused an increase in the volume of sedimented phase and decreased enrichment factor. We assume that salt addition increased the solubility of acetonitrile in chloroform. The analytical responses leading to the highest enrichment factors were obtained in the absence of salt. NaCl addition caused problems with extractive solvent phase sedimentation—instead of settling at the bottom of the centrifuge tube, it moved to the upper layer in the tube and the separation of phases was more difficult. Consequently, further experiments were performed without salt addition.

The effect of salt addition on the DLLME. Extraction conditions: 1 mL of MeCN extract spiked at the level corresponding to 100 μg/kg, 50 μL extractive solvent chloroform, and 3 mL water (n = 3)
Effect of Extraction Time
In DLLME, the extraction time is defined as the interval between the addition of the mixture of extractive and dispersive solvent to the sample and the start of centrifugation (Zhang et al. 2012). The effect of extraction time was examined in the range of 0 and 60 min at the increment interval of 15 min, while keeping other experimental conditions constant. The experimental results showed that the peak areas did not vary significantly with the extraction time. Based on these experiments, it is evident that DLLME is a time-independent process in the studied time interval.
Ternary System Formation
The criterion for the selection of a dispersive solvent is the complete miscibility with both extractive solvent and the aqueous solution. In this way, emulsion would be dispersed for maximal interfacial analyte exchange. In our case, acetonitrile extract after liquid–liquid extraction by QuEChERS method was used as a dispersive solvent. In preliminary studies (Hrouzková et al. in preparation), it was shown that out of the studied organic solvents, MeCN offers the highest efficiency for extraction of selected pesticides from oil seed samples. Increased volume of dispersive solvent would provide a greater portion of pesticides to the ternary system. The study was therefore carried out to minimize water addition, while allowing sufficient volume ratio dispersive solvent/water to achieve effective emulsification. A series of experiments were carried out using 50 μL extractive solvent while the added volumes of the dispersive solvents were 0.25, 0.5, 0.75, and 1 mL. In addition, the volume of water was examined in the range of 1–3 mL. For illustration, data for 1 and 3 mL water are shown in Fig. 4a and b. Our results showed that higher peak areas were recorded at higher volumes of dispersive solvent, keeping the volume of water at 3 mL. Finally, the volume of dispersive solvent of 1 mL was selected as optimum providing maximum efficiency (Fig. 4) for all tested water addition volumes. As shown in Fig. 4b, pesticide enrichment was important with the increasing water addition volume. It was observed that a portion of water/MeCN was solubilized in the extractive solvent when less than 3 mL water was added, resulting in a transfer of organic phase from the sedimented into the upper layer. The uptake of the upper layer is experimentally complicated; therefore, to minimize the analyte layer dilution, as a compromise, 3 mL of water added volume was used for the next experiment. With the increase of water volume, the volume of sedimented phase was decreased and higher analytical responses were obtained. Also, the peak areas were increased by increasing the volume of MeCN extract.

Comparison of peak area response using different volumes of the dispersive solvent at a concentration of 100 μg/kg with constant volume of water 1 mL (a) and 3 mL (b). The following conditions were chosen as the optimum parameters for DLLME procedure: 50 μL chloroform as extractive solvent, 1 mL MeCN extract as dispersive solvent, and 3 mL water addition
Method Evaluation
The validation of the proposed analytical method was carried out in order to establish the performance parameters which ensure the detection and quantification of pesticide residues in high lipid content samples. Validation of fast GC-MS method was realized following SANCO document (SANCO document 2013). Under the selected extraction conditions, the proposed method was applied to a series of matrix-matched standard solutions at various concentrations of pesticides in order to establish the accuracy, precision, linearity, limits of detection, and limits of quantification. The list of studied pesticides, chemical classes, polarity physical characteristics (expressed by molecular weight), retention characteristics (expressed by partition coefficients), and their target and qualifier ions used in SIM mode are shown in Table 1. A typical gas chromatogram at the concentration corresponding to 50 μg/kg in real sample under the conditions described in the “Experimental” section is shown in Fig. 5. The pesticide mixture was separated in 13.09 min.

Overlay chromatogram of target ions of pesticides after QuEChERS-DLLME sample preparation method (pumpkin seed) analyzed by GC-MS in SIM mode at a concentration of 50 μg/kg. Peak numbering corresponds with the numbers listed in Table 1
Linearity was evaluated at nine concentrations ranging from 0.1 to 500 μg/kg. Each standard was analyzed in triplicate. Good linearities ranging from 0.9842 to 0.9972 were obtained for all studied pesticides. The relative standard deviation (RSD) was calculated at the lowest calibration level (LCL) for each studied pesticide, and it ranged from 0.3 to 22 % with the majority of values below 12 %. The RSD values at the concentration of 50 μg/kg, representing the MRL values for pumpkin and similar (e.g., sunflower) seeds, varied in the range of 6–16 %. Limits of detection (LODs) were evaluated using a signal to noise (S/N) ratio of 3:1 and limits of quantification (LOQs) using S/N of 10:1. LODs of the proposed method are below the MRLs established by the European Union (EU pesticide database 2005) for pumpkin seeds. LODs of the proposed method ranged from 0.01 to 12.17 μg/kg and LOQ were in the range of 0.03–42.37 μg/kg. In one instance (tebuconazole), the LOQ was equal to the MRL value. For other pesticide LOQ values were lower than MRL up to a factor of 1000 (for bifenthrin). The enrichment factor (EF) was calculated (Eq. 1) as the ratio of the concentration of analyte in the collect phase (c sed) and the initial concentration in the sample (c 0), specifically in the QuEChERS extract. The value of c sed was calculated on the basis of matrix-matched injections of pesticide standards in chloroform with concentrations in the range of 0.01–0.25 mg/L.
Under the optimum conditions, the enrichment factor varied from 6 to 17. Validation parameters including LODs, LOQs, enrichment factor (EF), and determination coefficients (R 2) are summarized in Table 2.
The extraction recovery (ER) (Eq. 2) was defined as the percentage of analyte amount extracted in sediment phase (n sed) to the total amount of analyte (n 0). The enrichment factor (see above defined in Eq. 1) was used to calculate extraction recovery.
where V sed is the volume of the sedimented phase and V 0 is the volume of the acetonitrile extract.
Results of recoveries and RSD at different concentrations are shown in Table 3.
Recoveries at four concentrations varied from 70 to 117 % with RSD from 2 to 16 % except for pirimicarb which has the lowest enrichment factor. Pirimicarb with K o/w 1.7, which is the lowest value within the studied mixture, is the most polar analyte to which the proposed extraction method can be applied. Pirimicarb recovery was acceptable only at a higher concentration (250 μg/kg).
Analysis of Real Samples
The proposed method was applied to the analysis of 16 different samples with high lipid content acquired in local markets in Bratislava, Slovakia. Majority of analyzed samples including pumpkin seeds, sunflower seeds, sesame, and flax had no positive findings of studied pesticides above LOQ of the method. Residues of hexachlorobenzene were detected in one unshelled and one shelled pumpkin seed sample at concentrations of 0.74 and 1.46 μg/kg, respectively. Hexachlorobenzene belongs to the group of endocrine disrupting pesticides classified in Category 1 according to European Union classification, which means a compound with endocrinal effect recorded at least on one type of animal. For the illustration, Fig. 6 shows the extracted ion chromatogram of positive findings in the shelled pumpkin seed sample. Maximum residual limit established by the European Union for hexachlorobenzene in pumpkin seed, 50 μg/kg, was not exceeded. Compared with other reported methods (Table 4) for determination of pesticide residues in oil seeds, the present method provides lower LOD due to the enrichment of DLLME by a factor of approximately 10. All methods provide satisfactory recovery complying with the validation requirements. Moreover, the present method is performed in two simple extraction steps, omitting time-consuming purification utilizing sorbents.

Overlap ion chromatogram (m/z = 284, hexachlorobenzene) of positive shelled pumpkin seed sample (black line) and pure matrix (blue line) analyzed by GC-MS in SIM mode
Conclusions
In this work, a sensitive method for the determination of pesticides in pumpkin seed samples has been developed by combining the first step of QuEChERS with DLLME and GC-MS. DLLME was performed using chloroform as an extractive solvent and acetonitrile extract obtained by QuEChERS as dispersive solvent. After the isolation of the target pesticides from pumpkin seeds into acetonitrile by QuEChERS method, analytes were concentrated by DLLME and enriched by a factor of 10 or more. The developed method has been successfully applied to the analysis of real oilseed samples.
References
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Andraščíková M, Hrouzková S, Cunha SC (2013) Combination of QuEChERS and DLLME for GC-MS determination of pesticide residues in orange samples. Food Addit Contam Part A 30(2):286–297
Asensio-Ramos M, Ravelo-Pérez LM, González-Curbelo MÁ, Hernández-Borges J (2011) Liquid phase microextraction applications in food analysis. J Chromatogr A 1218:7415–7437
Boonchiangma S, Ngeontae W, Srijaranai S (2012) Determination of six pyrethroid insecticides in fruit juice samples using dispersive liquid–liquid microextraction combined with high performance liquid chromatography. Talanta 88:209–215
Colosio C, Tiramani M, Maroni M (2003) Neurobehavioral effects of pesticides: state of the art. NeuroToxicology 24:577–591
Cunha SC, Fernandes JO (2011) Multipesticide residue analysis in maize combining acetonitrile-based extraction with dispersive liquid–liquid microextraction followed by gas chromatography–mass spectrometry. J Chromatogr A 1218:7748–7757
Deme P, Azmeera T, Devi BLAP, Jonnalagadda PR, Prasad RBN, Sarathi UVRV (2014) An improved dispersive solid-phase extraction clean-up method for the gas chromatography-negative chemical ionisation tandem mass spectrometric determination of multiclass pesticide residues in edible oils. Food Chem 142:144–151
EU pesticide database (2005) Regulation (EC) No 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Off J Eur Union L 70:1–16. [cited 2012 June 15]. Available from: http://ec.europa.eu/sanco_pesticides/public/index.cfm
Farajzadeh MA, Vardast MBMR, Bamorowat M (2011) Dispersive liquid-liquid microextraction for the analysis of three organophosphorus pesticides in real samples by high performance liquid chromatography-ultraviolet detection and its optimization by experimental design. Microchim Acta 172:465–470
Farajzadeh MA, Khoshmaram L, Nabil AAA (2014) Determination of pyrethroid pesticides residues in vegetable oils using liquid–liquid extraction and dispersive liquid–liquid microextraction followed by gas chromatography–flame ionization detection. J Food Compos Anal 34:128–135
Fu L, Liu X, Hu J, Zhao X, Wang H, Wang X (2009) Application of dispersive liquid–liquid microextraction for the analysis of triazophos and carbaryl pesticides in water and fruit juice samples. Anal Chim Acta 632:289–295
Gilbert-López B, García-Reyes JF, Molina-Díaz A (2009) Sample treatment and determination of pesticide residues in fatty vegetable matrices: a review. Talanta 79:109–128
González-Curbelo MÁ, Herrera-Herrera AV, Ravelo-Pérez LM, Hernández-Borges J (2012) Sample-preparation methods for pesticide-residue analysis in cereals and derivatives. Trends Anal Chem 38:32–51
González-Rodríguez RM, Rial-Otero R, Cancho-Grande B, Simal-Gándara J (2008) Occurrence of fungicide and insecticide residues in trade samples of leafy vegetables. Food Chem 107:1342–1347
Huo X, Li Q, Lin X, Chen X, Bi K (2011) Application of dispersive liquid–liquid microextraction for the analysis of six fungicides in fruit samples by GC–ECD. Chromatographia 73:313–319
Kocúrová L, Balogh IS, Šandrejová J, Andruch V (2012) Recent advances in dispersive liquid–liquid microextraction using organic solvents lighter than water. A review. Microchem J 102:11–17
Koesukwiwat U, Lehotay SJ, Mastovska K, Dorweiler KD, Leepipatpiboon N (2010) Extension of the QuEChERS method for pesticide residues in cereals to flaxseed, peanuts and doughs. J Agric Food Chem 58:5950–5958
Lacina O, Zachariasova M, Urbanova J, Vaclavikova M, Cajka T, Hajslova J (2012) Critical assessment of extraction methods for the simultaneous determination of pesticide residues and mycotoxins in fruits, cereals, spices and oil seeds employing ultra-high performance liquid chromatography–tandem mass spectrometry. J Chromatogr A 1262:8–18
López-Fernández O, Rial-Otero R, González-Barreiro C, Simal-Gándara J (2012) Surveillance of fungicidal dithiocarbamate residues in fruits and vegetables. Food Chem 134:366–374
Lozowicka B, Jankowska M, Rutkowska E (2009) Comparison of two preparation procedures for determination of pesticides residues in oilseed rape by gas chromatography. Chem Anal 54(3):367–387
Matsadiq G, Hu H-L, Ren H-B, Zhou Y-W, Lu L, Cheng J (2011) Quantification of multi-residue levels in peach juices, pulps and peels using dispersive liquid–liquid microextraction based on floating organic droplet coupled with gas chromatography-electron capture detection. J Chromatogr B 879:2113–2118
Melo A, Cunha SC, Mansilha C, Aguiar A, Pinho O, Ferreira IMPLVO (2012) Monitoring pesticide residues in greenhouse tomato by combining acetonitrile-based extraction with dispersive liquid–liquid microextraction followed by gas-chromatography–mass spectrometry. Food Chem 135:1071–1077
Melo A, Mansilha C, Pinho O, Ferreira IMPLVO (2013) Analysis of pesticides in tomato combining quechers and dispersive liquid–liquid microextraction followed by high-performance liquid chromatography. Food Anal Methods 6:559–568
Nasreddine L, Parent-Massin D (2002) Food contamination by metals and pesticides in the European Union. Should we worry? Toxicol Lett 127:29–41
Ozkan A (2015) Determination of pesticide residues in some oilseeds and nuts using LC-MS/MS analysis. Fresenius Environ Bull 24(2a):615–620
Pose-Juan E, Cancho-Grande B, Rial-Otero R, Simal-Gándara J (2006) The dissipation rates of cyprodinil, fludioxonil, procymidone and vinclozoline during storage of grape juice. Food Control 17:1012–1017
Purdešová A, Hrouzková S, Andraščíková M, Krascsenits Z, Matisová E (2013) Evaluation of calibration approaches for quantification of pesticide residues in surface water by SPE with small-size cartridges followed by fast GC-MS. Anal Methods 5(13):3403–3409
Qiao F, Zhang X, Wang M, Kang Y (2010) Rapid extraction of imidacloprid in tomatoes by ultrasonic dispersion liquid–liquid microextraction coupled with LC determination. Chromatographia 72:331–335
Rajski Ł, Lozano A, Uclés A, Ferrer C, Fernández-Alba AR (2013) Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J Chromatogr A 1304:109–120
Rezaee M, Assadi Y, Milani Hosseini MR, Aghaee E, Ahmadi F, Berijani S (2006) Determination of organic compounds in water using dispersive liquid–liquid microextraction. J Chromatogr A 1116:1–9
SANCO document (2013) Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. Document No. SANCO/12571/2013 Available from: http://www.ec.europa.eufood/plant/pesticides/guidance_documents/docs/qualcontrol_en.pd
Shen Z, He Z, Wang P, Zhou Z, Sun M, Li J, Liu D (2013) Low-density magnetofluid dispersive liquid–liquid microextraction for the fast determination of organochlorine pesticides in water samples by GC-ECD. Anal Chim Acta 793:37–43
Sobhanzadeh E, Bakar NKA, Abas MRB, Nemati K (2011) Low temperature followed by matrix solid-phase dispersion-sonication procedure for the determination of multiclass pesticides in palm oil using LC-TOF-MS. J Hazard Mater 186:1308–1313
Wang P, Yang X, Wang J, Cui J, Dong AJ, Zhao HT, Zhang LW, Wang ZY, Xu RB, Li WJ, Zhang YC, Zhang H, Jing J (2012) Multi-residue method for determination of seven neonicotinoid insecticides in grains using dispersive solid-phase extraction and dispersive liquid–liquid micro-extraction by high performance liquid chromatography. Food Chem 134:1691–1698
Xiong J, Hu B (2008) Comparison of hollow fiber liquid phase microextraction and dispersive liquid–liquid microextraction for the determination of organosulfur pesticides in environmental and beverage samples by gas chromatography with flame photometric detection. J Chromatogr A 1193:7–18
Yang Z-H, Liu Y, Lu Y-L, Wu T, Zhou Z-Q, Liu D-H (2011) Dispersive suspended microextraction. Anal Chim Acta 706:268–274
Zang X, Wang J, Wang O, Wang M, Ma J, Xi G, Wang Z (2008) Analysis of captan, folpet, and captafol in apples by dispersive liquid–liquid microextraction combined with gas chromatography. Anal Bioanal Chem 392:749–754
Zang X-H, Wu Q-H, Zhang M-Y, Xi G-H, Wang Z (2009) Developments of dispersive liquid-liquid microextraction technique. Chin J Anal Chem 37(2):161–168
Zhang S, Yang X, Yin X, Wang C, Wang Z (2012) Dispersive liquid–liquid microextraction combined with sweeping micellar electrokinetic chromatography for the determination of some neonicotinoid insecticides in cucumber samples. Food Chem 133:544–550
Zhang Y, Zhang X, Jiao B (2014) Determination of ten pyrethroids in various fruit juices: comparison of dispersive liquid–liquid microextraction sample preparation and QuEChERS method combined with dispersive liquid–liquid microextraction. Food Chem 159:367–373
Zhao E, Zhao W, Han L, Jiang S, Zhou Z (2007) Application of dispersive liquid–liquid microextraction for the analysis of organophosphorus pesticides in watermelon and cucumber. J Chromatogr A 1175:137–140
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Scientific Grant Agency of the Ministry of Education of the Slovak Republic and the Academy of Sciences (VEGA, project no. 1/0503/14) and by the Slovak Research and Development Agency under contract no. APVV-0797-11.
Conflict of Interest
Mária Andraščíková declares that she has no conflict of interests. Svetlana Hrouzková declares that she has no conflict of interests.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Andraščíková, M., Hrouzková, S. Fast Preconcentration of Pesticide Residues in Oilseeds by Combination of QuEChERS with Dispersive Liquid–Liquid Microextraction Followed by Gas Chromatography-Mass Spectrometry. Food Anal. Methods 9, 2182–2193 (2016). https://doi.org/10.1007/s12161-016-0402-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0402-0