
Abstract
Mitochondria, essential organelles responsible for cellular energy production, emerge as a key factor in the pathogenesis of neurodegenerative disorders. This review explores advancements in mitochondrial biology studies that highlight the pivotal connection between mitochondrial dysfunctions and neurological conditions such as Alzheimer’s, Parkinson’s, Huntington’s, ischemic stroke, and vascular dementia. Mitochondrial DNA mutations, impaired dynamics, and disruptions in the ETC contribute to compromised energy production and heightened oxidative stress. These factors, in turn, lead to neuronal damage and cell death. Recent research has unveiled potential therapeutic strategies targeting mitochondrial dysfunction, including mitochondria targeted therapies and antioxidants. Furthermore, the identification of reliable biomarkers for assessing mitochondrial dysfunction opens new avenues for early diagnosis and monitoring of disease progression. By delving into these advancements, this review underscores the significance of understanding mitochondrial biology in unraveling the mechanisms underlying neurodegenerative disorders. It lays the groundwork for developing targeted treatments to combat these devastating neurological conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over 1.5 billion years ago, mitochondria (Mt) came into existence through the process of endosymbiosis, a process in which a eukaryotic ancestor cell incorporated a prokaryote resembling contemporary α-proteobacteria progenitors [1,2,3] that are derived from ocean dwelling clade [4]. Mt are the double membrane bound cell organelle that produce chemical energy as adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS) and empower the cell to carry on its functions and reactions [5,6,7]. Mt contain their own circular DNA or genomes of maternal origin [8], provided majority of mitochondrial proteins are powered by nuclear genome which are synthesized by cytosolic ribosomes and transferred to outer mitochondrial membrane (OMM), inner mitochondrial membrane (IMM), intermembrane space (IMS), and matrix [9]. The mutation in either mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) disrupts its functions and causes disorders such as cancer [10, 11], neurodegenerative diseases [12, 13], ageing [14, 15], and cardiovascular diseases [16]. Numerous mitochondrial and nuclear genes play specific roles in maintaining mitochondrial integrity and behavior, as detailed in Table 1. Understanding these roles is crucial for devising effective strategies in mitochondrial research for health and disease, extending beyond neurological disorders.
Structure and Function of Mitochondria (Mt)
When utilizing electron microscopy, the Mt exhibit a distinctive double-membrane structure comprised of essential phospholipids. These lipids play a critical role in various processes, including the regulation of membrane curvature, remodeling, and mitochondrial dynamics. Mt is integral to a multitude of cellular functions, such as phospholipid synthesis, hemoglobin biosynthesis, lipid synthesis, stem cell reprogramming, cell cycle progression, cellular proliferation, cell differentiation, ATP production, the citric acid cycle, fatty acid oxidation, innate immunity, iron-sulfur (Fe-S) cluster production, generation and maintenance of reactive oxygen species (ROS), redox signaling, calcium homeostasis, apoptosis, and autophagy [50, 70,71,72,73,74,75,76,77]. These vital cellular processes involve proteins distributed across four distinct mitochondrial compartments: the matrix, IMS, OMM, and IMM [78]. The OMM connects to the cytosol, while the IMM extends into the mitochondrial matrix, housing mtDNA [79]. MtDNA, consisting of approximately 1000–10,000 copies per cell, includes transfer RNAs (tRNAs) [74], two ribosomal RNAs (rRNAs) [13], and complex protein subunits (C1, C2, C3, C4, and C5) [80,81,82]. Over 1500 different proteins [83, 84], including 13 transported from the matrix to the oxidase assembly translocase (TOM complex), contribute to these processes.
The mitochondrial matrix hosts the tricarboxylic acid (TCA) cycle, housing essential enzymes, NADH, and FADH, utilized by the electron transport chain (ETC) to generate a mitochondrial membrane potential (Mtmp) crucial for OXPHOS [85]. OXPHOS facilitates significant ATP production in Mt. Mitochondrial NAD+ (MtNAD+), regulated by enzymes like nicotinamide phosphoribosyltransferase (NAMPT) and mitochondrial nicotinamide mononucleotide adenylyltransferase (NMNAT3), contributes to the intracellular NAD+ pool [86]. Mt NAD+ transporters, SLC25A51 and SLC25A52, aid in maintaining normal NAD+ levels in humans [87]. Disruption of NAMPT, for instance, can interfere with mitochondrial respiration in mammals[88, 89]. The IMM comprises the inner boundary membrane (located near the OMM) and the cristae membrane (found in the innermost regions of the IMM) [90]. The cristae membrane houses pro- and anti-apoptotic proteins, as well as regulators of mitochondrial fusion and fission.
Outer Membrane and Inner Membrane
Major phospholipids in the mitochondrial membrane include phosphatidylcholine, phosphoethanolamine, cardiolipin (CL), and phosphatidic acid (PA). PA, a saturated lipid, aids in the remodeling of the Mt membrane[91]. The OMM proteome comprises integral proteins grouped based on their structure, such as α-helical transmembrane segments and β-barrel proteins with multiple β-strands [92]. These proteins act as a physical barrier, restricting large molecule diffusion into the organelle while allowing the passage of small molecules through different import mechanisms. Outer membrane proteins are initially synthesized as precursors by cytosolic ribosomes, assisted by molecular chaperones in transit through the hydrophobic cytosol. Dedicated protein translocases facilitate their insertion into the Mt surface[9, 93,94,95]. The TOM complex and related membrane proteins mediate interactions between Mt and other cellular organelles, such as the endoplasmic reticulum (ER). These interactions facilitate the exchange of lipids and calcium ions, regulating Mt biogenesis and dynamics [96, 97]. The OMM appears adapted for storing charge with multi-spanning proteins like Ugo1, Mcp3, Ubx2, Om14, Scm4, mammalian PBR, and mammalian MITOL [98,99,100,101,102,103]. The 33-kDa protein Ayr1 functions as an ion channel in the OMM, also found in the ER. With around 200 proteins, the OMM acts as a specialized transport system with channel-like functions[104, 105]. Anion channels (ACs) on the OMM, classified as outer membrane AC (OMAC) and inner membrane AC (IMAC), can be anion selective (ASAC), cation selective (CSAC), or non-selective (NSAC).
Voltage-dependent anion channel (VDAC), porins on the OMM, controls metabolic communication between Mt and the cell[106, 107]. VDAC, comprising three isoforms (VDAC1-3)[23, 32, 108], has a 3D structure with antiparallel β-strands, a β-barrel transmembrane pore, and an N-terminal domain forming an α-helix [109]. VDAC1, positioned between cytosol and Mt, serves as the primary conduit for ions and metabolites, influencing cell bioenergetics and the flow of Krebs cycle intermediates[110,111,112,113]. Nine distinct channel-forming proteins transport metabolites, inorganic ions, and proteins across the OMM [114]. VDAC transports calcium to Mt [115]. VDAC1 is crucial for oxygen consumption and the function of ETC enzymes, while VDAC2 regulates cell death and survival through interactions with Bak and Bax [26]. Similarly, VDAC3 provides electrophysiological characteristics and undergoes post-translational modifications[116,117,118]. Figure 1 illustrates all the inbound and outbound activities of Mt and their association with neurodegenerative diseases.

Mitochondrial dynamics: Ugo1, Mcp3, and Ubx2 are the outer mitochondrial membrane (OMM) multi-spanning proteins for storing charge and 33-kDa protein Ayr1 act as ion channel. VDAC on OMM as a β-barrel transmembrane pore and an N terminal domain forming α-helix. In IMM, complex I accept electron from NADH and transferred to NADH and FADH2 and at the mt matrix, NADH and FADH2 carry electrons from TCA cycle to the ETC across the IMM. NADH: ubiquinone oxidoreductase subunit S4 (NDUFS4) is a subunit of C1 that releases four protons to IMS during NADH oxidation after transmitting two electrons to IMM to ubiquinone (UbQ) through flavin extending to centers of iron and sulfur (Fe–S). In C2, FAD and succinate undergo redox with SDHA (succinate dehydrogenase complex flavoprotein subunit A) and the electron transfer to UbQ is achieved by SDHB (succinate dehydrogenase complex flavoprotein subunit B). C2 modulates ROS along with C1 and C3, and loss in C2 function leads to severe ROS accumulation that is a basis of neurodegenerative disorders. C3 releases four protons to IMS catalyses ubiquinol (CoQH2) to Cyt C. C4 catalyzes electron transfer from Cyt C to molecular oxygen; the F1F0 (ATP synthase) produces ATP from ADP
Cristae
The organization and morphology of the IMM are intricate and can be divided into two compartments. One of these compartments is situated opposite to the OMM, while the other extends to the IMS through tubular projections known as cristae junctions (CJ)[119]. The IMM structure is established through the formation of protein-lipid complexes known as MICOS (Mitochondrial Contact Site and Cristae Organizing System), which have evolved from α-proteobacteria. Cristae, the folds within the IMM, house essential components such as ETC complexes, F0F1-ATP synthase, OPA1, and MICOS. Notably, the morphology of cristae undergoes changes during mitochondrial respiration [120, 121]. In the context of ferroptotic cells, modifications occur in the structure of cristae, marked by an increase in mitochondrial membrane content and a reduction in cristae structures [122]. OPA1, identified as a dynamin-related GTPase, plays a crucial role in maintaining cristae structure. It exists in two forms, i.e., L-OPA1 (long) and S-OPA1 (short), both of which act as anchors at CJ, preventing the release of cytochrome C (Cyt C) from the intercristae space. This information highlights the complexity of the IMM and cristae structure, underscoring the role of MICOS and OPA1 in maintaining mitochondrial integrity and function. The morphological changes observed in cristae during mitochondrial respiration and in ferroptotic cells further emphasize the dynamic nature of these structures.
Mt in Cellular Energetics
In the IMM, complex I serves as the exclusive electron acceptor from NADH, receiving electrons from the mitochondrial matrix. NADH and FADH2, generated in the TCA cycle, transport electrons across the IMM to the ETC, establishing a high positive potential in the mitochondrial matrix (mtmp). The ETC comprises five complexes, i.e., complex I (C1), complex II (C2), complex III (C3), complex IV (C4), and complex V (C5), encoded by both mitochondrial and nuclear genomes [123]. A vital subunit of C1, known as NADH—ubiquinone oxidoreductase subunit S4 (NDUFS4)—ensures the stability of C1 [124, 125]. During NADH oxidation, C1 releases four protons into the IMS while transferring electrons to ubiquinone (UbQ) through flavin, extending to Fe–S centers [124,125,126,127,128,129,130,131].
In C2, redox reactions occur with FAD and succinate catalyzed by SDHA, and the subsequent electron transfer to UbQ is facilitated by SDHB [132, 133]. C2, along with C1 and 3, plays a role in modulating ROS. Dysfunction in C2 can result in severe ROS accumulation, a contributing factor to neurodegenerative disorders [134,135,136,137]. Complex III releases four protons to the IMS and catalyzes the transfer of electrons from ubiquinol (CoQH2) to Cyt C [138, 139]. Complex IV facilitates electron transfer from Cyt C to molecular oxygen. The F1F0-ATP synthase, also known as ATP synthase, resides in the IMM. It consists of two domains: the hydrophobic F0 domain responsible for proton translocation and the hydrophilic F1 domain present in the matrix. This complex produces ATP from ADP and phosphate using the proton gradient[140,141,142]. Mutations in mitochondrial components can reduce the activity of F1F0-ATP synthase, resulting in diminished energy production[143,144,145,146].
Overview of Mitochondrial Dynamics and Biogenesis
Mt exhibit diverse shapes, ranging from tiny round structures to shorter lengths and larger tubular forms. The interplay between these morphologies involves the binding and rupturing of both the OMM and IMM, a phenomenon known as “Mt dynamics” that regulates the Mt network [147]. The dynamic nature of Mt enables them to adapt their shapes according to specific cellular functions. For instance, during the energy-intensive DNA replication phase (S phase), Mt can become hyperfused to enhance ATP production [148]. Proteins located on the OMM, including fission 1 (FIS1) and mitochondrial fission factor (MFF), assemble at specific locations. CL and PA, constituting 2% and 5% of total lipids in mammalian cells, respectively, play a role in this process. Although these lipids are enriched in Mt, the assembly of Dnm2, a GTPase involved in mt fission, occurs at membrane constrictions, resulting in individual Mt formation [149, 150].
The precise control of mt morphology is crucial for mitochondrial function and homeostasis (Fig. 2). Overexpression of Bif-1b/c enhances neuronal survival by promoting mt elongation, maintaining membrane potential, and reducing apoptosis [151]. The fusion of Mitofusin 1 (Mfn1) and Mitofusin 2 (Mfn2) in the OMM forms oligomers that expand the mitochondrial surface both within individual Mt and between nearby Mt [152]. Dynamins involved in division are thought to oligomerize in a GTP-dependent manner, forming helices that wrap around Mt [153]. Additionally, proteins like MFF, uniquely found in humans on the OMM, are essential for mt division [154]. These physical contacts persist under dynamic conditions, emphasizing the significance of the ER-Mitochondrial (ER-Mt) interface for proper functioning [155].

Mitochondrial fission and fusion: The intricate processes of mitochondrial dynamics, encompassing both fission and fusion events. Mitochondrial fission: The division of a mitochondrion into two separate entities, facilitated by the recruitment of dynamin-related protein (Drp1) to the outer mitochondrial membrane (OMM). This process ensures the maintenance of mitochondrial quality control and distribution. Mitochondrial fusion: The merging of two individual Mt, orchestrated by mitofusins (Mfn1 and Mfn2) on the OMM and optic atrophy 1 (OPA1) on the inner mitochondrial membrane (IMM). Fusion is crucial for the exchange of contents, complementation of damaged Mt, and the preservation of mitochondrial function. These dynamic processes collectively contribute to the regulation of mitochondrial morphology and function within the cell
Dynamics
Mitochondrial fusion is a process where two Mt merge to create healthier organelles, while mitochondrial fission involves the division of a single mitochondrion into several daughter organelles, facilitating the removal of damaged and fragmented Mt [156,157,158]. The term “mitochondrial dynamics” encompasses the interplay of mitochondrial translocation, fusion, and fission. This intricate process is regulated by nuclear-encoded enzymes, primarily big GTPases, as well as mitochondrial lipids, including CL and PA [50, 159]. Throughout the various cellular life processes, mitochondrial fusion and fission can occur rapidly, especially in response to external stress, leading to transient partial fusion events [150, 160].
At least five proteins play essential roles in regulating and maintaining mitochondrial structural dynamics. These include optic atrophy 1 (OPA1), Mfn1, and Mfn2, which facilitate mitochondrial fusion, FIS1, and dynamin-related protein 1 (DRP1), crucial for mitochondrial fission [161,162,163,164]. Mitochondrial dynamics are critical for the regulation of cell death [165]. The mitochondrion, as a dynamic network, plays a pivotal role in the cell by generating ROS, supplying energy, and controlling programmed cell death [166]. Elevated levels of Drp1, Fis1/Mfn1, and PINK1 suggest a shift in mitochondrial dynamics from fission to fusion, despite a reduction in ShcA, a protein regulating ROS [167]. Depletion of any fission-related proteins alters mitochondrial dynamics, leading to elongated mitochondrial morphology [149, 168].
To maintain a healthy mitochondrial network, Mt must achieve a stable state with balanced communication between fission and fusion events. Concurrent fusion and fission processes, controlled by proteins like Drp1, regulate the overall shape, size, and population of Mt [169]. This coordinated control of mitochondrial dynamics, synchronized with the cell cycle, ensures equal distribution of Mt to daughter cells. Drp1, in particular, plays a primary role in coordinating mitochondrial dynamics with mitosis [170]. Therefore, intricate and well-balanced regulatory mechanisms linking mitochondrial dynamics and mitochondrial quality control (mtQC) mechanisms are essential for maintaining the fitness of mitochondrial pools and networks in biological systems.
In the absence of Drp1, Fis1 can collaborate with Mfn2 and OPA1 to facilitate mitochondrial fission by reducing GTPase activity, thereby safeguarding against fusion-induced mitochondrial fragmentation [171]. The depletion of MFF results in a substantial decrease in mitochondrial fission in HeLa cells or MEFs, preventing the recruitment of Drp1 to the OMM. Conversely, an overexpression of MFF leads to the recruitment of Drp1 to the Mt, inducing hyper-fission in these cells [172]. Within mammals, the paralogs MiD51 and MiD49 serve as mitochondrial receptors, facilitating the cytosolic translocation of Drp1 to Mt [173, 174].
A proposed mechanism for the rapid exchange of metabolites, mtDNA, and membrane components is referred to as mitochondrial fusion [175,176,177,178,179,180,181]. Conversely, mitochondrial fission is believed to facilitate the separation of mtDNA and individual Mt from the network, allowing for their subsequent degradation [182,183,184,185,186]. These processes, mt fission and fusion, play a pivotal role in influencing various aspects of mitochondrial function, including respiration, calcium buffering, and apoptosis [28, 187,188,189,190].
The dynamics of mitochondrial fusion and fission are further regulated by specific phosphilipid, PA and CL that are promoting fusion and fission respectively [191]. The Miro-Milton complex, subject to calcium-dependent regulation, links Mt with kinesin motors, thereby controlling mitochondrial motility and the delicate balance between fission and fusion [192]. In the context of cellular transport, small, spherical Mt resulting from mitochondrial fission are crucial for axonal cell transport, whereas mitochondrial fusion provides protection against external stimuli [193].
Disruptions in the equilibrium between mitochondrial fission and fusion can have far-reaching consequences, impacting mitochondrial function and contributing to various diseases [194]. Enhanced expression of mitochondrial fission promotes fragmentation of the mitochondrial network, leads to the release of Cyt C from Mt, and increases apoptosis [27, 28]. Additionally, upon fracturing the mitochondrial network, FIS1 has been observed to reduce the abundance and survival of mitochondrial fusion proteins, including Mfn1, Mfn2, and OPA1 [171, 195].
Biogenesis
The process of mitochondrial biogenesis encompasses several vital steps, including the replication of mtDNA, synthesis of both IMM and OMM, production of proteins encoded by the Mt, and import as well as synthesis of nuclear-encoded mitochondrial proteins. Regulatory proteins nuclear respiratory factors 1 and 2 (NRF1 and NRF2) engage with the transcriptional coactivator peroxisome proliferator-activated receptor coactivator-1 (PGC-1), forming a crucial network that oversees mitochondrial biogenesis and energy metabolism [32, 196].
An essential player in this regulatory network is the mitochondrial transcription factor A (Tfam), which plays a pivotal role in mtDNA transcription and replication. Activation of Tfam is orchestrated by the concerted action of NRF1 and NRF2. These transcription factors not only govern the mtDNA processes but also regulate the import of nuclear-encoded mitochondrial proteins. Furthermore, they exert control over the five complexes constituting the mitochondrial ETC [33, 197]. In summary, the collaborative action of NRF1, NRF2, and PGC-1 orchestrates various aspects of mitochondrial biogenesis and function, influencing both mtDNA processes and the composition of the mitochondrial ETC.
Significance of Mt Dysfunction and mtDNA Alterations in Neurological Conditions
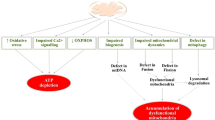
Mitochondrial dysfunction stands as a critical factor influencing both health and disease across a spectrum of physiological and pathological conditions [198] (Fig. 3). The mitochondrion, often referred to as the powerhouse of the cell, plays a pivotal role in energy production and serves as a hub for various cellular processes. In a state of optimal function, Mt orchestrate essential mechanisms such as OXPHOS, contributing to ATP production-the primary energy currency of the cell. Mt are also integral to metabolic pathways, including the citric acid cycle and fatty acid oxidation, crucial for maintaining cellular homeostasis [199]. However, when mitochondrial function falters, it becomes a contributing factor to the onset and progression of various diseases. Neurological disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis, are strongly linked to mitochondrial dysfunction. The repercussions extend beyond the nervous system, encompassing conditions like cardiovascular diseases, diabetes, and age-related degenerative disorders.

Mitochondrial dysfunction leading to different neurological disorders: The significance of Mt dysfunction and mtDNA modifications in various neurological disorders, emphasizing their crucial contribution to disease pathogenesis. The comprehensive overview underscores the unique insights into the molecular mechanisms underlying these conditions, highlighting the imperative need for targeted therapeutic interventions
Several key aspects contribute to mitochondrial dysfunction and subsequent health issues. Genetic mutations in mitochondrial and nDNA can compromise the integrity of proteins involved in mitochondrial function, leading to aberrant processes such as impaired OXPHOS and disrupted energy production. Environmental factors, including exposure to toxins and oxidative stress, further exacerbate mitochondrial damage. Mitochondrial dysfunction also plays a role in the aging process [107]. As cells age, mitochondria accumulate damage, leading to a decline in their function. This aging-associated mitochondrial dysfunction is implicated in a range of age-related diseases. Understanding and addressing mitochondrial dysfunction have become focal points in contemporary medical research. Therapeutic avenues include gene therapies targeting mtDNA, small molecules that enhance mitochondrial function, and strategies to promote mitochondrial biogenesis. Additionally, emerging technologies like mitochondrial transplantation hold promise for mitigating the effects of dysfunctional Mt. In the pursuit of overall health and the prevention of diseases linked to mitochondrial dysfunction, ongoing research aims to unravel the intricate molecular mechanisms governing mitochondrial function. As scientists delve deeper into these complexities, new diagnostic and therapeutic strategies will likely emerge, offering hope for improved treatments and preventive measures against diseases rooted in mitochondrial dysfunction.
The onset of neurodegeneration is prompted by the accumulation of diverse stressors, coupled with the simultaneous disruption of multiple cell-protective systems [47]. In neurodegenerative disorders, a shift in mitochondrial activity significantly contributes to the transition from a normal physiological state to a degenerative one. Pathological protein aggregation, reduced ATP synthesis, and the formation of plaques associated with dopaminergic neuronal death result from the adverse effects of several genetic abnormalities working in concert[200]. Mutations in Parkin and PINK1 exert their influence on Mt monitoring and cell biology[200]. PINK1 is initially translated into the outer OMM and subsequently translocated into Mt for proteolytic degradation in healthy Mt. This underscores the fact that PINK1 levels are typically low in normal mitochondrial conditions. However, when mitochondrial dysfunction occurs, such as membrane depolarization, PINK1 persists as a membrane-anchored component in the OMM. Parkin is activated in its new location through PINK1-mediated phosphorylation. Upon activation, Parkin-mediated ubiquitination signals trigger mitophagy, which is the selective elimination of Mt via the autophagosome [201]. This process leads to functional and anatomical transformations in Mt, impacting various cellular processes. These include excessive ROS generation, a decline in brain energy due to reduced ATP levels, alterations in calcium homeostasis, and the initiation of apoptosis[202, 203].
The circular mtDNA exhibits a mutation rate 10–17 times higher than that of nDNA, playing a crucial role in maintaining mitochondrial integrity[204,205,206]. Circulating mtDNA has been identified in human blood and serves as a potential biomarker for mitochondrial dysfunctions. Mutations in mtDNA, coupled with synaptic damage, result in the inhibition of transcription replication[207], increasing the likelihood of AD by 63% [136]. The impairment of synapses and mitochondrial dysfunction are key contributors to the development of AD[208]. Deletions and point mutations in mtDNA lead to compromised mitochondrial respiration [209,210,211,212,213,214]. LonPeptidase 1 (LONP1) is integral in orchestrating OXPHOS, mtDNA maintenance, and the expression of mitochondrial genes, forming a homo-hexameric complex in the mitochondrial matrix [215,216,217]. Mutations in LONP1 contribute to OXPHOS deficiencies [218], indirectly linking to pathophysiological disorders such as CODAS syndrome and Perrault syndrome. These disorders are associated with disruptions in CLPXP or ERAL1, sometimes manifesting as progressive cerebellar ataxia and intellectual deficit [219, 220].
Mutations in the YME1L gene lead to optic atrophy, developmental delay, and hearing loss, while DRP1 mutations can result in abnormal brain development, microcephaly, and optic atrophy. GDAP1 is implicated in Charcot Marie Tooth disease (CMT). Furthermore, mitochondrial proteins, including ATP5A, NDUFS3, SDHB, and other members such as tetraspanins CD9 and CD63, are found in decreased concentrations in small vesicles of PD patients. In summary, the heightened mutation rate of circular mtDNA, coupled with its interplay with nDNA, underscores its significance in mitochondrial integrity. Dysregulation of these processes contributes to various disorders, emphasizing the intricate connections within the mitochondrial network and their implications for neurodegenerative diseases.
Alzheimer’s Disease
The root cause of AD pathology is attributed to Mt cascade dysfunction [221, 222]. Two critical components in the course of AD are tangles and plaques [223, 224]. This involves the accumulation of β-amyloid in brain vessels [225, 226] and intracellular neurofibrillary tangles resulting from tau protein aggregation [198, 233]. The interaction between amyloid precursor protein (APP) and Aβ with Mt proteins leads to processes responsible for neurodegeneration [227, 228], induced by enhanced mitophagy and Mt defects. In AD patients, a reduction in the activity of Mt C4 has been observed in the hippocampus and platelets [229]. Suppression of communication between Aβ and Aβ-binding alcohol dehydrogenase (ABAD) has been shown to reduce Aβ-induced neuronal death and free radical production. Aβ inhibits two crucial Mt enzymes, α-ketoglutarate dehydrogenase and cytochrome oxidase, both found at low levels in the brains of AD patients. Aβ attaches to the Mt matrix protein, ABAD, following overwhelming complex IV and α-ketoglutarate dehydrogenase [230].
Overexpression of APP, including Nrf2, downregulates Mt fusion, biogenesis, and mitophagy [231]. Inactivated Nrf2 reduces ETC complexes’ activity and lowers NADH and FADH2 expression [232], contributing to the advancement of tau and amyloid in AD patients [233]. The tau protein, losing its physiological activities as AD progresses, reaches the dendrite soma, interacting with β-oligomers and enhancing excitotoxicity, forming neurofibrillary tangles [199, 234]. Aβ plaques, precipitated with high iron amounts, contribute to the development of hazardous Aβ oligomers and ROS, causing Mt malfunction and cell death [235,236,237]. Aberrant metal ion distribution or metabolism leads to synaptic dysfunction directly tied to Mt in the synapses [238]. Excess zinc, generated by increased metalloprotein release, stimulates Aβ synthesis and deposition, initiating a cascade reaction. Inhibition of protein phosphatase and tau hyper-phosphorylation, linked with toxicity related to N-methyl-d-aspartate channel activation and Aβ, is due to increased ROS production from soluble oligomers in the brain and cerebrospinal fluid of AD patients [226, 237, 239].
Chronic hypoxia reduces α-secretase expression, increasing Aβ formation and stimulating mt ROS development [240]. AD brains exhibit decreased fusion protein expression but increased fission protein expression or activity [241]. The increase in S-nitrosylation of dynamin-related protein 1 (Drp1) mediates Mt fission, contributing to AD pathogenesis [242, 243]. In AD brains, ryanodine receptor 2 (RyR2) expression levels are elevated [244], leading to excessive Ca2+ release affecting synaptic plasticity [243, 245, 246]. This induces iron-induced mt fission and stimulates mt Ca2+ uptake, indicating RyR malfunction and neurodegeneration [17, 247, 248].
Parkinson’s Disease
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons in the substantia nigra and the accumulation of α-synuclein (ASN) oligomers [223, 249], often referred to as Lewy bodies, making it the second most prevalent neurodegenerative condition after AD. The aggregation of ASN oligomers, coupled with disruptions in Ca2+ homeostasis, leads to Mt membrane permeabilization and the opening of the mitochondrial permeability transition pore (MPTP). This cascade results in the generation of ROS [250], release of Cyt C, and induction of apoptosis.
The manifestation of PD includes progressive muscle rigidity and tremors, attributed to a diminished dopaminergic modulation of striatal neurons, thereby modifying motor systems [251,252,253]. Several genetic mutations, including Parkin, PINK-1, LRRK2, DJ-1, and ASN, have been associated with familial PD. These gene products not only participate in mitophagy but also influence ER-Mt connections and signaling in PD [44, 254,255,256]. ASN and the PRKN gene, coding for the E3 ubiquitin-protein ligase parkin, are known to be mutated in early-onset PD, affecting around 10% of patients [257,258,259]. Autosomal recessive PD is linked to mutations in PINK1 and Parkin, resulting in striatal mitochondrial respiration deficiency, neuronal vulnerability, oxidative stress, and impaired mitophagy activation [221, 260,261,262,263,264,265].
Autosomal recessive PD is associated with mutations in PINK1 and Parkin, disrupting the degradation of damaged Mt through the activation of mitophagy [221, 263,264,265]. Both PINK1 and Parkin contribute to the degradation of the mitochondrial fusion proteins Mfn1/2 and induce fission by enhancing fission protein activity while reducing the trafficking proteins Miro 1/2. However, the inactivation of the PINK1-Parkin pathway halts the removal of damaged Mt, leading to a slowdown in mitochondrial protein turnover [266]. Genetic degradation of PINK1 results in deficiencies in striatal mitochondrial respiration and increased vulnerability of neuronal cells, ultimately causing oxidative stress [260,261,262]. The reduction in Mtmp leads to the accumulation of PINK1 at the OMM, where Parkin subsequently removes damaged Mt [186, 254]. Similarly, the absence of Parkin disrupts synaptic plasticity and causes dysfunction in striatal Mt [265].
Parkin ablation induces synaptic plasticity and striatal mitochondrial dysfunction [265]. Mutations in Parkin cause defective mitochondrial morphology in iPSC-derived neurons of PARK2 patients. A prevalent DNA lesion associated with oxidative stress is 8-hydroxy-deoxyguanine (8-oxo-dG), an oxidized form of guanine frequently observed in neurological illnesses like AD and PD [267]. PD patients exhibit elevated levels of oxidized CoQ-10 and 8-hydroxy-2-deoxyguanosine in their cerebrospinal fluid (CSF), implicating mitochondrial oxidative stress and DNA damage in PD pathogenesis [268]. A53T transgenic mice and the brains of PD patients also show mitochondrial degeneration with DNA damage [269]. The GBA gene, encoding the enzyme glucocerebrosidase (GCase) involved in lysosomal hydrolysis, plays a crucial role. GBA mutations cause mitochondrial defects and are associated with Gaucher disease (GD) and PD [270,271,272]. Approximately 5–15% of PD patients have mutations in the GBA gene, making it the most significant genetic risk factor for PD [273].
Huntington's Disease (HD)
Huntington’s disease (HD) is an autosomal dominant neurological disorder characterized by an accumulation of trinucleotide CAG repeats within the huntingtin (HTT) gene, leading to polyglutamine repeats in the huntingtin protein (mtHtt) [274, 275]. This mutation affects ion channels, induces oxidative and metabolic stress, and results in Mt malfunction. Mutant HTT inactivates GAPDH, impairing Mt protein transport, causing mtDNA degradation, and contributing to deletions in HD brains [276]. Neurodegeneration occurs through mutant HTT aggregates, disrupting Mt trafficking and altering neuronal movement [277]. Additionally, there is a reduction in mitophagosomes via mitophagy receptors, hindering mt clearance and leading to a buildup of damaged Mt [278].
MtQC dysfunction is evident in HD, with upregulated fusion proteins and downregulated fission protein expressions causing excessive mt fission [279]. HD pathophysiology includes mt dysfunction, impaired cellular antioxidants, and symptoms affecting motor coordination, cognition, and mental health [280, 281]. Stress induction in lymphoblast cell lines from HD patients reveals increased apoptotic cell death mediated by caspase-3, caspase-8, and caspase-9 activation [282,283,284]. Notably, exposure to stress induces apparent Mt differences and increased apoptosis in lymphoblasts from HD patients [204].
Mt failure is a pivotal factor in HD progression, with anomalies such as mtDNA errors, oxidative stress, calcium imbalance, and increased lipid peroxidation observed in HD mouse models [285,286,287,288] and human brains [281, 289]. These abnormalities are linked to disease progression [286, 288] and severity [281]. The antioxidant system’s inefficiency may result from the mtHtt protein, which reduces acetylase activity through CBP/p300 dimer interaction [290, 291] and affects Nrf2 stability and cellular localization [292]. The decrease in PGC1α, among other dysregulated proteins, contributes to HD pathogenesis by linking with transcriptional dysregulation and mt damage processes [293, 294].
Ischemic Stroke
During ischemia, intramitochondrial calcium levels increase, triggering the activation of mitochondrial phosphatases and subsequent dephosphorylation of the OXPHOS complexes, particularly Cyt c and Cyt c oxidase [295,296,297,298]. This leads to the loss of allosteric regulation by ATP. In the absence of oxygen as the final electron acceptor, OXPHOS is highly stimulated in a feed-forward manner [297, 299]. Simultaneously, due to the lack of cellular energy, the Na+/K+ ATPase pump fails, resulting in neuronal membrane depolarization and the release of excess excitatory neurotransmitters, particularly glutamate [300].
CL, a dimeric phospholipid in the IMM, interacts with various OXPHOS complexes and Cyt C, making it susceptible to oxidative damage [298, 301]. Its peroxidation results in the redistribution to the OMM, causing a 50% decrease in Cyt C oxidase activity. This leads to the release of mitochondrial apoptotic proteins, including Cyt C, apoptosis-inducing factor (AIF), Smac/DIABLO, and HtrA2/OMI, into the cytosol [53, 302,303,304]. These proteins contribute to cell death in the ischemia penumbra through various mechanisms.
During reperfusion, pro-apoptotic proteins from the Bcl-2 family, such as Bid and Bax, increase, with Bid being cleaved into truncated tBid by elevated mitochondrial calcium. tBid interacts with other pro-apoptotic proteins in the mitochondrial membrane. Activated Bad translocates to the OMM, suppressing antiapoptotic proteins [305, 306]. Upon opening of the mitochondrial permeability transition pore (MPTP), Cyt C is released into the cytosol, forming the apoptosome with APAF1 and procaspase-9, initiating apoptosis. SMAC/DIABLO and Omi/HtrA2, released from the mitochondrial IMS, enhance caspase-independent apoptosis by inhibiting inhibitor-of-apoptosis protein (IAP) family members, such as XIAP [55, 307].
Activation of autophagy has a protective effect in the early stages of ischemia by preventing defective Mt from producing harmful chemicals [308,309,310]. Mt normally undergo cellular recycling through autophagy, involving signaling pathways like beclin-1/class III PI3K, AMPK/mTOR, and PI3K/Akt/mTOR [56]. However, prolonged autophagy upregulation can lead to increased cell death.
Implications for Neurological Disorders and Potential Therapeutic Targets
The advancements in understanding mitochondrial function and its intricate involvement in neurological disorders have significant implications for the development of therapeutic interventions. The multifaceted nature of these disorders, ranging from PD and AD to traumatic brain injuries, necessitates a diverse and targeted approach to mitigate their impact on neuronal health. The identification of compounds, such as Szeto-Schiller peptides, Mt-penetrating peptides, and MitoQ, designed to enhance mitochondrial activity, opens up new avenues for therapeutic exploration. These compounds specifically target mitochondrial membranes, addressing the core issues of mitochondrial dysfunction observed in various neurological disorders.
Investigations on the present therapeutic approaches for AD show that among 30 agents at clinical trials, only one (caprylic triglyceride) focuses on their metabolism and its bioenergetics [311]. Similarly, in the case of PD, among 74 and 22 phase 2 and phase 1 clinical trials respectively, only 2 agents (nicotinamide riboside and terazosin) focus on Mt and the energy metabolisms [312]. There lies an inevitable need for mitochondrial therapies, and also the exploration of molecular targets needs to be expanded through research advancement [312].
Among the developing therapeutic approaches for the treatment of mitochondrial disorders, optogenetics marks its position. This technique is achieved by the ion channels/electron pumps/enzymes or transcription factors that are light-sensitive, allowing precise control of the biochemical signaling pathways. It is employed in a more advanced way, such that optogenetics controls mitochondrial fission through light-induced MLCs in many cell types, including HeLa cells, PC12, and SLC25A46−/− HDFn, where SLC25A46−/− HDFn affords to treat mitochondrial disorders [313].
Deep brain stimulation (DBS) is another technique used in the treatment of PD, targeting the subthalamic nucleus for symptomatic PD treatment. The hyperactivity in PD rodents was examined in the M1 pyramidal cells through DBS, where the study also sheds light on in vivo recording of intracellular and juxtacellular network recruiting the GABAergic networks. The activation of cortical SST interneurons by optogenetics mitigates the major symptoms of PD in mice [314]. Though it has promising research findings, DBS is still in the initial stages of medical application [315].
CRISPR-Cas9 is an intricate process to carry out mitochondrial gene editing as there is no guide to deliver the RNA and Cas9 enzyme complexes into the Mt. A recent study by Hussain et al. made a concept proof that the stem loop element sgRNA can be added [316], which will in turn help in precise travel to Mt and also interact functionally with Cas9, which mediates sequence-specific mtDNA cleavage, thus making a great system for targeted mitochondrial genome editing.
Another promising study revealed the set of genes impacting the mTORC1 pathway, which identifies mitochondrial dysfunction [317]. It targets the known leading genes at TORC1 pathway MIOS, RPTOR, WDR24, SEH1L, LAMTOR2/4, RHEB, RRAGA, and MTOR, where the ATF4 KO cells treated with oligomycin showed the induction of Sestrin2 and Redd1is essential to inhibit mTORC1 signaling [318].
Szeto-Schiller (SS) peptides
The Szeto-Schiller (SS) peptides, Mt-penetrating peptides, and MitoQ (ubiquinone covalently linked to lipophilic cation triphenylphosphonium) represent novel compounds designed to target Mt membranes and enhance mitochondrial activity, as reported by Jin et al. [319]. The respiratory chain’s complex II reduces MitoQ to active ubiquinol antioxidant, restoring its efficiency against lipid peroxidation in isolated Mt [320]. CERE120, a riluzole-containing drug with an adeno-associated virus, non-steroidal anti-inflammatory drugs, and caffeine A2A receptor antagonists, has shown promise in reducing the risk of neurodegenerative complications [321].
TIGAR
TIGAR, interacting with various signaling proteins and exhibiting significant mitochondrial functions and cell survival properties, emerges as a potential therapeutic target for conditions like cancer, cardiovascular, and neurological disorders. Despite incomplete understanding of its controls, the localization of TIGAR in subcellular organelles other than Mt, such as the ER and nucleus, warrants further investigation into the mechanisms governing its migration in response to stress [322].
Ursodeoxycholic Acid
Ursodeoxycholic acid (UDCA), an FDA-approved medication for biliary cirrhosis, has demonstrated neuroprotective effects in preclinical studies on PD models by preventing mitochondrial dysfunction [323, 324]. Managing glutathione levels with mitochondrial diseases and using mycophenolate mofetil (MMF) to activate Nrf2 represent promising therapeutic approaches in PD, with limited side effects [325]. Tecfidera, an oral formulation of dimethyl fumarate for multiple sclerosis, activates Nrf2, stimulating genes that promote anti-inflammatory, antioxidant, and mitochondrial biogenetic processes, protecting against MPTP-induced brain toxicity [326].
Niclosamide
Niclosamide’s ability to activate PINK1 and its regulatory enzyme suggests its potential as a treatment for PD [327]. Photobiomodulation, a low-level laser therapy, has been used to induce vascularization in injured muscle tissue with minimal side effects [328]. Treating AD with photobiomodulation aims to directly impact Mt by providing photons to Complex IV, reducing ROS generation from damaged Mt [328]. DNA methylation and transcription changes are explored as tools for reprogramming or differentiating induced pluripotent stem cells to treat neurodegenerative diseases [74, 329].
Edaravone
Edaravone, a drug scavenging free radicals, is approved for post-ischemic stroke and amyotrophic lateral sclerosis, but its effectiveness and safety in traumatic brain injury patients are still under investigation [330]. Apocynin, a NOX inhibitor, and TBHQ, an NRF2 activator, administered together show promising effects in rescuing white and gray matter in traumatic brain injury [331]. Mitoquinone (MitoQ), an antioxidant, leads to downstream effects, increasing NRF2 release and antioxidant enzyme gene expression, and uncouples mitochondrial respiration and phosphorylation to reduce ROS generation and prevent oxidative damage [330, 332, 333].
Mdivi (Mitochondrial Division Inhibitor-1)
Mdivi-1 is an inhibition molecule that suppresses the mitochondrial division by specifically targeting dynamins. The Mdivi-1 not only blocks the Cyt C [334] but also act on Drp1 in neurodegenerative diseases helps reducing the disease specific phenotypic appearance [182, 335]. The Mdivi prevents the Drp1 and GTPasey assembly by binding onto the GTPase and thus suppresses the GTPase activity [334]. In seizures, the death of hippocampal neuron was greatly saved by Mdivi-1 by preventing the Cyt C release and caspase 3 which are already activated [336]. Besides that, the enhanced mitochondrial fission and oxidative also got reduced drastically by Mdivi-1 in epileptic rat [337]. A condition of ischemia/reperfusion, i.e., cerebral damage, was sharply decreased by the Mdivi-1, and downregulated Drp1 and Cyt C was prevailed in ischemia/reperfusion mice [338]. In addition to the Cyt C blocking, Mdivi-1 significantly prevented the Bax from entering into the Mt in Rhabdomyolysis-induced rat [339]. In ischemic cases, Mdivi-1 increased the life of retinal ganglion cells [340].
Luteolin-Flavonoid
Luteolin enforces the mitochondrial respiration amd ATP production provided it depends on ER Ca2+ release channels. It has the hydrogen peroxide inducing property, and mitochondrial respiration increasing ability [341, 342]. It establishes the availability of nicotinamide adenine nucleotide (NADH) and electron carrier by activating the pyruvate dehydrogenase [343]. In mouse synaptosomes, enhanced ATP production was rendered by luteolin [344]. Luteolin facilitated the Nrf2 activation by translocating it to nucleus and thereby upregulated the heme oxygenase1 and NQO1 [345].
Others
Various flavonoids, such as 7,8-dihydroxyflavone, cudraflavone B, liquiritigenin, morachalcones, EGCG, procyanidins, huperzine A, geissoschizine methyl ether, sanguinarine, and fangchinoline, prevent mitochondrial oxidative injury and nerve cell death in HT22 cells induced by glutamate/erastin. Puerarin, derived from Pueraria lobata, exhibits protective effects against glutamate-induced toxicity in SH-SY5Y cells [346,347,348,349,350,351,352,353,354,355,356]. Coenzyme Q10 supplementation, involved in ATP formation, improves mitochondrial function, slowing motor deficits, atrophy, and improving survival in R6/2 mice [357,358,359]. Research on PMX500FI, a synthetic l-carnitine-conjugated alpha-lipoic acid (ALA) derivative, suggests its effective traversal of both the blood–brain and blood-retinal barriers. Additionally, it inhibits histone deacetylase activity, enhances mitochondrial function, and exhibits superior in vivo pharmacokinetics compared to traditional ALA [360,361,362,363,364].
The diverse array of compounds and strategies discussed here highlights the evolving landscape of potential therapeutic targets for neurological disorders. Further research and clinical trials are essential to validate these findings and translate them into effective treatments, offering hope for individuals affected by these challenging conditions.
Biomarkers of Mitochondrial Dysfunction in Neurological Conditions
Some of the present mitochondrial disease detection by laboratory tests are through lactate profiling, amino acid, and organic acid profiling and testing for species of acylcarnitine in mitochondrial diseased patients; and samples like blood, urine and CSF are the established means of detection. Many of the mitochondrial diseases still lie under the rare genetic disorders with approx. more than 350 gene mutations, yet do not contain the sensitive testing methods for the same [365]. The testing of serum creatine kinase levels, which is a muscular isoform, will be normal or only slightly higher in patients with mitochondrial disorders [366]. The identification of the peripheral vascular function in the mitochondrial diseased patients with a confirmed m.3243A > G mutation, which acts as a biomarker of mitochondrial function examined through flow mediated skin fluorescence testing [367]. The technique of near infrared spectroscopy (NMR) was employed in the examination of oxygenated and deoxygenated hemoglobin in skin and muscles at mitochondrial diseased patients, and it did not show significant changes with respect to oxygen consumption and blood flow in muscles [367]. The field of nuclear medicine also supports the diagnosis of some cases of mitochondrial diseases like PD with its single photon emission tomography study, expressing the mtDNA deletions at patients with tremor signs [368].
Focusing on the physical features, short stature is a well-established feature of mitochondrial diseases that are caused by both mtDNA and nDNA [369]. The mitochondrial disorders are the disorders that have a multivariant differential system diseases containing unique phenotypes which occur from changes in genetic makeup of Mt [370]. The most precise and direct way of approaching the mitochondrial identification is through the gene mutation and deletions identification that comprises of MT-TL1, MT-TK, LARS2, MTFMT, C12orf65, NDUFA4, SURF1, COX10, LRPPRC, OPA1, POLG, RRM2B, TWINK, and ESCH1gene mutations and mtDNA deletions [369].
The primary lowering of mitochondrial beta oxidation and 12–14 long-chain acylcarnitines (LCACs) serves as biomarker for PD. Among many diagnostic biomarkers for PD, LCACs serve to be the best tool for diagnosing PD with its high specificity for PD at early stage [371]. Mostly the neurodegenerative disorders are approached with nutrient supplements for treatment which comprises of CoQ10, Selenium, NADH/NAD/nicotinamide, vitamins B and D3, and alpha-lipoic acid [372]. CoQ10 is said to have significant effect on CSF biomarkers for treating AD [373]; selenium partially reversed the damaged dopaminergic neurotransmission in MPTP induced PD mice [374] and high-dose selenate showed improvement in mini mental state score in AD patients [375]; NADH/NAD administration for AD patients did not show any progressive cognitive impairment and also showed increased MDRS (Mattis Dementia Rating Scale) scores [375]; vitamin B supplementation showed increased cognitive function at AD patients [376]; vitamin D3 supplementation found to decrease the osteopenia risk in PD subjects [377] and alpha lipoic acid supplementation had good effects on developing cognitive function in AD patients [378].
Nanotechnology and its implications at therapeutic field makes the promising attempt to make a revolution at targeted drug delivery. This makes the way for delivering the CoQ10 by encapsulating inside nanocapsules and targeting the brain Mt which helps in oxidative stress reduction and enhancing the function of Mt [379]. Another application in nanomaterial delivery for treating dysfunction of AD is by conjugated liposomes which functions in aiming ligands such as transferrin or apolipoprotein E, and a Mt-derived cyclosporin A enhances the mitochondrial functioning and decreases cell death [380]. With many mitochondrial regulators at research, the direct inducers of mitophagy could be the key for its related pathways like PINK1/Parkin pathway in AD, which thus help improve the survival and functional property of glutamine and cholinergic neurons, amyloid beta, and tau pathologies [381].
In a recent study, the sFGF21 and sGDF15, the serum fibroblast growth factor 21 and serum growth differentiation factor 15, respectively, are employed in detection of mitochondrial disorders [382]. In AD, the ratio of L:P and hyperlactacidemia is used in the investigation of role of mitochondrial dysfunction [383]. In the study on hepatocerebral phenotype children, they were found to have complex 1 deficiency, depletion of mtDNA, and also POLG1 mutation [384]. The indicator of neuronal loss or dysfunction of neurons in mitochondrial encephalopathy is by the observation of N-acetylaspartate and choline, which tends to be the specific metabolic profile specific to mitochondrial dysfunction [385]. The lactic acid is neurotoxic, where the reduction of their levels is important but the research on the agents acting on lactic acidosis gave disappointing results [386, 387].
Mitochondrial Biology in Precision Medicine for Neurological Disorders
Mitochondrial mutations always occur in a heteroplasmy state which explains a cell with mitochondrial de novo mutation would also have a normal mtDNA in it [388]. They can be either inherited along generations or they can also be acquired through modifications by environmental changes as well as epigenetic factors, where distinguishing them into primary and secondary mitochondrial dysfunction and treating them accordingly is inevitable [389]. The need for personalized medicine is unavoidable as each mitochondrial dysfunction follows a distinct path of pathophysiology. Their specialized personalized therapies include the therapeutic approach by nucleotide supplementation, replacing the oocyte’s defective mtDNA and exogenous mitochondrial supplementing [390]. Mt being complex needing the demand of precision medicinal approach also shows that their unique dynamics allows them to be engineered for next generation of targeted therapy development [391].
Mitochondrial gene editing is the novel way of treating mitochondrial dysfunctions. Zinc finger deaminases have the potential ability of intrinsic cell penetration, which makes it suitable for gene editing both in nuclear mtDNA and cellular mtDNA paving the way for altering mtDNA mutations that are pathogenic [392]. There is a need for more precise mitochondrial gene editing and it can be achieved by the bacterial toxin DddA derived cytosine base editors (DdCBEs) made of cytosine deaminase, specific to dsDNA. The transcription activator which is similar to effector that is custom made with DNA binding proteins and inhibitor of uracil glycosylase enables the therapeutic modification of mt DNA possible in patients [393]. Achieving such a precise gene editing is further developed by adding the zinc finger base editors (ZnF-DdCBEs) to enhance the precision technology architecture as it contains N or C terminals that enable additional target options [394]. The screening of ZnF-DdCBEs are easy and they are cost effective, adding to the point ZnF are abundant endogeneous proteins of human cells which is much less receptive to factors that translate on reduced immunogenecity, making it more compatable [394]. This needs more cutting research to en-groove its potentiality, to improve methods for counter action for DddAtox deaminase enzyme that spontaneously splits during interactions of independent DNA binding [393]. Many optimized ZnF-DdCBEs have been employed in mtDNA and nDNA mutation specific diseases. Even this is aimed to efficiently discrtuct the mutational diseases at Mt by implication on post antal mice study by delivering a AAV9 to its heart, liver, and skeletal muscles [394].
Artificial Intelligence in Neurodegenerative Disorders
In the developing world, each and every field is empowered using artificial intelligence (AI) in different forms, which is even employed at the medical field. The computer systems using the interdisciplinary science, AI is applied to bring out automation at interfaces in recognition of visual, speech, decision-making, and also translating languages [395] which is applied to health care sector to provide patients, physicians, and lab technicians with time-efficient appointment books, and drug availability detailing, suggesting cost-effective alternative drugs and treatments. The three broad classifications of AI systems in the healthcare are majorly into patient oriented (AiCure), clinician oriented (Aidence, Bot MD), and administrative and operational oriented (Aiva Health, Babylon Health) [396] with the combinations of machine learning (ML) and deep learning (DL) algorithms [397]. The imaging techniques often support the neurodegenerative disorders for detecting the brain pathologies, with PET, SPECT, fMRI for the molecular imaging, fMRI and PET for functional imaging, and CT and MRI for structural imaging that are also employed with AI for accessing their different clinical data sources [398]. The neurodegenerative disease like AD has speech and language skills to be considered the most valuable clinical data as they will be reduced in the course of progression of the disease; thus, their collection in sources like voice data and implementing more of AI powered computational speech processing has been the new tool at processing of AD diagnosis and prediction of their disease progression [399]. The neurological disease diagnosis is achieved by AI mostly using either the ML or DL algorithms and by the elimination of interference factors of the data like unnecessary noises, redundancy factors, and variations which make it more accurate in measuring and analyzing the molecular gene analysis data like the major SNP reports obtained from patients and healthy controls. There are many ML studies carried out on PD, which compared the different biological pathways based on the different features of gene expression in PD diagnostic models with an accuracy rate of 93.8% [400]. There are also similar ML studies in AD with an accuracy of 97.8% which had ML employed to analyze the biomarkers at AD diagnosis which includes the clinical imaging, responsible genes, proteins, and the data of the cognitive tests [401] the ML algorithms also apply at the analysis of various gene-related variations that are found in many mitochondria-related genes [402]. Many generalized studies on neurodegenerative disorders involving ML and DL algorithms find its role in the comparing of the patient data from the control data using the deep analysis of multiple genes involving genes of neuron functioning, cell cycle, and immune responses with an accuracy of 95.2%[403] and the distinguishing of 68 different disease severity in neurological disorders with an accuracy of 88.6%[404]. There are many ways to research on the cognitive monitoring of the neurological disorders, in which AI is found to have the best base with the datasets developed by Gosh et al.[405] which had over 6400 MRI images where each were segregated into different stages (moderate dementia, non-dementia, very mild dementia, and mild dementia) of complexity in progression of the AD using the convolutional neural network technique using image data. Though there are many advances in the diagnosis techniques of ND using AI, as each has its own limitations, AI also has its own way of limitations. The limitations include the availability of data set which may have discrepancies in versions of the data taken, the training data set which has the chances to be small and fragmented, the biased model making which arises when the research set is focused on a single aspect of data, and processing the large datasets may lead to loss in accuracy, but can be eventually achieved when the training data set achieves the best in data volume. With the development of research in neurodegenerative disorders, each aspect of the research development needs its role in development of the diagnosis, where AI would definitely give its hands for future diagnosis of ND with nearing perfect accuracy.
Conclusion
Mt dysfunction is a significant contributor to the pathogenesis of many neurological diseases like AD, PD, HD, ischemic stroke, sepsis, POAG, ALS, multiple sclerosis, LGS, and prion disease. Mt is the essential organelle for neuronal function and survival, containing about 1500 proteins of which mutations in them lead to malfunctioning of the Mt. They perform a broad spectrum of functions comprising of fusion, fission, mitophagy, biogenesis, maintenance of homeostasis, regulation of apoptosis, cell cycle progression, cellular proliferation, and cell differentiation; also comprising of physiological functions like innate immunity, autophagy, redox signalling, calcium homeostasis, and stem cell reprogramming; and other crucial cellular process like production of ATP through OXPHOS, citric acid cycle, fatty acid oxidation, phospholipid synthesis, hemoglobin biosynthesis, generation, and maintenance of ROS. The five complexes of ETC are encoded by the mt and nuclear genomes, where mutation or chemical inhibition in them causes Mt-related diseases and also results in low energy production. The defects in proteins of mtDNA maintenance or repair machinery leads to secondary multiple deletions, duplications or depletion of mtDNA which leads to poor mt respiration, and dysfunction linking to broad spectrum of mt and age-related diseases. There are various mitochondrial and nuclear genes that have its specific role in the maintenance of Mt and its behavior that is discussed (Table 1) which will be the best approaching strategy for mitochondrial research for health and disease, and not only for neurological disorders.
Data Availability
No datasets were generated or analysed during the current study.
References
Zimorski V, Ku C, Martin WF, Gould SB (2014) Endosymbiotic theory for organelle origins. Curr Opin Microbiol 22:38–48. https://doi.org/10.1016/j.mib.2014.09.008
Archibald JM (2015) Endosymbiosis and eukaryotic cell evolution. Curr Biol 25(19):R911–R921. https://doi.org/10.1016/j.cub.2015.07.055
Gray MW, Burger G, Lang BF (1999) Mitochondrial evolution. Science 283(5407):1476–1481. https://doi.org/10.1126/science.283.5407.1476
Brindefalk B, Ettema TJG, Viklund J, Thollesson M, Andersson SGE (2011) A Phylometagenomic exploration of oceanic alphaproteobacteria reveals mitochondrial relatives unrelated to the SAR11 clade. PLoS ONE 6(9):e24457. https://doi.org/10.1371/journal.pone.0024457
Cooper GM (2000) The Cell: A Molecular Approach, 2nd edn. Sinauer Associates, Sunderland (MA)
Cogliati S, Enriquez JA, Scorrano L (2016) mitochondrial cristae: where beauty meets functionality. Trends Biochem Sci 41(3):261–273. https://doi.org/10.1016/j.tibs.2016.01.001
Formosa LE, Ryan MT (2018) Mitochondrial OXPHOS complex assembly lines. Nat Cell Biol 20(5):511–513. https://doi.org/10.1038/s41556-018-0098-z
Chial H, Craig J (2008) mtDNA and mitochondrial diseases. Nat Educ 1(1):217
Wiedemann N, Pfanner N (2017) Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86(1):685–714. https://doi.org/10.1146/annurev-biochem-060815-014352
Gasparre G, Porcelli AM, Lenaz G, Romeo G (2013) Relevance of mitochondrial genetics and metabolism in cancer development. Cold Spring Harb Perspect Biol 5(2):a011411–a011411. https://doi.org/10.1101/cshperspect.a011411
Dumas J-F, Peyta L, Couet C, Servais S (2013) Implication of liver cardiolipins in mitochondrial energy metabolism disorder in cancer cachexia. Biochimie 95(1):27–32. https://doi.org/10.1016/j.biochi.2012.07.009
McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2006) Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients. J Mol Biol 361(3):462–469. https://doi.org/10.1016/j.jmb.2006.06.057
McKenzie M, Lazarou M, Thorburn DR, Ryan MT (2007) Analysis of mitochondrial subunit assembly into respiratory chain complexes using blue native polyacrylamide gel electrophoresis. Anal Biochem 364(2):128–137. https://doi.org/10.1016/j.ab.2007.02.022
Genova ML, Lenaz G (2015) The interplay between respiratory supercomplexes and ROS in aging. Antioxid Redox Signal 23(3):208–238. https://doi.org/10.1089/ars.2014.6214
Kauppila TES, Kauppila JHK, Larsson N-G (2017) Mammalian mitochondria and aging: an update. Cell Metab 25(1):57–71. https://doi.org/10.1016/j.cmet.2016.09.017
Rosca M, Minkler P, Hoppel CL (2011) Cardiac mitochondria in heart failure: normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochimica et Biophysica Acta (BBA)-Bioenergetics 1807(11): 1373–1382s https://doi.org/10.1016/j.bbabio.2011.02.003.
Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, McCaffery JM, Cappelletti M et al (2017) OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep 19(12):2557–2571. https://doi.org/10.1016/j.celrep.2017.05.073
Chapa-Dubocq XR, Rodríguez-Graciani KM, García-Báez J, Vadovsky A, Bazil JN, Javadov S (2023) The role of swelling in the regulation of OPA1-mediated mitochondrial function in the heart in vitro. Cells 12(16):2017. https://doi.org/10.3390/cells12162017
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy–inflammation–cell death axis in organismal aging. Science 333(6046):1109–1112. https://doi.org/10.1126/science.1201940
Czabotar PE, Lessene G, Strasser A, Adams JM (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 15(1):49–63. https://doi.org/10.1038/nrm3722
Salvador-Gallego R, Mund M, Cosentino K, Schneider J, Unsay J, Schraermeyer U, Engelhardt J, Ries J et al (2016) Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. The EMBO J 35(4):389–401 https://doi.org/10.15252/embj.201593384
Li J, Ren P, Chen Z, Ren Z, Lian T, Ma J (2017) Neural attentive session-based recommendation. In Proceedings of the 2017 ACM on Conference on Information and Knowledge Management.s ACM: Singapore Singapore pp 1419–1428. https://doi.org/10.1145/3132847.3132926.
Messina A, Reina S, Guarino F, De Pinto V (2012) VDAC isoforms in mammals. Biochimica et Biophysica Acta (BBA) Biomembranes 1818(6):1466–1476 https://doi.org/10.1016/j.bbamem.2011.10.005
De Stefani D, Rizzuto R, Pozzan T (2016) Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem 85(1):161–192. https://doi.org/10.1146/annurev-biochem-060614-034216
Zinghirino F, Pappalardo XG, Messina A, Guarino F, De Pinto V (2020) Is the Secret of VDAC isoforms in their gene regulation? Characterization of human VDAC genes expression profile, promoter activity, and transcriptional regulators. IJMS 21(19):7388. https://doi.org/10.3390/ijms21197388
Cheng EH-Y, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ (2003) VDAC2 Inhibits BAK activation and mitochondrial apoptosis. Science 301(5632):513–517. https://doi.org/10.1126/science.1083995
Qin S-L, Deng J, Lou D-D, Yu W-F, Pei J, Guan Z-Z (2015) The decreased expression of mitofusin-1 and increased fission-1 together with alterations in mitochondrial morphology in the kidney of rats with chronic fluorosis may involve elevated oxidative stress. J Trace Elem Med Biol 29:263–268. https://doi.org/10.1016/j.jtemb.2014.06.001
Lee Y, Jeong S-Y, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. MBoC 15(11):5001–5011. https://doi.org/10.1091/mbc.e04-04-0294
Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH (2020) Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech Ageing Dev 186:111212. https://doi.org/10.1016/j.mad.2020.111212
Truban D, Hou X, Caulfield TR, Fiesel FC, Springer W (2017) PINK1, Parkin, and mitochondrial quality control: what can we learn about Parkinson’s disease pathobiology? JPD 7(1):13–29. https://doi.org/10.3233/JPD-160989
Venediktova N, Solomadin I, Starinets V (2023) Effect of thyroxine on the structural and dynamic features of cardiac mitochondria and mitophagy in rats. Cells 12(3):396. https://doi.org/10.3390/cells12030396
Wu S, Sampson MJ, Decker WK, Craigen WJ (1999) Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochimica et Biophysica Acta (BBA) Molecular Cell Research 1452(1):68–78s
Kelly DP, Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18(4):357–368. https://doi.org/10.1101/gad.1177604
Sakowska P, Jans DC, Mohanraj K, Riedel D, Jakobs S, Chacinska A (2015) The oxidation status of Mic19 regulates MICOS assembly. Mol Cell Biol 35(24):4222–4237. https://doi.org/10.1128/MCB.00578-15
Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L, Gong L, Sun L et al (2016) Mic60/mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization. Cell Death Differ 23(3):380–392. https://doi.org/10.1038/cdd.2015.102
Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138(4):628–644. https://doi.org/10.1016/j.cell.2009.08.005
Avila-Rodriguez M, Garcia-Segura LM, Hidalgo-lanussa O, Baez E, Gonzalez J, Barreto GE (2016) Tibolone protects astrocytic cells from glucose deprivation through a mechanism involving estrogen receptor beta and the upregulation of neuroglobin expression. Mol Cell Endocrinol 433:35–46. https://doi.org/10.1016/j.mce.2016.05.024
Ji W, Hatch AL, Merrill RA, Strack S, Higgs HN (2015) Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife 4:e11553
Chen Y, Guo S, Tang Y, Mou C, Hu X, Shao F, Yan W, Wu Q (2020) Mitochondrial fusion and fission in neuronal death induced by cerebral ischemia-reperfusion and its clinical application: a mini-review. Med Sci Monit 26s https://doi.org/10.12659/MSM.928651.
Khayati F, Pérez-Cano L, Maouche K, Sadoux A, Boutalbi Z, Podgorniak M-P, Maskos U, Setterblad N et al (2015) EMMPRIN/CD147 is a novel coreceptor of VEGFR-2 mediating its activation by VEGF. Oncotarget 6(12):9766–9780ss
Ong S-B, Kalkhoran SB, Hernández-Reséndiz S, Samangouei P, Ong S-G, Hausenloy DJ (2017) Mitochondrial-shaping proteins in cardiac health and disease – the long and the short of it! Cardiovasc Drugs Ther 31(1):87–107. https://doi.org/10.1007/s10557-016-6710-1
Hardie DG, Pan DA (2002) Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30(6):1064–1070. https://doi.org/10.1042/bst0301064
Pan T, Kondo S, Le W, Jankovic J (2008) The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 131(8):1969–1978. https://doi.org/10.1093/brain/awm318
Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37(6):315–324. https://doi.org/10.1016/j.tins.2014.03.004
Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD + Depletion is necessary and sufficient forpoly(ADP-Ribose) polymerase-1-mediated neuronal death. J Neurosci 30(8):2967–2978. https://doi.org/10.1523/JNEUROSCI.5552-09.2010
Abeti R, Abramov AY, Duchen MR (2011) β-Amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134(6):1658–1672. https://doi.org/10.1093/brain/awr104
Wang P, Deng J, Dong J, Liu J, Bigio EH, Mesulam M, Wang T, Sun L et al (2019) TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet 15(5):e1007947. https://doi.org/10.1371/journal.pgen.1007947
Wang W, Arakawa H, Wang L, Okolo O, Siedlak SL, Jiang Y, Gao J, Xie F et al (2017) Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol Ther 25(1):127–139. https://doi.org/10.1016/j.ymthe.2016.10.013
Salvatori I, Ferri A, Scaricamazza S, Giovannelli I, Serrano A, Rossi S, D’Ambrosi N, Cozzolino M et al (2018) Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J Neurochem 146(5):585–597. https://doi.org/10.1111/jnc.14465
Yu R, Lendahl U, Nistér M, Zhao J (2020) Regulation of mammalian mitochondrial dynamics: opportunities and challenges. Front Endocrinol 11:374. https://doi.org/10.3389/fendo.2020.00374
Downward J (1999) How BAD phosphorylation is good for survival. Nat Cell Biol 1(2):E33–E35. https://doi.org/10.1038/10026
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. In International Review of Cell and Molecular Biology. Elsevier 298: pp 229–317. https://doi.org/10.1016/B978-0-12-394309-5.00006-7.
Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA et al (2005) Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol 1(4):223–232. https://doi.org/10.1038/nchembio727
Webster KA, Graham RM, Thompson JW, Spiga M-G, Frazier DP, Wilson A, Bishopric NH (2006) Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal 8(9–10):1667–1676. https://doi.org/10.1089/ars.2006.8.1667
Galluzzi L, Morselli E, Kepp O, Kroemer G (2009) Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1787(5):402–413
Soares ROS, Losada DM, Jordani MC, Évora P, Castro-e-Silva O (2019) Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. IJMS 20(20):5034. https://doi.org/10.3390/ijms20205034
Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222–230. https://doi.org/10.1038/ni.1980
Wang Z, Lu M, Zhang Y, Ji W, Lei L, Wang W, Fang L, Wang L et al (2019) Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 18(1):e12860. https://doi.org/10.1111/acel.12860
De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau K-F, Ackerley S, Warley A, Shaw CE et al (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostsasis. Hum Mol Genet 21(6):1299–1311. https://doi.org/10.1093/hmg/ddr559
Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K-F, Vizcay-Barrena G, Lin W-L et al (2014) ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5(1):3996. https://doi.org/10.1038/ncomms4996
Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ (2009) Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48(9):2045–2052. https://doi.org/10.1021/bi8019178
Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A et al (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331(6016):456–461. https://doi.org/10.1126/science.1196371
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741. https://doi.org/10.1016/j.cell.2011.10.026
Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, Kohda H, Nakagawa I (2017) Rab35 GTPase recruits NDP52 to autophagy targets. The EMBO Journal 36(18):2790–2807
Pickles S, Vigié P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28(4):R170–R185. https://doi.org/10.1016/j.cub.2018.01.004
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14(2):177–185. https://doi.org/10.1038/ncb2422
Wei Y, Chiang W-C, Sumpter R, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168(1–2):224-238.e10. https://doi.org/10.1016/j.cell.2016.11.042
Chai R, Chen G, Shi HOW, Martin-DeLeon PA, Chen H (2017) Prohibitin involvement in the generation of mitochondrial superoxide at Complex I in human sperm. J Cellular Molecular Medi 21(1):121–129
Li X-H, Chai R-R, Chen G-W, Zhang L-F, Tan-Tai W-J, Shi H-J, Martin-DeLeon P et al (2020) Prohibitin (PHB) Interacts with AKT in mitochondria to coordinately modulate sperm motility. Asian J Androl 22(6):583. https://doi.org/10.4103/aja.aja_46_20
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11(12):872–884. https://doi.org/10.1038/nrm3013
Kamer KJ, Mootha VK (2015) The molecular era of the mitochondrial calcium uniporter. Nat Rev Mol Cell Biol 16(9):545–553. https://doi.org/10.1038/nrm4039
Nikoletopoulou V, Markaki M, Palikaras K (1833) Tavernarakis N (2013) Crosstalk between apoptosis, necrosis and autophagy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 12:3448–3459
Rambold AS, Pearce EL (2018) Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol 39(1):6–18. https://doi.org/10.1016/j.it.2017.08.006
Takashima Y, Guo G, Loos R, Nichols J, Ficz G, Krueger F, Oxley D, Santos F et al (2014) Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell 158(6):1254–1269. https://doi.org/10.1016/j.cell.2014.08.029
Spinelli JB, Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20(7):745–754. https://doi.org/10.1038/s41556-018-0124-1
Osellame LD, Blacker TS, Duchen MR (2012) Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab 26(6):711–723. https://doi.org/10.1016/j.beem.2012.05.003
Rangaraju V, Lewis TL, Hirabayashi Y, Bergami M, Motori E, Cartoni R, Kwon S-K, Courchet J (2019) Pleiotropic mitochondria: the influence of mitochondria on neuronal development and disease. J Neurosci 39(42):8200–8208. https://doi.org/10.1523/JNEUROSCI.1157-19.2019
Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014) The protein import machinery of mitochondria—a regulatory hub in metabolism, stress, and disease. Cell Metab 19(3):357–372. https://doi.org/10.1016/j.cmet.2014.01.010
Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89(3):799–845. https://doi.org/10.1152/physrev.00030.2008
Chocron ES, Munkácsy E (1865) Pickering AM (2019) Cause or casualty: the role of mitochondrial DNA in aging and age-associated disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2:285–297ss
Clayton DA (2000) Transcription and replication of mitochondrial DNA. Hum Reprod 15(suppl 2):11–17. https://doi.org/10.1093/humrep/15.suppl_2.11
Park CB, Larsson N-G (2011) Mitochondrial DNA mutations in disease and aging. J Cell Biol 193(5):809–818. https://doi.org/10.1083/jcb.201010024
Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong S-E, Walford GA, Sugiana C et al (2008) A Mitochondrial protein compendium elucidates complex I disease biology. Cell 134(1):112–123. https://doi.org/10.1016/j.cell.2008.06.016
Morgenstern M, Stiller SB, Lübbert P, Peikert CD, Dannenmaier S, Drepper F, Weill U, Höß P et al (2017) Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep 19(13):2836–2852. https://doi.org/10.1016/j.celrep.2017.06.014
Srere PA, Sumegi B (1986) Organization of the mitochondrial matrix. In: Brautbar N (ed) Myocardial and Skeletal Muscle Bioenergetics. Advances in Experimental Medicine and Biology; Springer, US: Boston, MA, 194: pp 13–25 https://doi.org/10.1007/978-1-4684-5107-8_2.
Berger F, Lau C, Dahlmann M, Ziegler M (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280(43):36334–36341. https://doi.org/10.1074/jbc.M508660200
Luongo TS, Eller JM, Lu M-J, Niere M, Raith F, Perry C, Bornstein MR, Oliphint P et al (2020) SLC25A51 is a mammalian mitochondrial NAD+ transporter. Nature 588(7836):174–179. https://doi.org/10.1038/s41586-020-2741-7
Agerholm M, Dall M, Jensen BAH, Prats C, Madsen S, Basse AL, Graae A-S, Risis S et al (2018) Perturbations of NAD + salvage systems impact mitochondrial function and energy homeostasis in mouse myoblasts and intact skeletal muscle. American Journal of Physiology-Endocrinology and Metabolism 314(4):E377–E395. https://doi.org/10.1152/ajpendo.00213.2017
Frederick DW, Loro E, Liu L, Davila A, Chellappa K, Silverman IM, Quinn WJ, Gosai SJ et al (2016) Loss of NAD homeostasis leads to progressive and reversible degeneration of skeletal muscle. Cell Metab 24(2):269–282. https://doi.org/10.1016/j.cmet.2016.07.005
Giacomello M, Pyakurel A, Glytsou C, Scorrano L (2020) The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21(4):204–224. https://doi.org/10.1038/s41580-020-0210-7
Vance JE (2015) Phospholipid synthesis and transport in mammalian cells. Traffic 16(1):1–18. https://doi.org/10.1111/tra.12230
Burri L, Vascotto K, Gentle IE, Chan NC, Beilharz T, Stapleton DI, Ramage L, Lithgow T (2006) Integral membrane proteins in the mitochondrial outer membrane of Saccharomyces Cerevisiae. FEBS J 273(7):1507–1515. https://doi.org/10.1111/j.1742-4658.2006.05171.x
Becker T, Song J, Pfanner N (2019) Versatility of preprotein transfer from the cytosol to mitochondria. Trends Cell Biol 29(7):534–548. https://doi.org/10.1016/j.tcb.2019.03.007
Hansen KG, Herrmann JM (2019) Transport of proteins into mitochondria. Protein J 38(3):330–342. https://doi.org/10.1007/s10930-019-09819-6
Kutik S, Guiard B, Meyer HE, Wiedemann N, Pfanner N (2007) Cooperation of translocase complexes in mitochondrial protein import. J Cell Biol 179(4):585–591. https://doi.org/10.1083/jcb.200708199
Helle SCJ, Kanfer G, Kolar K, Lang A, Michel AH (1833) Kornmann B (2013) Organization and function of membrane contact sites. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 11:2526–2541s
Eisenberg-Bord M, Shai N, Schuldiner M, Bohnert M (2016) A tether is a tether is a tether: tethering at membrane contact sites. Dev Cell 39(4):395–409. https://doi.org/10.1016/j.devcel.2016.10.022
Sesaki H, Jensen RE (2001) UGO1 Encodes an outer membrane protein required for mitochondrial fusion. J Cell Biol 152(6):1123–1134. https://doi.org/10.1083/jcb.152.6.1123
Sinzel M, Tan T, Wendling P, Kalbacher H, Özbalci C, Chelius X, Westermann B, Brügger B et al (2016) Mcp3 is a novel mitochondrial outer membrane protein that follows a unique IMP-dependent biogenesis pathway. EMBO Reports 17(7):965–981
Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N et al (2019) Mitochondrial protein translocation-associated degradation. Nature 569(7758):679–683. https://doi.org/10.1038/s41586-019-1227-y
Lesnik C, Cohen Y, Atir-Lande A, Schuldiner M, Arava Y (2014) OM14 is a mitochondrial receptor for cytosolic ribosomes that supports co-translational import into mitochondria. Nat Commun 5(1):5711. https://doi.org/10.1038/ncomms6711
Joseph-Liauzun E, Delmas P, Shire D, Ferrara P (1998) Topological analysis of the peripheral benzodiazepine receptor in yeast mitochondrial membranes supports a five-transmembrane structure. J Biol Chem 273(4):2146–2152. https://doi.org/10.1074/jbc.273.4.2146
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S (2006) MARCH-V is a novel Mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7(10):1019–1022. https://doi.org/10.1038/sj.embor.7400790
Pfanner N, Warscheid B, Wiedemann N (2019) Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol 20(5):267–284. https://doi.org/10.1038/s41580-018-0092-0
O’Rourke B (2007) Mitochondrial ion channels. Annu Rev Physiol 69(1):19–49. https://doi.org/10.1146/annurev.physiol.69.031905.163804
Vijayan M, Reddy PH (2022) Reduced VDAC1, Maintained mitochondrial dynamics and enhanced mitochondrial biogenesis in a transgenic tau mouse model of Alzheimer’s disease. Int J Mol Sci 23(15):8561. https://doi.org/10.3390/ijms23158561
Vijayan M, Alvir RV, Alvir RV, Bunquin LE, Pradeepkiran JA, Reddy PH (2022) A partial reduction of VDAC1 enhances mitophagy, autophagy, synaptic activities in a transgenic tau mouse model. Aging Cell 21(8):e13663. https://doi.org/10.1111/acel.13663
Sampson MJ, Lovell RS, Craigen WJ (1996) Isolation, characterization, and mapping of two mouse mitochondrial voltage-dependent anion channel isoforms. Genomics 33(2):283–288. https://doi.org/10.1006/geno.1996.0193
Manzo G, Serra I, Magrí A, Casu M, De Pinto V, Ceccarelli M, Scorciapino MA (2018) Folded structure and membrane affinity of the N-terminal domain of the three human isoforms of the mitochondrial voltage-dependent anion-selective channel. ACS Omega 3(9):11415–11425. https://doi.org/10.1021/acsomega.8b01536
Magri A, Messina A (2018) Interactions of VDAC with proteins involved in neurodegenerative aggregation: an opportunity for advancement on therapeutic molecules. CMC 24(40):4470–4487. https://doi.org/10.2174/0929867324666170601073920
Benz R (1994) Permeation of hydrophilic solutes through mitochondrial outer membranes: review on mitochondrial porins. Biochimica et Biophysica Acta - Reviews on Biomembranes 1197(2):167–196sss
Rostovtseva T, Colombini M (1997) VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J 72(5):1954–1962. https://doi.org/10.1016/S0006-3495(97)78841-6
Gincel D, Shoshan-Barmatz V (2004) Glutamate interacts with VDAC and modulates opening of the mitochondrial permeability transition pore. J Bioenerg Biomembr 36(2):179–186. https://doi.org/10.1023/B:JOBB.0000023621.72873.9e
Krüger V, Becker T, Becker L, Montilla-Martinez M, Ellenrieder L, Vögtle F-N, Meyer HE, Ryan MT et al (2017) Identification of new channels by systematic analysis of the mitochondrial outer membrane. J Cell Biol 216(11):3485–3495. https://doi.org/10.1083/jcb.201706043
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225. https://doi.org/10.1038/nature09663
Reina S, Checchetto V, Saletti R, Gupta A, Chaturvedi D, Guardiani C, Guarino F, Scorciapino MA et al (2016) VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: the putative role of cysteine residue modifications. Oncotarget 7(3):2249–2268
Queralt-Martín M, Bergdoll L, Teijido O, Munshi N, Jacobs D, Kuszak AJ, Protchenko O, Reina S et al (2020) A lower affinity to cytosolic proteins reveals VDAC3 isoform-specific role in mitochondrial biology. J Gen Physiol 152(2):e201912501. https://doi.org/10.1085/jgp.201912501
Saletti R, Reina S, Pittalà MGG, Belfiore R, Cunsolo V, Messina A, De Pinto V (1859) Foti S (2017) High resolustion mass spectrometry characterization of the oxidation pattern of methionine and cysteine residues in rat liver mitochondria voltage-dependent anion selective channel 3 (VDAC3). Biochimica et Biophysica Acta (BBA) - Biomembranes 3:301–311
Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V et al (2013) Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155(1):160–171. https://doi.org/10.1016/j.cell.2013.08.032
Hackenbrock CR (1966) Ultrastructural bases for metabolically linked mechanical activity in mitochondria. J Cell Biol 30(2):269–297. https://doi.org/10.1083/jcb.30.2.269
Hackenbrock CR (1968) Ultrastructural bases for metabolically linked mechanical activity in mitochondria. J Cell Biol 37(2):345–369. https://doi.org/10.1083/jcb.37.2.345
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ et al (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149(5):1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283(5407):1482–1488. https://doi.org/10.1126/science.283.5407.1482
Scacco S, Petruzzella V, Budde S, Vergari R, Tamborra R, Panelli D, Van Den Heuvel LP, Smeitink JA, et al (2003) Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. Journal of Biological Chemistry 278(45): 44161–44167. https://doi.org/10.1074/jbc.M307615200
Kahlhöfer F, Kmita K, Wittig I, Zwicker K (1858) Zickermann V (2017) Accessory Subunit NUYM (NDUFS4) is required for stability of the electron input module and activity of mitochondrial complex I. Biochimica et Biophysica Acta (BBA) - Bioenergetics 2:175–181
Walker JE (1992) The NADH:ubiquinone oxidoreductase (complex I) of respiratory chains. Quart Rev Biophys 25(3):253–324. https://doi.org/10.1017/S003358350000425X
Papa S, De Rasmo D (2013) Complex I deficiencies in neurological disorders. Trends Mol Med 19(1):61–69. https://doi.org/10.1016/j.molmed.2012.11.005
Rodenburg RJ (2016) Mitochondrial complex I-linked disease. Biochimica et Biophysica Acta - Bioenergetics 1857(7):938–945
Abramov AY, Angelova PR (2019) Cellular mechanisms of complex I-associated pathology. Biochem Soc Trans 47(6):1963–1969. https://doi.org/10.1042/BST20191042
Holper L, Ben-Shachar D, Mann JJ (2019) Psychotropic and neurological medication effects on mitochondrial complex I and IV in rodent models. Eur Neuropsychopharmacol 29(9):986–1002. https://doi.org/10.1016/j.euroneuro.2019.06.010
González-Rodríguez P, Zampese E, Stout KA, Guzman JN, Ilijic E, Yang B, Tkatch T, Stavarache MA et al (2021) Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 599(7886):650–656. https://doi.org/10.1038/s41586-021-04059-0
Cecchini G (2003) Function and structure of complex II of the respiratory chain. Annu Rev Biochem 72(1):77–109. https://doi.org/10.1146/annurev.biochem.72.121801.161700
Miyadera H, Shiomi K, Ui H, Yamaguchi Y, Masuma R, Tomoda H, Miyoshi H, Osanai A et al (2003) Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc Natl Acad Sci USA 100(2):473–477. https://doi.org/10.1073/pnas.0237315100
Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, Léger C, Byrne B, Cecchini G et al (2003) Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 299(5607):700–704. https://doi.org/10.1126/science.1079605
Hadrava Vanova K, Kraus M, Neuzil J, Rohlena J (2020) Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep 25(1):26–32. https://doi.org/10.1080/13510002.2020.1752002
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795. https://doi.org/10.1038/nature05292
Iverson TM, Maklashina E, Cecchini G (2012) Structural basis for malfunction in complex II. J Biol Chem 287(42):35430–35438. https://doi.org/10.1074/jbc.R112.408419
Zhang Z, Huang L, Shulmeister VM, Chi Y-I, Kim KK, Hung L-W, Crofts AR, Berry EA et al (1998) Electron transfer by domain movement in cytochrome Bc1. Nature 392(6677):677–684. https://doi.org/10.1038/33612
Meunier B, Fisher N, Ransac S, Mazat J-P (1827) Brasseur G (2013) Respiratory complex III dysfunction in humans and the use of yeast as a model organism to study mitochondrial myopathy and associated diseases. Biochimica et Biophysica Acta (BBAs) - Bioenergetics 11–12:1346–1361https://doi.org/10.1016/j.bbabio.2012.11.015
Mitchell P (1961) Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191(4784):144–148. https://doi.org/10.1038/191144a0
Zhou L, Sazanov LA (2019) Structure and conformational plasticity of the intact Thermus Thermophilus V/A-Type ATPase. Science 365(6455):eaaw9144 https://doi.org/10.1126/science.aaw9144
Spikes TE, Montgomery MG, Walker JE (2020) Structure of the dimeric ATP synthase from bovine mitochondria. Proc Natl Acad Sci USA 117(38):23519–23526. https://doi.org/10.1073/pnas.2013998117
Kucharczyk R, Salin B, Di Rago J-P (2009) Introducing the human Leigh syndrome mutation T9176G into Saccharomyces cerevisiae mitochondrial DNA leads to severe defects in the incorporation of Atp6p into the ATP synthase and in the mitochondrial morphology. Hum Mol Genet 18(15):2889–2898. https://doi.org/10.1093/hmg/ddp226
Ebanks B, Ingram TL, Chakrabarti L (2020) ATP synthase and Alzheimer’s disease: putting a spin on the mitochondrial hypothesis. Aging 12(16):16647–16662 https://doi.org/10.18632/aging.103867
Mnatsakanyan N, Jonas EA (2020) The new role of F1Fo ATP synthase in mitochondria-mediated neurodegeneration and neuroprotection. Exp Neurol 332:113400. https://doi.org/10.1016/j.expneurol.2020.113400
Patro S, Ratna S, Yamamoto HA, Ebenezer AT, Ferguson DS, Kaur A, McIntyre BC, Snow R et al (2021) ATP synsthase and mitochondrial bioenergetics dysfunction in Alzheimer’s disease. IJMS 22(20):11185. https://doi.org/10.3390/ijms222011185
Jin J, Wei X, Zhi X, Wang X, Meng D (2021) Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin 42(5):655–664. https://doi.org/10.1038/s41401-020-00518-y
Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009) A Hyperfused mitochondrial state achieved at G 1 –S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA 106(29):11960–11965. https://doi.org/10.1073/pnas.0904875106
Ferguson SM, De Camilli P (2012) Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol 13(2):75–88. https://doi.org/10.1038/nrm3266
Tilokani L, Nagashima S, Paupe V, Prudent J (2018) Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem 62(3):341–360. https://doi.org/10.1042/EBC20170104
Wang T, Sha H, Ji D, Zhang HL, Chen D, Cao Y, Zhu J (2014) Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 157(7):1591–1604. https://doi.org/10.1016/j.cell.2014.04.042
El-Hattab AW, Suleiman J, Almannai M, Scaglia F (2018) Mitochondrial dynamics: biological roles, molecular machinery, and related diseases. Mol Genet Metab 125(4):315–321. https://doi.org/10.1016/j.ymgme.2018.10.003
Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J (2005) Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170(7):1021–1027. https://doi.org/10.1083/jcb.200506078
Gandre-Babbe S, Van Der Bliek AM (2008) The novel tail-anchored membrane protein mff controls mitochondrial and peroxisomal fission in mammalian cells. MBoC 19(6):2402–2412. https://doi.org/10.1091/mbc.e07-12-1287
Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK (2010) ER sliding dynamics and ER–mitochondrial contacts occur on acetylated microtubules. J Cell Biol 190(3):363–375. https://doi.org/10.1083/jcb.200911024
Mendl N, Occhipinti A, Müller M, Wild P, Dikic I, Reichert AS (2011) Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci 124(8):1339–1350. https://doi.org/10.1242/jcs.076406
Adaniya H, Rudek B, Osipov T, Haxton DJ, Weber T, Rescigno TN, McCurdy CW, Belkacem A, et al (2011) Reply: Phys. Rev. Lett. 106(4): 049302 https://doi.org/10.1103/PhysRevLett.106.049302.
Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i -AAA Protease YME1L and OMA1 Cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204(6):919–929. https://doi.org/10.1083/jcb.201308006
Adachi Y, Itoh K, Yamada T, Cerveny KL, Suzuki TL, Macdonald P, Frohman MA, Ramachandran R et al (2016) Coincident phosphatidic acid interaction restrains Drp1 in mitochondrial division. Mol Cell 63(6):1034–1043. https://doi.org/10.1016/j.molcel.2016.08.013
Bereiter-Hahn J (1990) Behavior of mitochondria in the living cell. In International Review of Cytology; Elsevier 122: pp 1–63 https://doi.org/10.1016/S0074-7696(08)61205-X.
Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22(1):79–99. https://doi.org/10.1146/annurev.cellbio.22.010305.104638
Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E et al (2004) Mutations in the mitochondrial GTPase Mitofusin 2 cause charcot-marie-tooth neuropathy type 2A. Nat Genet 36(5):449–451. https://doi.org/10.1038/ng1341
Bereiter-Hahn J, Vöth M (1994) Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microscopy Res & Technique 27(3):198–219. https://doi.org/10.1002/jemt.1070270303
Frazier AE, Kiu C, Stojanovski D, Hoogenraad NJ, Ryan MT (2006) Mitochondrial morphology and distribution in mammalian cells. Biol Chem 387(12):1551–1558. https://doi.org/10.1515/BC.2006.193
Jeong S-Y, Seol D-W (2008) The role of mitochondria in apoptosis. BMB Rep 41(1):11–22. https://doi.org/10.5483/BMBRep.2008.41.1.011
Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27(2):105–117. https://doi.org/10.1016/j.tem.2015.12.001
Hwang J-A, Shin N, Shin HJ, Yin Y, Kwon HH, Park H, Shin J, Kim SI et al (2021) Protective effects of ShcA protein silencing for photothrombotic cerebral infarction. Transl Stroke Res 12(5):866–878. https://doi.org/10.1007/s12975-020-00874-1
Losón OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 Mediate Drp1 recruitment in mitochondrial fission. MBoC 24(5):659–667. https://doi.org/10.1091/mbc.e12-10-0721
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8(11):870–879. https://doi.org/10.1038/nrm2275
Chen H, Chan DC (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab 26(1):39–48. https://doi.org/10.1016/j.cmet.2017.05.016
Yu R, Jin S, Lendahl U, Nistér M, Zha J (2019) Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. The EMBO Journal 38(8):e99748 https://doi.org/10.15252/embj.201899748
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191(6):1141–1158. https://doi.org/10.1083/jcb.201007152
Zhao W, Varghese M, Yemul S, Pan Y, Cheng A, Marano P, Hassan S, Vempati P et al (2011) Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol Neurodegeneration 6(1):51. https://doi.org/10.1186/1750-1326-6-51
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT (2011) MiD49 and MiD51, New components of the mitochondrial fission machinery. EMBO Rep 12(6):565–573. https://doi.org/10.1038/embor.2011.54
Busch KB, Bereiter-Hahn J, Wittig I, Schagger H, Jendrach M (2006) Mitochondrial dynamics generate equal distribution but patchwork localization of respiratory complex I. Mol Membr Biol 23(6):509–520. https://doi.org/10.1080/09687860600877292
Jakobs S (2006) High resolution imaging of live mitochondria. Biochimica et Biophysica Acta (BBA)- Molecular Cell Research 1763(5–6):561–575https://doi.org/10.1016/j.bbamcr.2006.04.004
Jakobs S, Schauss AC, Hell SW (2003) Photoconversion of matrix targeted GFP enables analysis of continuity and intermixing of the mitochondrial lumen. FEBS Lett 554(1–2):194–200. https://doi.org/10.1016/S0014-5793(03)01170-0
Karbowski M, Arnoult D, Chen H, Chan DC, Smith CL, Youle RJ (2004) Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J Cell Biol 164(4):493–499. https://doi.org/10.1083/jcb.200309082
Nakada K, Inoue K, Ono T, Isobe K, Ogura A, Goto Y-I, Nonaka I, Hayashi J-I (2001) Inter-mitochondrial complementation: mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat Med 7(8):934–940. https://doi.org/10.1038/90976
Ono T, Isobe K, Nakada K, Hayashi J-I (2001) Human cells are protected from mitochondrial dysfunction by complementation of DNA products in fused mitochondria. Nat Genet 28(3):272–275. https://doi.org/10.1038/90116.(181)
Twig G, Graf SA, Wikstrom JD, Mohamed H, Haigh SE, Elorza A, Deutsch M, Zurgil N et al (2006) Tagging and tracking individual networks within a complex mitochondrial web with photoactivatable GFP. Am J Physiol Cell Physiol 291(1):C176–C184. https://doi.org/10.1152/ajpcell.00348.2005
Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Gräber S, Kovacs I, Lee WD et al (2006) Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25(16):3900–3911. https://doi.org/10.1038/sj.emboj.7601253
Gomes LC, Scorrano L (2008) High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochimica et Biophysica Acta (BBA) - Bioenergetics 1777(7–8):860–866 https://doi.org/10.1016/j.bbabio.2008.05.442
Malena A, Loro E, Di Re M, Holt IJ, Vergani L (2009) Inhibition of mitochondrial fission favours mutant over wild-type mitochondrial DNA. Hum Mol Genet 18(18):3407–3416. https://doi.org/10.1093/hmg/ddp281
Suen D-F, Narendra DP, Tanaka A, Manfredi G, Youle RJ (2010) Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA 107(26):11835–11840. https://doi.org/10.1073/pnas.0914569107
Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE et al (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27(2):433–446. https://doi.org/10.1038/sj.emboj.7601963
Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB (1988) Coupling Membranes as Energy-Transmitting Cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. The Journal of cell biology 107(2):481–495 https://doi.org/10.1083/jcb.107.2.481
Aon MA, Cortassa S, O’Rourke B (2004) Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci USA 101(13):4447–4452. https://doi.org/10.1073/pnas.0307156101
Frieden M, James D, Castelbou C, Danckaert A, Martinou J-C, Demaurex N (2004) Ca2+ Homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J Biol Chem 279(21):22704–22714. https://doi.org/10.1074/jbc.M312366200
Skulachev VP (2001) Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci 26(1):23–29. https://doi.org/10.1016/S0968-0004(00)01735-7
Kameoka S, Adachi Y, Okamoto K, Iijima M, Sesaki H (2018) Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol 28(1):67–76. https://doi.org/10.1016/j.tcb.2017.08.011
Liu X, Hajnóczky G (2009) Ca2+-dependent regulation of mitochondrial dynamics by the Miro-Milton complex. Int J Biochem Cell Biol 41(10):1972–1976. https://doi.org/10.1016/j.biocel.2009.05.013
Meyer JN, Leuthner TC, Luz AL (2017) Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391:42–53. https://doi.org/10.1016/j.tox.2017.07.019
Chan DC (2020) Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol Mech Dis 15(1):235–259. https://doi.org/10.1146/annurev-pathmechdis-012419-032711
Pernas L, Scorrano L (2016) Mito-Morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol 78(1):505–531. https://doi.org/10.1146/annurev-physiol-021115-105011
Sanchis-Gomar F, Garcia-Gimenez J, Gomez-Cabrera M, Pallardo F (2014) Mitochondrial biogenesis in health and disease. Molecular and Therapeutic Approaches CPD 20(35):5619–5633. https://doi.org/10.2174/1381612820666140306095106
Uittenbogaard M, Chiaramello A (2014) Mitochondrial biogenesis: a therapeutic target for neurodevelopmental disorders and neurodegenerative diseases. CPD 20(35):5574–5593. https://doi.org/10.2174/1381612820666140305224906
Vijayan M, Yin L, Reddy PH, Benamar K (2022) Behavioral evidence for a Tau and HIV-Gp120 interaction. IJMS 23(10):5514. https://doi.org/10.3390/ijms23105514
Vijayan M, George M, Bunquin LE, Bose C, Reddy PH (2022) protective effects of a small-molecule inhibitor DDQ against Tau-induced toxicities in a transgenic Tau mouse model of Alzheimer’s disease. Hum Mol Genet 31(7):1022–1034. https://doi.org/10.1093/hmg/ddab285
Wu Y, Chen M, Jiang J (2019) Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 49:35–45s https://doi.org/10.1016/j.mito.2019.07.003
Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139(S1):216–231. https://doi.org/10.1111/jnc.13731
Hroudová J, Singh N, Fišar Z (2014) Mitochondrial dysfunctions in neurodegenerative diseases: relevance to Alzheimer’s disease. Biomed Res Int 2014:1–9. https://doi.org/10.1155/2014/175062
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40(3):151–166. https://doi.org/10.1016/j.tins.2017.01.002
Annesley SJ, Fisher PR (2021) Lymphoblastoid cell lines as models to study mitochondrial function in neurological disorders. IJMS 22(9):4536. https://doi.org/10.3390/ijms22094536
Zinovkina LA (2018) Mechanisms of mitochondrial DNA repair in mammals. Biochemistry Moscow 83(3):233–249. https://doi.org/10.1134/S0006297918030045
Allkanjari K, Baldock RA (2021) Beyond base excision repair: an evolving picture of mitochondrial DNA repair. Bioscience Reports 41(10):BSR20211320 https://doi.org/10.1042/BSR20211320
Coskun PE, Beal MF, Wallace DC (2004) Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA 101(29):10726–10731. https://doi.org/10.1073/pnas.0403649101
Reddy PH, Manczak M, Mao P, Calkins MJ, Reddy AP, Shirendeb U (2010) Amyloid-β and mitochondria in aging and Alzheimer’s disease: implications for synaptic damage and cognitive decline. JAD 20(s2):S499–S512. https://doi.org/10.3233/JAD-2010-100504
Marcelino LA, Thilly WG (1999) Mitochondrial mutagenesis in human cells and tissues. Mutation Research/DNA Repair 434(3):177–203. https://doi.org/10.1016/S0921-8777(99)00028-2
Su B, Wang X, Zheng L, Perry G, Smith MA (1802) Zhu X 2010 Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1:135–142ssshttps://doi.org/10.1016/j.bbadis.2009.09.013
Tuppen HAL, Blakely EL, Turnbull DM, Taylor RW (2010) Mitochondrial DNA mutations and human disease. Biochimica et Biophysica Acta (BBAss) - Bioenergetics 1797(2):113–128 https://doi.org/10.1016/j.bbabio.2009.09.005
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS et al (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38(5):515–517. https://doi.org/10.1038/ng1769
Larsson N-G (2010) Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem 79(1):683–706. https://doi.org/10.1146/annurev-biochem-060408-093701
Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148(6):1145–1159. https://doi.org/10.1016/j.cell.2012.02.035
Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, Grutter P, Shoubridge EA (2007) The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. MBoC 18(9):3225–3236. https://doi.org/10.1091/mbc.e07-05-0404
Gustafsson CM, Falkenberg M, Larsson N-G (2016) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85(1):133–160. https://doi.org/10.1146/annurev-biochem-060815-014402
Litonin D, Sologub M, Shi Y, Savkina M, Anikin M, Falkenberg M, Gustafsson CM, Temiakov D (2010) Human mitochondrial transcription revisited. J Biol Chem 285(24):18129–18133. https://doi.org/10.1074/jbc.C110.128918
Peter B, Waddington CL, Oláhová M, Sommerville EW, Hopton S, Pyle A, Champion M, Ohlson M et al (2018) Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum Mol Genet 27(10):1743–1753. https://doi.org/10.1093/hmg/ddy080
Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, Drummond MC, Khan SN et al (2013) Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. The American Journal of Human Genetics 92(4):605–613. https://doi.org/10.1016/j.ajhg.2013.02.013
Chatzispyrou IA, Alders M, Guerrero-Castillo S, Zapata Perez R, Haagmans MA, Mouchiroud L, Koster J, Ofman R et al (2017) A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet 26(13):2541–2550. https://doi.org/10.1093/hmg/ddx152
Monzio Compagnoni G, Di Fonzo A, Corti S, Comi GP, Bresolin N, Masliah E (2020) The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol Neurobiol 57(7):2959–2980. https://doi.org/10.1007/s12035-020-01926-1
Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17(3):157–172. https://doi.org/10.1038/s41582-020-00435-y
Tan SH, Karri V, Tay NWR, Chang KH, Ah HY, Ng PQ, Ho HS, Keh HW et al (2019) Emerging pathways to neurodegeneration: dissecting the critical molecular mechanisms in Alzheimer’s disease. Parkinson’s disease Biomed Pharmacother 111:765–777. https://doi.org/10.1016/j.biopha.2018.12.101
Vijayan M, Reddy PH (2016) Stroke, vascular dementia, and Alzheimer’s disease: molecular links. J Alzheimers Dis 54(2):427–443. https://doi.org/10.3233/JAD-160527
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 330(6012):1774–1774. https://doi.org/10.1126/science.1197623
Wiederholt R, Stainback GA, Paudel R, Khare Y, Naja M, Davis SE, Van Lent T (2020) Economic valuation of the ecological response to hydrologic restoration in the greater everglades ecosystem. Ecol Ind 117:106678. https://doi.org/10.1016/j.ecolind.2020.106678
Chaturvedi RK, Flint Beal M (2013) Mitochondrial diseases of the brain. Free Radical Biol Med 63:1–29. https://doi.org/10.1016/j.freeradbiomed.2013.03.018
Roy S, Rauk A (2005) Alzheimer’s disease and the ‘ABSENT’ hypothesis: mechanism for amyloid β endothelial and neuronal toxicity. Med Hypotheses 65(1):123–137. https://doi.org/10.1016/j.mehy.2004.08.031
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, ShiDu Yan S (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci USA 107(43):18670–18675. https://doi.org/10.1073/pnas.1006586107
Herholz K (2012) Use of FDG PET as an imaging biomarker in clinical trials of Alzheimer’s disease. Biomark Med 6(4):431–439. https://doi.org/10.2217/bmm.12.51
Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum Mol Genet 27(8):1332–1342. https://doi.org/10.1093/hmg/ddy042
Holmström KM, Kostov RV, Dinkova-Kostova AT (2016) The multifaceted role of Nrf2 in mitochondrial function. Current Opinion in Toxicology 1:80–91. https://doi.org/10.1016/j.cotox.2016.10.002
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q et al (2007) Reducing endogenous Tau ameliorates amyloid ss-induced deficits in an Alzheimer’s disease mouse model. Science 316(5825):750–754. https://doi.org/10.1126/science.1141736
Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, Tyagi N (2016) Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: understanding the therapeutics strategies. Mol Neurobiol 53(1):648–661. https://doi.org/10.1007/s12035-014-9053-6
Mani C, Acharya G, Kshirsagar S, Vijayan M, Khan H, Reddy PH, Palle K (2022) A novel role for BRIP1/FANCJ in neuronal cells health and in resolving oxidative stress-induced DNA lesions. J Alzheimers Dis 85(1):207–221. https://doi.org/10.3233/JAD-215305
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA (2002) β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80(1):91–100. https://doi.org/10.1046/j.0022-3042.2001.00681.x
Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14:450–464. https://doi.org/10.1016/j.redox.2017.10.014
Wang L, Yin Y-L, Liu X-Z, Shen P, Zheng Y-G, Lan X-R, Lu C-B, Wang J-Z (2020) Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl Neurodegener 9(1):10. https://doi.org/10.1186/s40035-020-00189-z
Abu-Hassan DW, Li X, Ryan EI, Acott TS, Kelley MJ (2015) Induced pluripotent stem cells restore function in a human cell loss model of open-angle glaucoma. Stem Cells 33(3):751–761. https://doi.org/10.1002/stem.1885
Kelleher RJ, Soiza RL (2013) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3(4):197–226
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. Journal of Neuroscience 29(28):9090–9103s https://doi.org/10.1523/JNEUROSCI.1357-09.2009
Cho D-H, Nakamura T, Lipton SA (2010) Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci 67(20):3435–3447. https://doi.org/10.1007/s00018-010-0435-2
Vijayan M, Bose C, Reddy PH (2021) Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 59:17–29. https://doi.org/10.1016/j.mito.2021.04.005
Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE (2012) Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 33(5):1001.e1-1001.e6. https://doi.org/10.1016/j.neurobiolaging.2011.03.011
Paula-Lima AC, Adasme T, SanMartín C, Sebollela A, Hetz C, Carrasco MA, Ferreira ST, Hidalgo C (2011) Amyloid β-peptide oligomers stimulate RyR-mediated Ca 2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid Redox Signal 14(7):1209–1223. https://doi.org/10.1089/ars.2010.3287
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE (2009) Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 29(30):9458–9470. https://doi.org/10.1523/JNEUROSCI.2047-09.2009
Sun S, Zhang H, Liu J, Popugaeva E, Xu N-J, Feske S, White CL, Bezprozvanny I (2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82(1):79–93. https://doi.org/10.1016/j.neuron.2014.02.019
Zhang J, Yu J, Chen Y, Liu L, Xu M, Sun L, Luo H, Wang Y et al (2018) Exogenous hydrogen sulfide supplement attenuates isoproterenol-induced myocardial hypertrophy in a sirtuin 3-dependent manner. Oxid Med Cell Longev 2018:1–17. https://doi.org/10.1155/2018/9396089
Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ (1999) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154(5):1423–1429. https://doi.org/10.1016/S0002-9440(10)65396-5
Puspita L, Chung SY, Shim J (2017) Oxidative stress and cellular pathologies in Parkinson’s disease. Mol Brain 10(1):53. https://doi.org/10.1186/s13041-017-0340-9
Sanjari Moghaddam H, Valitabar Z, Ashraf-Ganjouei A, Mojtahed Zadeh M, Ghazi Sherbaf F, Aarabi MH (2018) Cerebrospinal fluid C-reactive protein in Parkinson’s disease: associations with motor and non-motor symptoms. Neuromol Med 20(3):376–385. https://doi.org/10.1007/s12017-018-8499-5
Moreira ELG, Rial D, Aguiar AS, Figueiredo CP, Siqueira JM, DalBó S, Horst H, De Oliveira J et al (2010) Proanthocyanidin-rich fraction from croton Celtidifolius Baill confers neuroprotection in the intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine rat model of Parkinson’s disease. J Neural Transm 117(12):1337–1351. https://doi.org/10.1007/s00702-010-0464-x
Rani L, Mondal AC (2020) Emerging concepts of mitochondrial dysfunction in Parkinson’s disease progression: pathogenic and therapeutic implications. Mitochondrion 50:25–34. https://doi.org/10.1016/j.mito.2019.09.010
Narendra D, Tanaka A, Suen D-F, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803. https://doi.org/10.1083/jcb.200809125
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and P62/SQSTM1. Nat Cell Biol 12(2):119–131. https://doi.org/10.1038/ncb2012
Gómez-Suaga P, Bravo-San Pedro JM, González-Polo RA, Fuentes JM, Niso-Santano M (2018) ER–mitochondria signaling in Parkinson’s disease. Cell Death Dis 9(3):337. https://doi.org/10.1038/s41419-017-0079-3
Oczkowska A, Kozubski W, Lianeri M, Dorszewska J (2014) Mutations in PRKN and SNCA genes important for the progress of Parkinson’s disease. CG 14(8): 502–517 https://doi.org/10.2174/1389202914666131210205839.
Pandi S, Chinniah R, Sevak V, Ravi PM, Vijayan M, Vellaiappan NA, Karuppiah B (2020) Association of slow acetylator genotype of N-acetyltransferase 2 with Parkinson’s disease in south Indian population. Neurosci Lett 735:135260. https://doi.org/10.1016/j.neulet.2020.135260
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12(1):9–14. https://doi.org/10.1038/nrm3028
Inamdar N, Arulmozhi D, Tandon A, Bodhankar S (2007) Parkinsons disease: genetics and beyond. CN 5(2):99–113 https://doi.org/10.2174/157015907780866893
Abou-Sleiman PM, Muqit MMK, Wood NW (2006) Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci 7(3):207–219. https://doi.org/10.1038/nrn1868
Martin I, Dawson VL, Dawson TM (2011) Recent advances in the genetics of Parkinson’s disease. Annu Rev Genom Hum Genet 12(1):301–325. https://doi.org/10.1146/annurev-genom-082410-101440
Desai S, Juncker M, Kim C (2018) Regulation of Mitophagy by the ubiquitin pathway in neurodegenerative diseases. Exp Biol Med (Maywood) 243(6):554–562. https://doi.org/10.1177/1535370217752351
Butler D, Bahr BA (2006) Oxidative stress and lysosomes: CNS-related consequences and implications for lysosomal enhancement strategies and induction of autophagy. Antioxid Redox Signal 8(1–2):185–196. https://doi.org/10.1089/ars.2006.8.185
Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT (2009) Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem 284(20):13843–13855. https://doi.org/10.1074/jbc.M808515200
Pryde KR, Smith HL, Chau K-Y, Schapira AHV (2016) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol 213(2):163–171. https://doi.org/10.1083/jcb.201509003
Wang M, Hattori N, Matsumine H, Kobayashi T, Yoshino H, Morioka A, Kitada T, Asakawa S et al (1999) Polymorphism in Theparkin gene in sporadic Parkinson’s disease. Ann Neurol 45(5):655–658. https://doi.org/10.1002/1531-8249(199905)45:5%3c655::AID-ANA15%3e3.0.CO;2-G
Isobe C, Abe T, Terayama Y (2010) Levels of reduced and oxidized coenzymeQ-10 and 8-hydroxy-2′-deoxyguanosine in the cerebrospinal fluid of patients with living Parkinson’s disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. Neurosci Lett 469(1):159–163. https://doi.org/10.1016/j.neulet.2009.11.065
Reeve AK, Ludtmann MH, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, Gandhi S, Turnbull DM et al (2015) Aggregated α-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis 6(7):e1820–e1820. https://doi.org/10.1038/cddis.2015.166
Diao X, Wang F, Becerra-Calixto A, Soto C, Mukherjee A (2021) Induced pluripotent stem cell-derived dopaminergic neurons from familial Parkinson’s disease patients display α-synuclein pathology and abnormal mitochondrial morphology. Cells 10(9):2402. https://doi.org/10.3390/cells10092402
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. The Lancet Neurology 11(11):986–998. https://doi.org/10.1016/S1474-4422(12)70190-4
Osellame LD, Rahim AA, Hargreaves IP, Gegg ME, Richard-Londt A, Brandner S, Waddington SN, Schapira AHV et al (2013) mitochondria and quality control defects in a mouse model of Gaucher disease—links to Parkinson’s disease. Cell Metab 17(6):941–953. https://doi.org/10.1016/j.cmet.2013.04.014
Smith L, Schapira AHV (2022) GBA Variants and Parkinson disease: mechanisms and treatments. Cells 11(8):1261. https://doi.org/10.3390/cells11081261
Reddy KR, Kadlec RH, Flaig E, Gale PM (1999) Phosphorus retention in streams and wetlands: a review. Crit Rev Environ Sci Technol 29(1):83–146. https://doi.org/10.1080/10643389991259182
Squitieri F, Falleni A, Cannella M, Orobello S, Fulceri F, Lenzi P, Fornai F (2010) Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J Neural Transm 117(1):77–83. https://doi.org/10.1007/s00702-009-0328-4
Jha SK, Jha NK, Kumar D, Ambasta RK (1863) Kumar, P (2017) Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in neurodegeneration. Biochimica et Biophysica Acta - Molecular Basis of Disease 5:1132–1146ss https://doi.org/10.1016/j.bbadis.2016.06.015
Chang DTW, Rintoul GL, Pandipati S, Reynolds IJ (2006) Mutant Huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis 22(2):388–400. https://doi.org/10.1016/j.nbd.2005.12.007
Franco-Iborra S, Plaza-Zabala A, Montpeyo M, Sebastian D, Vila M, Martinez-Vicente M (2021) Mutant HTT (Huntingtin) impairs mitophagy in a cellular model of huntington disease. Autophagy 17(3):672–689. https://doi.org/10.1080/15548627.2020.1728096
Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH (2012) Mutant Huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet 21(2):406–420. https://doi.org/10.1093/hmg/ddr475
Vonsattel J-P, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP (1985) Neuropathological classification of Huntingtonʼs disease. J Neuropathol Exp Neurol 44(6):559–577. https://doi.org/10.1097/00005072-198511000-00003
Browne SE, Beal MF (2006) Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal 8(11–12):2061–2073. https://doi.org/10.1089/ars.2006.8.2061
Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Greenamyre JT, Snyder SH et al (1999) Increased apoptosis of huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med 5(10):1194–1198. https://doi.org/10.1038/13518
Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG, Piacentini M, Frati L et al (2006) Genotype-dependent priming to self- and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with Huntington’s disease. J Neurochem 98(4):1090–1099. https://doi.org/10.1111/j.1471-4159.2006.03998.x
Maglione V, Cannella M, Gradini R, Cislaghi G, Squitieri F (2006) Huntingtin fragmentation and increased caspase 3, 8 and 9 activities in lymphoblasts with heterozygous and homozygous Huntington’s disease mutation. Mech Ageing Dev 127(2):213–216. https://doi.org/10.1016/j.mad.2005.09.011
Chen C-M, Wu Y-R, Cheng M-L, Liu J-L, Lee Y-M, Lee P-W, Soong B-W, Chiu DT-Y (2007) Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem Biophys Res Commun 359(2):335–340. https://doi.org/10.1016/j.bbrc.2007.05.093
Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V (2007) Oxidative stress parameters in plasma of Huntington’s disease patients, asymptomatic Huntington’s disease gene carriers and healthy subjects: a cross-sectional study. J Neurol 254(12):1676–1683. https://doi.org/10.1007/s00415-007-0611-y
Lee J, Kosaras B, Del Signore SJ, Cormier K, McKee A, Ratan RR, Kowall NW, Ryu H (2011) Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol 121(4):487–498. https://doi.org/10.1007/s00401-010-0788-5
Perez-Severiano F, Rios C, Segovia J (2000) Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington’s disease. Brain Research 862(1–2):234–237 https://doi.org/10.1016/S0006-8993(00)02082-5
Li S, Li X-J (2006) Optical burst switching with large switching overhead. Mol Neurodegeneration 1(1):19. https://doi.org/10.1186/1750-1326-1-19
Jin YN, Yu YV, Gundemir S, Jo C, Cui M, Tieu K, Johnson GVW (2013) Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE 8(3):e57932. https://doi.org/10.1371/journal.pone.0057932
Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E et al (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413(6857):739–743. https://doi.org/10.1038/35099568
Ganner A, Pfeiffer Z-C, Wingendorf L, Kreis S, Klein M, Walz G, Neumann-Haefelin E (2020) The acetyltransferase P300 regulates NRF2 stability and localization. Biochem Biophys Res Commun 524(4):895–902. https://doi.org/10.1016/j.bbrc.2020.02.006
Intihar TA, Martinez EA, Gomez-Pastor R (2019) Mitochondrial dysfunction in Huntington’s disease; interplay between HSF1, P53 and PGC-1α transcription factors. Front Cell Neurosci 13:103. https://doi.org/10.3389/fncel.2019.00103
Bano D, Zanetti F, Mende Y, Nicotera P (2011) Neurodegenerative processes in Huntington’s disease. Cell Death Dis 2(11):e228–e228. https://doi.org/10.1038/cddis.2011.112
Zaidan E, Sims NR (1994) The calcium content of mitochondria from brain subregions following short-term forebrain ischemia and recirculation in the rat. J Neurochem 63(5):1812–1819. https://doi.org/10.1046/j.1471-4159.1994.63051812.x
Vijayan M, Alamri FF, Al Shoyaib A, Karamyan VT, Reddy PH (2019) Novel miRNA PC-5P-12969 in ischemic stroke. Mol Neurobiol 56(10):6976–6985. https://doi.org/10.1007/s12035-019-1562-x
Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M (2013) Molecular mechanisms of ischemia–reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol 47(1):9–23. https://doi.org/10.1007/s12035-012-8344-z
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2018) Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: implications for pharmacological cardioprotection. American Journal of Physiology-Heart and Circulatory Physiology 315(5):H1341–H1352. https://doi.org/10.1152/ajpheart.00028.2018
Vijayan M, Reddy PH (2020) Non-coding RNAs based molecular links in type 2 diabetes, ischemic stroke, and vascular dementia. J Alzheimers Dis 75(2):353–383. https://doi.org/10.3233/JAD-200070
Bender E, Kadenbach B (2000) The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett 466(1):130–134. https://doi.org/10.1016/S0014-5793(99)01773-1
Kagan VE, Bayır HA, Belikova NA, Kapralov O, Tyurina YY, Tyurin VA, Jiang J, Stoyanovsky DA et al (2009) Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radical Biol Med 46(11):1439–1453. https://doi.org/10.1016/j.freeradbiomed.2009.03.004
Vijayan M, Kumar S, Yin X, Zafer D, Chanana V, Cengiz P, Reddy PH (2018) Identification of novel circulatory microRNA signatures linked to patients with ischemic stroke. Hum Mol Genet 27(13):2318–2329. https://doi.org/10.1093/hmg/ddy136
Fernández P (2002) Shareholder value creation, basic concepts. In Valuation Methods and Shareholder Value Creation. Elsevier, pp 3–20. https://doi.org/10.1016/B978-012253841-4.50002-0.
Vijayan M, Chinniah R, Ravi PM, Sivanadham R, Mosses Joseph AK, Vellaiappan NA, Krishnan JI, Karuppiah B (2016) MTHFR (C677T) CT genotype and CT-apoE3/3 genotypic combination predisposes the risk of ischemic stroke. Gene 591(2):465–470. https://doi.org/10.1016/j.gene.2016.06.062
Vijayan M, Reddy PH (2016) Peripheral biomarkers of stroke: focus on circulatory microRNAs. Biochim Biophys Acta 1862(10):1984–1993. https://doi.org/10.1016/j.bbadis.2016.08.003
Carinci M, Vezzani B, Patergnani S, Ludewig P, Lessmann K, Magnus T, Casetta I, Pugliatti M et al (2021) Different roles of mitochondria in cell death and inflammation: focusing on mitochondrial quality control in ischemic stroke and reperfusion. Biomedicines 9(2):169. https://doi.org/10.3390/biomedicines9020169
Murali V, Rathika C, Ramgopal S, Padma Malini R, Arun Kumar MJ, Neethi Arasu V, Jeyaram Illiayaraja K, Balakrishnan K (2016) Susceptible and protective associations of HLA DRB1*/DQB1* alleles and haplotypes with ischaemic stroke. Int J Immunogenet 43(3):159–165. https://doi.org/10.1111/iji.12266
Ma H, Folmes CDL, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J et al (2015) Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature 524(7564):234–238. https://doi.org/10.1038/nature14546
Vijayan M, Chinniah R, Ravi PM, Mosses Joseph AK, Vellaiappan NA, Krishnan JI, Karuppiah B (2014) ACE-II genotype and I allele predicts ischemic stroke among males in South India. Meta Gene 2:661–669. https://doi.org/10.1016/j.mgene.2014.09.003
Zhang Y, Marsboom G, Toth PT, Rehman J (2013) Mitochondrial respiration regulates adipogenic differentiation of human mesenchymal stem cells. PLoS ONE 8(10):e77077. https://doi.org/10.1371/journal.pone.0077077
Cummings J, Lee G, Nahed P, Kambar MEZN, Zhong K, Fonseca J, et al. Alzheimer’s disease drug development pipeline: 2022. A&D Transl Res & Clin Interv [Internet]. 2022 Jan [cited 2024 Mar 11];8(1):e12295. Available from: https://alz-journals.onlinelibrary.wiley.com/doi/10.1002/trc2.12295
McFarthing K, Rafaloff G, Baptista M, Mursaleen L, Fuest R, Wyse RK, et al. Parkinson’s disease drug therapies in the clinical trial pipeline: 2022 Update. JPD [Internet]. 2022 May 24 [cited 2024 Mar 11];12(4):1073–82. Available from: https://www.medra.org/servlet/aliasResolver?alias=iospress&doi/10.3233/JPD-229002
Plascencia-Villa G, Perry G. Exploring molecular targets for mitochondrial therapies in neurodegenerative diseases. IJMS [Internet]. 2023 Aug 6 [cited 2024 Mar 11];24(15):12486. Available from: https://www.mdpi.com/1422-0067/24/15/12486
Qiu K, Zou W, Fang H, Hao M, Mehta K, Tian Z, et al. Light-activated mitochondrial fission through optogenetic control of mitochondria-lysosome contacts. Nat Commun [Internet]. 2022 Jul 25 [cited 2024 Mar 11];13(1):4303. Available from: https://www.nature.com/articles/s41467-022-31970-5
Valverde S, Vandecasteele M, Piette C, Derousseaux W, Gangarossa G, Aristieta Arbelaiz A, et al. Deep brain stimulation-guided optogenetic rescue of parkinsonian symptoms. Nat Commun [Internet]. 2020 May 13 [cited 2024 Mar 11];11(1):2388. Available from: https://www.nature.com/articles/s41467-020-16046-6
Magno LAV, Tenza-Ferrer H, Collodetti M, Aguiar MFG, Rodrigues APC, Da Silva RS, et al. Optogenetic stimulation of the M2 cortex reverts motor dysfunction in a mouse model of Parkinson’s disease. J Neurosci [Internet]. 2019 Apr 24 [cited 2024 Mar 11];39(17):3234–48. Available from: https://www.jneurosci.org/lookup/doi/10.1523/JNEUROSCI.2277-18.2019
Hussain SRA, Yalvac ME, Khoo B, Eckardt S, McLaughlin KJ. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front Genet [Internet]. 2021 Apr 6 [cited 2024 Mar 11];12:627050. Available from: https://www.frontiersin.org/articles/10.3389/fgene.2021.627050/full
Condon KJ, Orozco JM, Adelmann CH, Spinelli JB, Van Der Helm PW, Roberts JM, et al. Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc Natl Acad Sci USA [Internet]. 2021 Jan 26 [cited 2024 Mar 11];118(4):e2022120118. Available from: https://pnas.org/doi/full/10.1073/pnas.2022120118
Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B (1842) Kanthasamy AG (2014) Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 8:1282–1294https://doi.org/10.1016/j.bbadis.2013.09.007
James AM, Cochemé HM, Smith RAJ, Murphy MP (2005) Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. J Biol Chem 280(22):21295–21312. https://doi.org/10.1074/jbc.M501527200
Chen X, Pan W (2015) The treatment strategies for neurodegenerative diseases by integrative medicine. Integr Med Int 1(4):223–225. https://doi.org/10.1159/000381546
Tang J, Chen L, Qin Z, Sheng R (2021) Structure, regulation, and biological functions of TIGAR and its role in diseases. Acta Pharmacol Sin 42(10):1547–1555. https://doi.org/10.1038/s41401-020-00588-y
Abdelkader NF, Safar MM, Salem HA (2016) Ursodeoxycholic acid ameliorates apoptotic cascade in the rotenone model of Parkinson’s disease: modulation of mitochondrial perturbations. Mol Neurobiol 53(2):810–817. https://doi.org/10.1007/s12035-014-9043-8
Mortiboys H, Furmston R, Bronstad G, Aasly J, Elliott C, Bandmann O (2015) UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2 G2019S carriers and in vivo. Neurology 85(10):846–852. https://doi.org/10.1212/WNL.0000000000001905
Ammal Kaidery N, Thomas B (2018) Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem Int 117:91–113. https://doi.org/10.1016/j.neuint.2018.03.001
Ahuja M, Ammal Kaidery N, Yang L, Calingasan N, Smirnova N, Gaisin A, Gaisina IN, Gazaryan I et al (2016) Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease. J Neurosci 36(23):6332–6351. https://doi.org/10.1523/JNEUROSCI.0426-16.2016
Barini E, Miccoli A, Tinarelli F, Mulholland K, Kadri H, Khanim F, Stojanovski L, Read KD et al (2018) The anthelmintic drug niclosamide and its analogues activate the Parkinson’s disease associated protein kinase PINK1. ChemBioChem 19(5):425–429. https://doi.org/10.1002/cbic.201700500
Yang L, Youngblood H, Wu C, Zhang Q (2020) Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation. Transl Neurodegener 9(1):19. https://doi.org/10.1186/s40035-020-00197-z
Gal A, Balicza P, Weaver D, Naghdi S, Joseph SK, Várnai P, Gyuris T, Horváth A, Nagy L, Seifert EL, Molnar MJ, Hajnóczky G (2017) MSTO 1 is a cytoplasmic pro-mitochondrial fusion protein. EMBO Mol Med 9(7):967–984https://doi.org/10.15252/emmm.201607058
Ismail H, Shakkour Z, Tabet M, Abdelhady S, Kobaisi A, Abedi R, Nasrallah L, Pintus G et al (2020) Traumatic brain injury: oxidative stress and novel anti-oxidants such as mitoquinone and edaravone. Antioxidants 9(10):943. https://doi.org/10.3390/antiox9100943
Ünal İ, Çalışkan-Ak E, Üstündağ ÜV, Ateş PS, Alturfan AA, Altinoz MA, Elmaci I, Emekli-Alturfan E (2020) Neuroprotective effects of mitoquinone and oleandrin on Parkinson’s disease model in zebrafish. Int J Neurosci 130(6):574–582. https://doi.org/10.1080/00207454.2019.1698567
Zhou J, Wang H, Shen R, Fang J, Yang Y, Dai W, Zhu Y, Zhou M (2018) Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am J Transl Res 10(6):1887–1899
Aghili-Mehrizi S, Williams E, Yan S, Willman M, Willman J, Lucke-Wold B (2022) Secondary mechanisms of neurotrauma: a closer look at the evidence. Diseases 10(2):30. https://doi.org/10.3390/diseases10020030
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT et al (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Xie N, Wang C, Lian Y, Zhang H, Wu C, Zhang Q (2013) A selective inhibitor of Drp1, mdivi-1, protects against cell death of hippocampal neurons in pilocarpine-induced seizures in rats. Neurosci Lett 545:64–68
Qiu X, Cao L, Yang X, Zhao X, Liu X, Han Y, Xue Y, Jiang H et al (2013) Role of mitochondrial fission in neuronal injury in pilocarpine-induced epileptic rats. Neuroscience 245:157–165
Zhang N, Wang S, Li Y, Che L, Zhao Q (2013) A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett 535:104–109
Tang WX, Wu WH, Qiu HY, Bo H, Huang SM (2013) Amelioration of rhabdomyolysis-induced renal mitochondrial injury and apoptosis through suppression of Drp-1 translocation. J Nephrol 26:1073–1082
Park SW, Kim KY, Lindsey JD, Dai Y, Heo H, Nguyen DH, Ellisman MH, Weinreb RN et al (2011) A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest Ophthalmol Vis Sci 52:2837–2843
Tam EW, Feigenbaum A, Addis JB, Blaser S, Mackay N, Al-Dosary M, Taylor RW, Ackerley C et al (2008) A novel mitochondrial DNA mutation in COX1 leads to strokes, seizures, and lactic acidosis. Neuropediatrics 39:328–334
Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Schimmer AD (2011) Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell 20(5):674–688
Oliveira AM, Cardoso SM, Ribeiro M, Seixas RSGR, Silva AMS, Rego AC (2015) Protective effects of 3-alkyl luteolin derivatives are mediated by Nrf2 transcriptional activity and decreased oxidative stress in Huntington’s disease mouse striatal cells. Neurochem Int 91:1–12. https://doi.org/10.1016/j.neuint.2015.10.004
Xu J, Wang H, Ding K, Zhang L, Wang C, Li T et al (2014) Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2–ARE pathway. Free Radic Biol Med 71:186–195. https://doi.org/10.1016/j.freeradbiomed.2014.03.009
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta Bioenerg 1787:1309–1316
Chua K, Laurent F, Coombs G, Grayson ML, Howden B et al (2011) Clin Infect Dis 52(12):1472–1472. https://doi.org/10.1093/cid/cir250
Ko A-R, Kang T-C (2017) TRPC6-mediated ERK1/2 phosphorylation prevents dentate granule cell degeneration via inhibiting mitochondrial elongation. Neuropharmacology 121:120–129. https://doi.org/10.1016/j.neuropharm.2017.05.004
Yang E-J, Park GH, Song K-S (2013) Neuroprotective effects of liquiritigenin isolated from licorice roots on glutamate-induced apoptosis in hippocampal neuronal cells. Neurotoxicology 39:114–123. https://doi.org/10.1016/j.neuro.2013.08.012
Wen L, Shi D, Zhou T, Tu J, He M, Jiang Y, Yang B (2020) Identification of two novel prenylated flavonoids in mulberry leaf and their bioactivities. Food Chem 315:126236. https://doi.org/10.1016/j.foodchem.2020.126236
He J, Xu L, Yang L, Sun C (2019) Anti-oxidative effects of catechins and theaflavins on glutamate-induced HT22 cell damage. RSC Adv 9(37):21418–21428. https://doi.org/10.1039/C9RA02721A
Song JH, Lee H-J, Kang KS (2019) Procyanidin C1 Activates the Nrf2/HO-1 signaling pathway to prevent glutamate-induced apoptotic HT22 cell death. IJMS 20(1):142. https://doi.org/10.3390/ijms20010142
Mao X-Y, Zhou H-H, Li X, Liu Z-Q (2016) Huperzine A alleviates oxidative glutamate toxicity in hippocampal HT22 cells via activating BDNF/TrkB-dependent PI3K/Akt/mTOR signaling pathway. Cell Mol Neurobiol 36(6):915–925. https://doi.org/10.1007/s10571-015-0276-5
Sun J, Ren X, Qi W, Yuan D, Simpkins JW (2016) Geissoschizine methyl ether protects oxidative stress-mediated cytotoxicity in neurons through the “Neuronal Warburg Effect.” J Ethnopharmacol 187:249–258. https://doi.org/10.1016/j.jep.2016.04.034
Park SY, Jin ML, Kim YH, Kim C-M, Lee SJ, Park G (2014) Involvement of heme oxygenase-1 in neuroprotection by sanguinarine against glutamate-triggered apoptosis in HT22 neuronal cells. Environ Toxicol Pharmacol 38(3):701–710. https://doi.org/10.1016/j.etap.2014.08.022
Bao F, Tao L, Zhang H (2019) Neuroprotective effect of natural alkaloid fangchinoline against oxidative glutamate toxicity: involvement of Keap1-Nrf2 axis regulation. Cell Mol Neurobiol 39(8):1177–1186. https://doi.org/10.1007/s10571-019-00711-6
Zhu X, Wang K, Zhang K, Lin X, Zhu L, Zhou F (2016) Puerarin protects human neuroblastoma SH-SY5Y cells against glutamate-induced oxidative stress and mitochondrial dysfunction. J Biochem Mol Toxicol 30(1):22–28. https://doi.org/10.1002/jbt.21736
Andrich J, Saft C, Gerlach M, Schneider B, Arz A, Kuhn W, Müller Th (2004) Coenzyme Q10 serum levels in Huntington’s disease. In Focus on Extrapyramidal Dysfunction; Müller, Th., Riederer, P., Eds.; Journal of Neural Transmission. Supplementa; Springer Vienna: Vienna, 68: pp 111–116. https://doi.org/10.1007/978-3-7091-0579-5_13.
Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF (2002) Therapeutic effects of coenzyme Q 10 and remacemide in transgenic mouse models of Huntington’s disease. J Neurosci 22(5):1592–1599. https://doi.org/10.1523/JNEUROSCI.22-05-01592.2002
Yang L, Calingasan NY, Wille EJ, Cormier K, Smith K, Ferrante RJ, Flint Beal M (2009) Combination therapy with coenzyme Q 10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J Neurochem 109(5):1427–1439. https://doi.org/10.1111/j.1471-4159.2009.06074.x
Steliou K. Mitochondria-Targeting Antioxidant Therapeutics. 8,741,853, 2014. https://patents.google.com/patent/US8741853B2/en.
Parameshwaran K, Irwin MH, Steliou K, Pinkert CA (2010) D -Galactose effectiveness in modeling aging and therapeutic antioxidant treatment in mice. Rejuvenation Res 13(6):729–735. https://doi.org/10.1089/rej.2010.1020
Moos WH, Pinkert CA, Irwin MH, Faller DV, Kodukula K, Glavas IP, Steliou K (2017) Epigenetic treatment of persistent viral infections. Drug Dev Res 78(1):24–36. https://doi.org/10.1002/ddr.21366
Hoffman R, Sultan LD, Saada A, Hirschberg J, Osterzetser-Biran O, Gruenbaum Y (2019) Astaxanthin Extends Lifespan via Altered Biogenesis of the Mitochondrial Respiratory Chain Complex III; preprint; Developmental Biology https://doi.org/10.1101/698001.
Irwin MH, Moos WH, Faller DV, Steliou K, Pinkert CA (2016) Epigenetic treatment of neurodegenerative disorders: Alzheimer and Parkinson diseases. Drug Dev Res 77(3):109–123. https://doi.org/10.1002/ddr.21294
Paredes-Fuentes AJ, Oliva C, Urreizti R, Yubero D, Artuch R (2023) Laboratory testing for mitochondrial diseases: biomarkers for diagnosis and follow-up. Critical Reviews in Clinical Laboratory Sciences [Internet] [cited 2024 Mar 11];60(4):270–89. Available from: https://www.tandfonline.com/doi/full/https://doi.org/10.1080/10408363.2023.2166013
Mancuso M, Orsucci D, Coppede F, Nesti C, Choub A, Siciliano G (2009) Diagnostic approach to mitochondrial disorders: the need for a reliable biomarker. CMM [Internet] [cited 2024 Mar 11];9(9):1095–107. Available from: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1566-5240&volume=9&issue=9&spage=1095
Van Kraaij SJW, Pereira DR, Smal B, Summo L, Konkel A, Lossie J, et al (2023) Identification of peripheral vascular function measures and circulating biomarkers of mitochondrial function in patients with mitochondrial disease. Clinical Translational Sci [Internet] [cited 2024 Mar 11];16(7):1258–71. Available from: https://ascpt.onlinelibrary.wiley.com/doi/https://doi.org/10.1111/cts.13530
Mancuso M, Orsucci D, Gori S, Ceravolo R, Siciliano G (2008) Mitochondrial DNA single deletion in a patient with postural tremor. Movement Disorders [Internet] [cited 2024 Mar 11];23(14):2098–100. Available from: https://movementdisorders.onlinelibrary.wiley.com/doi/https://doi.org/10.1002/mds.22050
Romo L, Gold NB, Walker MA (2024) Endocrine features of primary mitochondrial diseases. Current Opinion in Endocrinology, Diabetes & Obesity [Internet] [cited 2024 Mar 11];31(1):34–42. Available from: https://journals.lww.com/https://doi.org/10.1097/MED.0000000000000848
Ng YS, Lim AZ, Panagiotou G, Turnbull DM, Walker M (2022) Endocrine manifestations and new developments in mitochondrial disease. Endocrine Reviews [Internet]. [cited 2024 Mar 11];43(3):583–609. Available from: https://academic.oup.com/edrv/article/43/3/583/6396126
Saiki S, Hatano T, Fujimaki M, Ishikawa KI, Mori A, Oji Y, et al. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci Rep [Internet]. 2017 Aug 4 [cited 2024 Mar 11];7(1):7328. Available from: https://www.nature.com/articles/s41598-017-06767-y
Mantle D, Hargreaves IP (2022) Mitochondrial dysfunction and neurodegenerative disorders: role of nutritional supplementation. IJMS [Internet] [cited 2024 Mar 11];23(20):12603. Available from: https://www.mdpi.com/1422-0067/23/20/12603
Khan HA (2010) Selenium partially reverses the depletion of striatal dopamine and its metabolites in MPTP-treated C57BL mice. Neurochemistry International [Internet] [cited 2024 Mar 11];57(5):489–91. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0197018610002159
Brakedal B, Dölle C, Riemer F, Ma Y, Nido GS, Skeie GO, et al (2022) The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metabolism [Internet] s[cited 2024 Mar 11];34(3):396–407.e6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1550413122000456
Cardoso BR, Roberts BR, Malpas CB, Vivash L, Genc S, Saling MM, et al (2019) Supranutritional sodium selenate supplementation delivers selenium to the central nervous system: results from a randomized controlled pilot trial in Alzheimer’s disease. Neurotherapeutics [Internet] [cited 2024 Mar 11];16(1):192–202. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1878747923010097
Mimori Y, Katsuoka H, Nakamura S (1996) Thiamine therapy in Alzheimer’s disease. Metab Brain Dis [Internet] [cited 2024 Mar 11];11(1):89–94. Available from: http://springerlink.bibliotecabuap.elogim.com/https://doi.org/10.1007/BF0208s0934
Jia J, Hu J, Huo X, Miao R, Zhang Y, Ma F (2019) Effects of vitamin D supplementation on cognitive function and blood Aβ-related biomarkers in older adults with Alzheimer’s disease: a randomised, double-blind, placebo-controlled trial. J Neurol Neurosurg Psychiatry [Internet]s [cited 2024 Mar 11];jnnp-2018–320199. Available from: https://jnnp.bmj.com/lookup/doi/https://doi.org/10.1136/jnnp-2018-320199
Fava A, Pirritano D, Plastino M, Cristiano D, Puccio G, Colica C, et al (2013) The effect of lipoic acid therapy on cognitive functioning in patients with Alzheimer’s disease. Journal of Neurodegenerative Diseases [Internet] [cited 2024 Mar 11];2013:1–7. Available from: https://www.hindawi.com/journals/jnd/2013/454253/
Liu Z, Li Y, Li C, Yu L, Chang Y, Qu M (2021) Delivery of coenzyme Q10 with mitochondria-targeted nanocarrier attenuates renal ischemia-reperfusion injury in mice. Materials Science and Engineering: C [Internet] [cited 2024 Mar 11];131:112536. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0928493121006767
Buchke S, Sharma M, Bora A, Relekar M, Bhanu P, Kumar J (2022) Mitochondria-targeted, nanoparticle-based drug-delivery systems: therapeutics for mitochondrial disorders. Life [Internet] [cited 2024 Mar 11];12(5):657. Available from: https://www.mdpi.com/2075-1729/12/5/657
Xie C, Zhuang XX, Niu Z, Ai R, Lautrup S, Zheng S, et al (2022) Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Biomed Eng [Internet] [cited 2024 Mar 11];6(1):76–93. Available from: https://www.nature.com/articles/s41551-021-00819-5
Boenzi S, Diodato D (2018) Biomarkers for mitochondrial energy metabolism diseases. Garone C, Minczuk M, editors. Essays in Biochemistry [Internet] [cited 2024 Mar 11];62(3):443–54. Available from: https://portlandpress.com/essaysbiochem/article/62/3/443/78628/Biomarkers-for-mitochondrial-energy-metabolism
Mancuso M, Filosto M, Bosetti F, Ceravolo R, Rocchi A, Tognoni G, et al (2003) Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Experimental Neurology [Internet] [cited 2024 Mar 11];182(2):421–6. Available from: https://linkinghub.elsevier.com/retrieve/pii/S001448860300092X
Wortmann SB, Rodenburg RJT, Jonckheere A, De Vries MC, Huizing M, Heldt K, et al (2009) Biochemical and genetic analysis of 3-methylglutaconic aciduria type IV: a diagnostic strategy. Brain [Internet] [cited 2024 Mar 11];132(1):136–46. Available from: https://academic.oup.com/brain/article-lookup/doi/https://doi.org/10.1093/brain/awn296
Bianchi MC, Tosetti M, Siciliano G, Battini R, Leuzzi V, Mancuso M, et al (2000) La spettroscopia protonica nello studio delle malattie metaboliche in età pediatrica. Rivista di Neuroradiologia [Internet] [cited 2024 Mar 11];13(1):45–50. Available from: http://journals.sagepub.com/doi/10.1177/197140090001300108
Chinnery P, Majamaa K, Turnbull D, Thorburn D (2006) Treatment for mitochondrial disorders. In: The Cochrane Collaboration, editor. Cochrane Database of Systematic Reviews [Internet]. Chichester, UK: John Wiley & Sons, Ltd; [cited 2024 Mar 11]. p. CD004426.pub2. Available from: https://doi.wiley.com/doi.org/10.1002/14651858.CD004426.pub2
Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, et al (2006) Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology [Internet] [cited 2024 Mar 11];66(3):324–30. Available from: https://www.neurology.org/doi/10.1212/01.wnl.0000196641.05913.27
Shoop WK, Bacman SR, Barrera-Paez JD, Moraes CT (2023) Mitochondrial gene editing. Nat Rev Methods Primers [Internet] [cited 2024 Mar 11];3(1):19. Available from: https://www.nature.com/articles/s43586-023-00200-7
Niyazov DM, Kahler SG, Frye RE (2016) Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol [Internet] [cited 2024 Mar 11];7(3):122–37. Available from: https://www.karger.com/Article/FullText/446586
McCann MR, George De La Rosa MV, Rosania GR, Stringer KA (2021) L-Carnitine and acylcarnitines: mitochondrial biomarkers for precision medicine. Metabolites [Internet] [cited 2024 Mar 11];11(1):51. Available from: https://www.mdpi.com/2218-1989/11/1/51
Li L, Goel A, Wang X (2022) Novel paradigms of mitochondrial biology and function: potential clinical significance in the era of precision medicine. Cell Biol Toxicol [Internet] [cited 2024 Mar 11];38(3):371–5. Available from: https://springerlink.bibliotecabuap.elogim.com/doi.org/10.1007/s10565-022-09721-5
Lim K, Cho SI, Kim JS (2022) Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat Commun [Internet] [cited 2024 Mar 11];13(1):366. Available from: https://www.nature.com/articles/s41467-022-27962-0
Lee S, Lee H, Baek G, Kim JS (2023) Precision mitochondrial DNA editing with high-fidelity DddA-derived base editors. Nat Biotechnol [Internet] [cited 2024 Mar 11];41(3):378–86. Available from: https://www.nature.com/articles/s41587-022-01486-w
Willis JCW, Silva-Pinheiro P, Widdup L, Minczuk M, Liu DR (2022) Compact zinc finger base editors that edit mitochondrial or nuclear DNA in vitro and in vivo. Nat Commun [Internet] [cited 2024 Mar 11];13(1):7204. Available from: https://www.nature.com/articles/s41467-022-34784-7
Laura Craft (2017) Emerging Applications of Ai for Healthcare Providers GARTNER.s Available from: https://www.gartner.com/en/documents/3753763
Basu K, Sinha R, Ong A, Basu T (2020) Artificial intelligence: how is it changing medical sciences and its future? Indian J Dermatol 65(5):365–370. https://doi.org/10.4103/ijd.IJD_421_20
Mancuso R et al (2020) Artificial intelligence for Alzheimer’s disease- promise or challenge. Front Neurol 11:1019
Dabbaghi KG, Khosravirad Z, Jamalnia S, GhorbaniNia R, Mahmoudikohani F, Zakeri H, Khastehband S (2023) The use of artificial intelligence in the management of neurodegenerative disorders; focus on Alzheimer’s disease. Galen Medical Journal 12:1
Garcia DLF, Ritchie CW, Luz S (2020) Artificial intelligence, speech, and language processing approaches to monitoring Alzheimer’s disease: a systematic review. J Alzheimer’s Dis 78(4):1547–1574
Liu Y et al (2021) Integrative gene expression analysis for the diagnosis of Parkinson’s disease using machine learning and explainable AI. Sci Rep 11:102
Li X et al (2020) Use of magnetic resonance imaging and artificial intelligence in studies of diagnosis of Parkinson’s disease. Front Neurol 11:1040
Lu Y et al (2021) Amelioration of Alzheimer’s disease pathology by mitophagy inducers identified via machine learning and a cross-species workflow. Nat Commun 12:2118
Serra-Mestres J et al (2020) Multi-layer picture of neurodegenerative diseases- lessons from the use of Big Data through artificial intelligence. Int J Mol Sci 21(23):9083
McMillan CT, Irwin DJ (2020) Decoding degeneration- the implementation of machine learning for clinical detection of neurodegenerative disorders. J Neurol Neurosurg Psychiatry 91(10):1067–1076
Ghosh R, Cingreddy AR, Melapu V, Joginipelli S, Kar S (2021) Application of artificial intelligence and machine learning techniques in classifying extent of dementia across alzheimer’s image data. IJQSPR 6(2):29–46
Author information
Authors and Affiliations
Contributions
M.V contributed to the conceptualization of the article. M.V and V.M.P designed the framework of the paper. D.S.K, R.B, K.M, P.J, N.R, and S.G.S drafted the manuscript and created the illustrations. M.V and V.M.P revised the manuscript critically. The authors collectively reviewed the manuscript and provided their consent for its publication in its current form.
Corresponding authors
Ethics declarations
Consent to Participate
Not applicable.
Consent to Publication
All authors agreed to publish the contents.
Competing Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kathiresan, D.S., Balasubramani, R., Marudhachalam, K. et al. Role of Mitochondrial Dysfunctions in Neurodegenerative Disorders: Advances in Mitochondrial Biology. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04469-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04469-x