
Abstract
Systemic oxidative stress (SOS) has an important role in the mechanisms activation of neuronal death, involved in the neurodegenerative disease (ND) etiology. Brain is susceptible to oxidative stress injuries due to its high energy and metabolic request, therefore minimal imbalances of the redox state, as occurs in mitochondrial dysfunction, favour tissue injury and neuroinflammatory mechanisms activation. ND affect around the world about a billion people, without distinction of sex, educational level and economic status. Public measures generation that prevent ND from the SOS are possible promising therapeutic targets that could reduce the ND incidence. We discuss here the effects and mechanisms of SOS derived neurodegeneration, as well as the neuroinflammation repercussions for some cerebral structures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Neurodegenerative disease — A central matter
Neurodegenerative diseases (NDs) form a heterogeneous group of diseases that mainly have their point of injury in the nervous system (NS). They are characterised by the progressive loss of neurons, known as neurodegeneration, in regions that compromise NS functions (1).
According to the World Health Organization (WHO) report, ND affects around a billion people worldwide. The report, “Neurological Disorders: Public health challenges” cites the global prevalence of ND to be 50 million cases of epilepsy and 24 million of Alzheimer’s and other dementias. Neurological disorders affect people from all countries irrespective of sex, educational level and economic status. The WHO advocates that neurological care be integrated into primary health care in order to prevent ND through strategies that can also directly affect metabolic disturbance aspects (2, 3).
NDs share a common binomial with other diseases such as diabetes, cardiovascular disease and adiposity-based chronic disease (obesity). The oxidative stress and chronic inflammation binomial (OS-I) is much more than the consequence of oxidative-reductive alterations that lead to neuronal death (4, 5). OS-I is an important aetiological factor in the development of metabolic disease (6), that lately has gained greater prominence in the study of ND, to elucidate the various mechanisms involved between alterations in the cellular redox state and the injury to NS structural components (7). However, many questions remain to be resolved; for example: ¿Is oxidative stress the cause or a consequence of the development of ND?, What are the implications of systemic oxidative stress (SOS) in the oxidative alterations development at the NS level? Is chronic oxidative stress an early ND trigger? In the present review, we discuss the effects and mechanisms of SOS-derived neurodegeneration, as well as the repercussions of neuroinflammation for some NS structures.
An updated context of oxidative stress based on the problems of the 21st century
The first studies of oxidative stress were reported in 1956 by Dr. Harman, who was the first to describe ageing as a phenomenon characterised by the progressive decline of cellular function and what is known as his oxidative stress theory (or the involvement of free radicals), describing the circumstances under which cells age and die (8). In recent decades, research into oxidative stress has increased, associating it with the development of metabolic disorders such as adiposity-based chronic disease (ABCD) (9), diabetes (10), cardiovascular diseases (CVD) (11), cancer (12) and recently, ND (13).
Society has undergone great economic, political and sociocultural changes that have influenced people’s lifestyles (14). People live in faster societies, with less time to consume fresh and natural foods, with greater work stress and economic crises that compromise the nutritional quality of their food, favouring the consumption of high ultra-processed products (UPPs) (15).
Twenty-first century society changes quickly and unpredictably. Currently, people live in an uncertain society, experiencing change and excess; these characteristics are also reflected in the attitudes to health systems, leading to important implications in the development of SOS, associated with the current incidence of chronic metabolic diseases (16). This is a reflection of the change in eating guidelines, regarding diets such as those high in simple carbohydrates (DHSCs) and in saturated fatty acids (DHSFs), characteristics of the Western diet (15). Food consumption guidelines play an important role in the increase in the levels of systemic pro-oxidants and inflammatory mediators, which result in neuroinflammatory responses in the central nervous system (CNS) structures such as the hypothalamus, choroid plexus, cerebellum, cerebral cortex and hippocampus (17).
Mitochondrial dysfunction: Key point kick-off from systemic oxidative stress
Oxygen is a vital element for the correct every human operation being. Cell uses oxygen in oxidative-reductive reactions, in order to obtain energy and intermediate metabolites necessary for its growth, development, survival, homeostasis and adaptability (18). Specifically, the mitochondrion is the central subcellular organelle in the process of internal respiration (oxidative-reductive reactions) where the complete nutrients oxidation such as glucose and fatty acids is carried out in the intermediate metabolites production: ATP, H2O y CO2 (19).
Usually, mitochondria use 95 to 98% of O2 get into oxidative processes. Another 2% of O2 reacts with H2O molecules to produce hydrogen peroxide (H2O2) or other species (O2-, OH-), called reactive oxygen species (ROS) (20, 21). Several factors are related to the exacerbated ROS production, among them the diet quality. DHSC and DHSF they correlate with the intracellular increase pro-oxidant substances. Initially, skeletal muscle and adipose tissue are two tissues involved in oxidative-reductive alterations, in response to an increase in intracellular glucose concentrations due to changes in the diet quality. The intracellular increase glucose leads to a greater substrate amount for oxidative metabolism enzymes.
As the substrate concentration increases, the initial enzymes velocity increases to a saturation point (22). The enzymes saturation involved in oxidative processes, conditions the decrease in the physiological O2 use, producing an increase in free O2, and ROS overproduction (21, 18), decrease in oxidative phosphorylation and decrease in ATP production. These alterations in the oxidative processes, result in a higher ROS production and lower ATP. These mitochondrial perturbations oxide-reductive processes are known as mitochondrial dysfunction (23).
Disturbances to the redox state in the mitochondria are considered the main triggering OS-I factor, a situation that compromises oxide-reductive cell functions, after the exacerbated ROS increase and free radicals (FR) and lower antioxidant production substances. ROS and FR, are atoms or atoms groups with an unpaired electron, such compounds are characterised by sequestering electrons from electrochemically stable molecules, such as proteins, lipids and nucleic acids (24). The ability to sequester electrons depends on the atom electronegativity; because oxygen and nitrogen are highly electronegative elements, ROS and reactive nitrogen species (RNS) are produced more easily, respectively. Reactive species are produced as part of everyday physiological processes and have important roles in cell signalling, gene transcription and immune response (25). However, the FR overproduction, generates oxidative damage on cellular structures (26).
ROS accumulation in mitochondrial matrix, induces oxidation lipids and proteins membrane, which contribute to mitochondrial dysfunction (21). Additionally, disturbances in mitochondrial DNA due to oxidation by ROS, is correlated with a lower genes expression responsible for coding mitochondrial protection proteins against oxidative stress. Although the mechanisms involved are still unclear, up to 30 mutations in mitochondrial DNA have been identified, related to the pro-oxidative environment (27). DNA oxidation and mitochondrial membranes, with their subsequent fragmentation, implies an irreversible cellular lesion. Finally, mitochondria death and the decrease in ATP production, activate cell death signals that trigger proinflammatory processes (26).
Oxidative stress perpetuation, leads to a chronic state that propagation to other tissues. ROS and RNS in blood are capable of triggering proinflammatory and oxidative processes in organs such as liver (28), kidney (29), enterocyte (30), endocrine glands (6), encephalon (31), among others, associated with the metabolic diseases development (Figure 1). For the purposes of this review, the following sections will focus on the SOS effects and its affections on the NS, as a potential trigger of ND.

Systemic dissemination by mitochondrial dysfunction
Blood-Brain barriers (BBB) alteration due to the SOS
Dissemination of pro-oxidant compounds in the blood and inflammatory mechanisms activation compromises CNS functions through different communication channels, such as the vagus nerve, the choroid plexus and the BBB (32). NS has protection from the pro-oxidants accumulation in blood, through the BBB, therefore, its disruption implies the components filtration that injury the NS (33). BBB is a physical barrier located between blood vessels and brain tissue, which selectively limits the access of molecules to protect the brain from any pathogen type or toxic substance. It is an active structure that reacts to stimuli by modifying its permeability and uptake capacities (34).
SOS favours a chronic oxidative environment that leads to changes related to the cells epigenetics of the neurovascular unit (NVU) that alter the BBB integrity (33). NVU includes endothelial cells, capillaries and pericytes surrounded by basal lamina, which are surrounded by astrocytes very close to the neurons and microglia. So, BBB expresses a large ion number channels and transporters, has a low pinocytosis rate and tight junction proteins (TJ) intercellular, such as occludin, claudins and cell adhesion molecules, which limit paracellular permeability (34).
As a protection, endothelial cells of the BBB have a high antioxidant activity rate. They have significant reduced glutathione (GSH) amounts, glutathione peroxidase (Gpx), glutathione reductase and catalase (CAT), compared to the brain rest. Specifically, GSH plays a crucial role in the BBB integrity, reducing the damage caused by ROS and FR (33). However, antioxidant activity is compromised in a chronic oxidation environment (35), as is the DHSC and DHSF case.
Among the most widely studied factors causing the BBB disruption are the DHSC and DHSF (36, 37). ROS contributes to brain injury by reacting with proteins, lipids and nucleic acids, as well as by activating a number of signalling pathways sensitive to redox. ROS overproduction affects the BBB permeability by a mechanisms variety, including the modulation of binding proteins, as is the case with occludin and claudin (38).
Occludin is a transmembrane protein present in the oligomers form in the plasma membrane of the BBB. Multiple domains of occludin regulate the solutes diffusion through binding proteins (39). Ocludin oligomers are held together covalently by disulfide bonds, sensitive to ROS oxidation. Changes in the structure of the occludin oligomers, leads to a lower capacity of the binding protein complexes to limit the paracellular diffusion of the blood molecules to the brain (40).
A chronic oxidation environment is associated with lower genes expression cytoskeletal proteins, chaperones, enzymes, transport-related proteins and regulators for transcriptional and translational of brain microvessel cells, causing changes in the regulation of the peptides and substrates transport that cross the BBB. DHSC and DHSF it is associated with lower mRNA expression of tight binding proteins, particularly claudin-5 and -12, in the choroid plexus and BBB (41).
All the organism cell, including NVU, present a defence mechanism against the OS called adaptive response (AR) (42). In response to the OS, the cell expresses antioxidant enzymes genes, activating the transcription factor Nrf2, at the same time that it inhibits enzymes genes involved in the ROS production, such as NADPH oxidase, anti-inflammatory mediators increase, proteasome activity and other transcription factors involved in mitochondrial biogenesis (43).
Although the SNC has a protective defence mechanism against the OS, in addition to the BBB (33), the generalised and chronic oxidative environment is a factor that leads to the loss of the AR (24). Lower genes expression involved in the NVU integrity, causes a hypometabolic state that influences the oxidative processes of the cells that make BBB (37). The mitochondrial dysfunction triggered, favours cellular injury that culminate in endothelial cell necrosis and loss of BBB integrity.
Chronic oxidative stress and neurodegeneration
With a 2 kg weight (3% of body composition) the NS is one of the smallest and most complex organs of the body. It consists of an intricate, specialised and highly organised network of cells called neurons and glial cells. The NS is subdivided into CNS, consisting of encephalon and spinal cord, and the peripheral nervous system (PNS), which includes all nerve tissues located in the periphery. The brain is the main constituent organ of the CNS, whose cognitive function involves the participation of cell networks that include neurons, one of the most important NS cell populations (44).
Neurons are the main functional units of the CNS and have the ability to communicate with each other quickly and efficiently through electrical or chemical signals that are translated as action potentials. Structurally, they are composed of a cell body (soma) where the nucleus is located and where several extensions are projected (appendices or protuberances), which include many short branching extensions, known as dendrites and a separate extension that is usually longer than dendrites, known as axon (45).
Unlike other cells, neurons use a lot of oxygen (20% of the O2 that enters the organism), so they have a high mitochondrial activity rate that allows them to cover high ATP demands; however, the above is a crucial point also for increased ROS production. Especially, neurons are more sensitive to the accumulation and damage induced by ROS (5).
Not all nerve cells produce action potentials. Glial cells differ from neurons, since they do not have synaptic contacts and have the ability to divide throughout life. The main glial cells functions are: 1) Maintain an ionic medium in nerve cells; 2) Modulate the propagation speed of nerve signals; 3) Modulate the synaptic action by controlling the neurotransmitters uptake; 4) Provide a foundation for neural development; and 5) Contribute (or prevent, in some cases) the survival of a neuronal lesion (46). The glial cells are together with the NVU, responsible for the BBB integrity and neuronal metabolism, by preventing / responding to nerve tissue injury (47).
Microglia constitute part of the macrophages and constitute between 5 and 10% of the neuronal cell population (47). Under basal conditions, it is at rest; however, in response to NS injury, the microglia becomes polarised (morphological transformation of an inactive branched state to an active phagocytic amiboide) and triggers mechanisms that promote neurotoxic activities, including the ROS overproduction, RNS, chemokines and proinflammatory cytokines. The above is called microgliosis, whose main characteristic is an increase in the microglias number activated at the site of the lesion, which leads to a complex cascade of physiological acute inflammation responses (48, 49).
The chronic state of OS in the organism, systemic ROS dissemination and BBB disruption, causes a chronic microgliosis response that promotes a chronic state of inflammation. Activation of the microglia remains until it becomes chronic, which leads to an imbalance in the inflammatory response, generating a neuroinflammation cycle and tissue damage (46).
Neuroinflammation is the set of mechanisms and physiopathological phenomena mediated by alterations in the glial cell morphology. It is a reactive state of the immunological component of the CNS (50). The damage responsible for triggering neuroinflammation can be: a) Physical, such as trauma; b) Biological, such as bacterial or parasitic infections in NS; or c) Chemical, as is the case of an exacerbated ROS production, FR, and RNS, present in the SOS (19).
In response to the damage and the consequent microglial activation, involved proinflammatory molecules that mediate the response at the damage site. IL1β induces the expression of the inducible nitric oxide synthase gene (iNOS), responsible for triggering the nitric oxide (NO-) release, on glial cells and cause nitrosative stress. An exacerbated RNS production, as it happens in the chronic neuroinflammatory response, increases the oxidation of cell membrane lipids (48).
Likewise, astrogliosis (activation of the inflammatory response by astrocytes), increases the number and size of the fibrillary acid protein (FAP) of the glia, cytoskeletal protein that contributes to hypertrophy and hyperplasia of astrocytes. Active astrocytes produce a molecules variety, which are involved in the initiation, progression and regulation of the inflammatory response. In addition to the pro and anti-inflammatory cytokines production, there is a greater expression of the genes of the cyclooxygenase — 2 (COX-2) enzymes e iNOS, as well as higher RNS production. Neuroinflammatory response, initially triggered by ROS and FR, contributes to oxidative stress in the CNS, generating a perpetuated state between neuroinflammation and chronic oxidative stress (51).
OS plays a critical role in the neuronal activation death mechanisms, implicated in the several aetiology ND (52). Brain is susceptible to EO lesions due to its high energy use and metabolic demand, therefore minimal imbalances of the redox state, as occurs in mitochondrial dysfunction, favour tissue injury, damage and neuroinflammatory mechanisms activation (53).
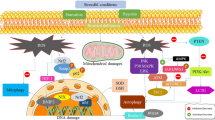
OS perpetuation, produces the genes activation involved in the recovery and cell death, one of the most studied is the FOXO transcription factors. The FOXO transcription factors have a central role in oxidative stress-induced neuronal cell death. FOXO promotes neuronal apoptosis in response to oxidative stress through inducing the expression of pro-apoptotic downstream genes including Bim and FasL (54). Likewise, exacerbated ROS production intracellular and DNA damage induces the expression of the p53 gene, activating the Bcl2 pro-apoptotic protein family, Bax and Bad permeabilisation of the mitochondrial outer membrane with translocation and release of cytochrome C that forms a complex with apoptotic protease activating factor 1 (Apaf-1). Result is the 9 caspase activation, leading to a proteolytic caspase activation cascade 3 and 7, and execution of the cell death process, as well as changes in the plasma membrane (blistering and phosphatidyl-serine exposure on the cell surface which is a signal that stimulates cellular phagocytosis by macrophages / microglia) that culminates in the condensation and nuclear chromatin fragmentation (55, 56).
Increase in ROS concentrations in the neuron, induces a rapid increase in intracellular Ca+2, from: 1) extracellular spaces, after the rupture of the membrane and, 2) release of the endoplasmic reticulum, causing cytochrome C translocation from the mitochondrial outer membrane to the cytosol, triggering cell death signals (56). Likewise, another mechanism involved in cell death, is the BBB disruption, which dissipates the ROS passage and proinflammatory cytokines to NS. Disengagement microgliosis, increases the inflammation mediators production, ROS and RNS, causing cell death signs, by recognising the FAS receptor or cell death receptor and TNF receptor 1 (TNFR1), activating the necroptotic pathway, also related to the ND pathogenesis (Figure 2) (57).

Systemic oxidative stress effect on the neurodegenerative disease development
Unlike other organism cells, neurons have a low regenerative activity, apoptotic pathway is highly restricted, to allow neurons to survive during the life of an organism (58). Regions such as the adult hippocampus do not present a neurogenesis in the dentate gyrus, associated with the early cognitive decline of the ND (59).
Therapeutic perspectives
Advances in medical and biotechnology sciences have redirected the way nutrition is done. Currently the implementation of a feed from the bioactive compounds present in them, whose biological activity has shown beneficial effects on the SOS biochemical-metabolic alterations (60–62).
The antioxidants use to mitigate the OS alterations, it is an alternative in the modulation of the oxidative environment. An antioxidant is any substance that is present at low concentrations with respect to those of an oxidisable substrate (biomolecule), retards or prevents the oxidation of the substrate (63). Currently, a role of antioxidants is proposed as neuroprotective agents in ND. Polyphenols such as quercetin, catechin and resveratrol that have exhibited neuroprotective capacity in several animal models with neurological disorder (64). Epidemiological research support the potential polyphenols use in the diet, on better neuronal health (65). The proposed mechanisms include the blocking of specific enzymes that generate FR, metals chelation and neutralising ROS, anti-inflammatory effects and microglia inhibition recruitment (66).
Fruits and beverages such as tea, red wine, cocoa and coffee are the main polyphenols dietary sources (65). Its neuroprotective effects are correlated through the potential to protect neurons against lesions induced by neurotoxins, the ability to suppress neuroinflammation and the potential to promote memory, learning and cognitive function (67). Recent evidence suggests that its beneficial effects involve decreases in OS signalling and modulation of gene expression of antioxidant enzymes, neurotrophic factors and cytoprotective protein (68).
Other compounds of interest such as caffeine (69), Ginkgo biloba (70), α-tocoferol (71) and curcumin (72), they have also shown important protective activities and suggest a potential use in the ND treatment. The therapeutic strategies development based on the potential antioxidant and anti-inflammatory effect of food are useful for modulating the oxidative environment and mitigating OS alterations and inflammation (73).
Conclusion
SOS it is a complex reversible entity, whose chronicity can lead to other systemic disturbances such as neurodegeneration. Its early detection would be a window of opportunity for the ND prevention. The use of dietary antioxidants is important to mitigate the biochemical-metabolic alterations product of the oxidative environment generated by the OS. The search for new strategies of functional nutrition, based on the functional potential of antioxidants are allies in the prevention of diseases with an oxidative base.
References
Torrell G. Enfermedades neurodegenerativas. Actualización en Medicina de Familia. 2015; 11(7): 374–383.
National Institute of Neurological Disorders and Stroke. Neurodegenerative diseases. 2017.
Noncommunicable Diseases Progress Monitor, 2017. Geneva: World Health Organization; 2017. Licence: CC BY-NC-SA 3.0 IGO. https://apps.who.int/iris/bitstream/handle/10665/258940/9789241513029-eng.pdf?sequence=1
Andersen J. (2004). Oxidative stress in neurodegeneration: cause or consequence? Nat Rev Neurosci. 10: S18–S25 https://doi.org/10.1038/nrn1434
Johri A, Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther. 2012; 342(3): 619–30 https://doi.org/10.1124/jpet.112.192138
Vitale G, Salvioli S, Franceschi C. Oxidative stress and the ageing endocrine system. Nat Rev Endocrinol. 2013; 9: 228–240 https://doi.org/10.1038/nrendo.2013.29
Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017; 13: 477–491 https://doi.org/
Harman D. Aging: a theory based on free radical and radiation chemistry. Int J Gerontol. 1956; 11: 298–300.
Mechanick J, Hurley D, Garvey W. Adiposity-based chronic disease as a new diagnostic term: The American Association of Clinical Endocrinologists and American College of Endocrinology position statement. Endocrine Practice. 2017; 23(3): 372–378 https://doi.org/10.4158/EP161688.PS
Oliveira C, Villar-Delfino P, Dos Anjos P, Nogueira A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018; 9(119) https://doi.org/10.1038/s41419-017-0135-z.
Vaziri N, Rodríguez-Iturbide B. Mechanisms of Disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Prac Nephrol. 2006; 2: 582–593 https://doi.org/10.1038/ncpneph0283
Paschos A, Pandya R, Duivenvoorden WCM, Pinthus JH. Oxidative stress in prostate cancer: changing research concepts towards a novel paradigm for prevention and therapeutics. Prostate Cancer Prostatic Dis. 2013; 16: 217–225 https://doi.org/10.1038/pcan.2013.13
Roy-Sarkar S, Banerjee S. Gut microbiota in neurodegenerative disorders. J Neuroimmunol. 2019; 328(15): 98–104 https://doi.org/10.1016/jjneuroim.2019.01.004
Martine G, Diniz J. Economy, society and environment in the 21st century: three pillars or trilemma of sustainability?. Revista Brasileira de Estudos de População. 2015; 32(3): 433–459 https://doi.org/10.1590/S0102-3098201500000027
Monteiro C, Moubarac J, Cannon G, Ng S, Popkin B. Ultra-processed products are becoming dominant in the global food system. 2014; Obesity rev. 2: 21–28 https://doi.org/10.1111/obr.12107
Jovell AJ. The XXI century patient. Anales del Sistema Sanitario de Navarra. 2006; 29(3): 85–90.
Parimisetty A. Dorsemans AC, Awada R, Ravanan P, Diotel N, Lefebvre d’Hellencourt C. Secret talk between adipose tissueand central nervous system via secreted factors — an emerging frontier in the neurodegenerative research. J. Neuroinflammation. 2016; 13: 67 https://doi.org/10.1186/s12974-016-0530-x
Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015; 4: 180–183 https://doi.org/10.1016/j.redox.2015.01.002
Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007; 47:143–183 https://doi.org/10.1146/annurev.pharmtox.47.120505.105122
Hernández A, Rull A, Rodríguez E, Riera M, Luciano F, Camps J, Menéndez J, Joven J. Mitochondrial Dysfunction: A Basic Mechanism in Inflammation-Related Non-Communicable Diseases and Therapeutic Opportunities. Mediators Inflamm. 2013; 1–13. https://doi.org/10.1155/2013/135698.
Schieber M, Chandel N. ROS Function in Redox Signaling and Oxidative Stress. Current Biol. 2014; 24(10): R453–R462 https://doi.org/10.1016/jxub.2014.03.034
Davis RE, Williams M. Mitochondrial function and dysfunction: an update. J Pharmacol Exp Ther. 2012; 342: 598–607 https://doi.org/10.1124/jpet.112.192104
Area-Gomez E, De Groof A, Bonilla E, Montesinos J, Tanji K, et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018; 9(335): 1–10 https://doi.org/10.1038/s41419-017-0215-0
Martínez-Leo E, Acevedo J, Segura M. Biopeptides with antioxidant and anti-inflammatory potential in the prevention and treatment of diabesity disease. Biomed Pharmacother. 2016; 83: 816–826 https://doi.org/10.1016/j.biopha.2016.07.051
Sies H, Berndt C, Jones D. Oxidative stress. Annu. Rev. Biochem. 2017; 86: 25.1–25.34 https://doi.org/10.1146/annurev-biochem-061516-045037
Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur J Med Chem. 2015; 97(5): 55–74 https://doi.org/10.1016/j.ejmech.2015.04.040
Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018; 592(5): 728–742 https://doi.org/10.1002/1873-3468.12956.
Kim SY, Jeong JM, Kim S, Seo W, Kim MH, Choi WM, et al. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun. 2017; 8 (2247) https://doi.org/10.1038/s41467-017-02325-2.
Kong MJ, Han SJ, Kim JI, Park JW, Park KM. Mitochondrial NADP+-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. 2018; 9(488) https://doi.org/10.1038/s41419-018-0537-6.
Hu Q, Ren J, Li G, Wu J, Wu X, Wang G, Gu G, et al. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018; 9(403) https://doi.org/10.1038/s41419-018-0436-x.
Mattson M, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006; 7: 278–294.
Guillemot-Legris O, Muccioli G. Obesity- induced neuroinflammation: Beyond the hypothalamus. Trends Neurosci. 2017; 40(4): 237–253.
Obermeier O, Daneman R, Ransohoff R. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013; 19: 1584–1596.
Nzou G, Wicks RT, Wicks EE, Seale SA, Sane CH, Chen A, Murphy SV, et al. Human Cortex Spheroid with a Functional Blood Brain Barrier for High-Throughput Neurotoxicity Screening and Disease Modeling. Sci Rep. 2018; 8 (7413) https://doi.org/10.1038/s41598-018-25603-5
Forsberg SL, Ilieva M, Michel T. Epigenetics and cerebral organoids: promising directions in autism spectrum disorders. Transl Psychiatry. 2018; 8(14) https://doi.org/10.1038/s41398-017-0062-x
Baufeld C, Osterloh A, Prokop S, Miller K, Heppner F. High-fat diet-induced brain region specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016; 132: 361–375 https://doi.org/10.1007/s00401-016-1595-4
Rhea E, Salameh T, Logsdon A, Hanson A, Erickson M, Banks W. Blood-Brain Barriers in Obesity. The AAPS Journal. 2017; 19(4): 921–930 https://doi.org/10.1208/s12248-017-0079-3
Lochhead J, McCaffrey G, Quigley C, Finch J, De Marco K. et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occluding during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010; 30: 1625–1636 https://doi.org/10.1038/jcbfm.2010.29
McCaffrey G, Staatz WD, Quigley CA, Nametz N, Seelbach MJ, Campos CR, et al. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. J Neurochem. 2007; 103(6): 2540–55 https://doi.org/10.1111/j.1471-4159.2007.04943.x
Walter JK, Rueckert C, Voss M, Mueller SL, Piontek J, Gast K, Blasig IE. The oligomerization of the coiled coil-domain of occludin is redox sensitive. Ann NY Acad Sci. 2009; 1165:19–27 https://doi.org/10.1111/j.1749-6632.2009.04058.x
Ouyang S, Hsuchou H, Kastin A, Wang Y, Yu C, Pan W. Diet-induced obesity suppresses expression of many proteins at the blood-brain barrier. J Cereb Blood Flow Metab. 2014; 34(1): 43–51.
Morel Y, Barouki R. Repression of gene expression by oxidative stress. Biochem J. 1999; 342: 481–496.
Tufekci K.U, Civi Bayin E, Genc S, Genc K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson’s Disease, Parkinsons Dis. 2011; 314082 https://doi.org/10.4061/2011/314082
Shrestha R, Millington O, Brewer J, Bushell T. Is Central Nervous System an Immune-Privileged site? Kathmandu University Med J. 2013; 41(1): 102–107.
Avena-Koenigsberger A, Misic B, Sporns O. Communication dynamics in complex brain networks. Nat Rev Neurosci. 2018; 19: 17–33 https://doi.org/10.1038/nrn.2017.149
Mizuno T. Neuron-microglia interactions in neuroinflammation. Clin Exp Neuroimmunol. 2015; 6: 225–231.
Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, Pauli JR, et al. High-fat diet induces apoptosis of hypothalamic neurons. PLoS One. 2009; 4(4): e5045.
Chen W, Zhang X, Huang W. Role of neuroinflammation in neurodegenerative diseases. Mol Med Rep. 2016; 13: 3391–3396 https://doi.org/10.3892/mmr.2016.4948
Li T, Zhang S. Microgliosis in the Injured Brain: Infiltrating Cells and Reactive Microglia Both Play a Role. Neuroscientist. 2016; 22(2): 165–70 https://doi.org/10.1177/1073858415572079
Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. 2016; https://doi.org/10.1038/nri.2016.123
Zhang D, Hu X, Qian L, O’Callaghan J, Hong J. Astrogliosis in CNS pathologies: Is there a role for microglia?. Mol neurobiol. 2010; 41: 232–241 https://doi.org/10.1007/s12035-010-8098-4
Zhu W. Methylation of FOXO3 regulates neuronal cell death. Acta Pharmacol Sinica. 2012; 33: 577 https://doi.org/10.1038/aps.2012.48
Cai L, Wu X, Lv Y, Xu Y, Mi G, Li J. The neuroprotective and antioxidant activities of protein hydrolysates from grass carp (Ctenopharyngodon idella) skin. JFST. 2015; 52(6), 3750–3755 https://doi.org/10.1007/s13197-014-1438-z
Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, et al. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO Rep. 2012; 13: 371–377 https://doi.org/10.1038/embor.2012.25
Xin YJ, Yuan B, Yu B, Wang YQ, Wu JJ, Zhou WH, Qiu Z. Tet1-mediated DNA demethylation regulates neuronal cell death induced by oxidative stress. Sci Rep. 2015; 5 (7645): 1–9 https://doi.org/10.1038/srep07645
Mattson M. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000; 1: 120–130 https://doi.org/10.1038/35040009
Zhang S, Tang M, Luo H, Shi C, Xu Y. Necroptosis in neurodegenerative diseases: a potential therapeutic target. Cell Death Dis. 2017; 8: e2905. https://doi.org/10.1038/cddis.2017.286.
Cusack C, Swahari V, Henley W, Ramsey M, Deshmukh M. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun. 2013; 4(1876): 1–11 https://doi.org/10.1038/ncomms2910
Sorrells S, Paredes M, Cebrian-Silla A, Sandoval K, Qi D, Kelley K. et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Letter. 2018; 1–5 https://doi.org/10.1038/nature25975.
Pelcastre D, Martínez-Leo E, Segura-Campos M. Functional and Biological Potential of Bioactive Compounds in Foods for the Dietary Treatment of Type 2 Diabetes Mellitus. In: Functional Food — Improve Health through Adequate Food, 1st edition. Ed. InTech; 2017.
Martínez-Leo E, Villavicencio T, Segura-Campos M. Functional Foods and Chemoprevention in Cancer. In: Grumezescu A, Holban AM, editors. Therapeutic Foods, 1st edition. Elsevier; 2017.
Martínez-Leo E, Rojas R, Segura-Campos M. Protective Effect of Omega 3 Fatty Acids EPA and DHA in the Neurodegenerative Disease. In Mérillon JM, Ramawat KG, editors. Bioactive Molecules in Food, 1st edition. Ed. Springer International. 2018.
Halliwell B. Free radicals and antioxidants — quo vadis?. Trends Pharmacol Sci. 2011; 32. 125–130 https://doi.org/10.1016/j.tips.2010.12.002
Potter A, Buck AC, Self WK, Callanan ME, Sunil S, Capadona JR. The effect of resveratrol on neurodegeneration and blood brain barrier stability surrounding intracortical microelectrodes. Biomaterials. 2013; 34:7001–7015 https://doi.org/10.1016/j.biomaterials.2013.05.035
Rodríguez C, Segura A, Del Mar M. Phenolic compounds as natural and multifunctional anti-obesity agents: A review. Crit Rev Food Sci Nutr. 2017; 20:1–18 https://doi.org/10.1080/10408398.2017.1399859
Dong W, Gao D, Lin H, Zhang X, Li N, Li F. New insights into mechanism for the effect of resveratrol preconditioning against cerebral ischemic stroke: possible role of matrix metalloprotease-9. Med Hypotheses. Medical Hypotheses. 2008; 70:52–55 https://doi.org/10.1016/j.mehy.2007.04.033
Shukitt-Hale B, Lau FC, Carey AN, Galli RL, Spangler EL, Ingram DK, Joseph JA. Blueberry polyphenols attenuate kainic acid-induced decrements in cognition and alter inflammatory gene expression in rat hippocampus. Nutr Neurosci. 2008; 11: 172–182 https://doi.org/10.1179/147683008X301487
Vauzour D. Dietary polyphenols as modulators of brain functions: biological actions and molecular mechanisms underpinning their beneficial effects. Oxid Med Cell Longev. 2012; 914273 https://doi.org/10.1155/2012/914273
Góngora JL. Caffeine as a preventive drug for Parkinson’s disease: epidemiologic evidence and experimental support. Rev Neurol. 2010; 50(4), 221–9.
Fehske CJ, Leuner K, Müller WE. Ginkgo biloba extract (EGb761®) influences monoaminergic neurotransmission via inhibition of NE uptake, but not MAO activity after chronic treatment. Pharmacol Res. 2009; 60: 68–77 https://doi.org/10.1016/j.phrs.2009.02.012
Grimm MO, Stahlmann CP, Mett J, Haupenthal VJ, Zimmer VC, et al. Vitamin E: Curse or benefit in Alzheimer’s disease? A systematic investigation of the impact of α-, γ- and δ-tocopherol on Aβ generation and degradation in neuroblastoma cells. J Nutr Health Aging. 2015; 19:646–654 https://doi.org/10.1007/s12603-015-0506-z.
Zbarsky V, Datla K, Parkar S, Rai D, Aruoma O, Dexter D. Neuroprotective properties of the natural phenolic antioxidants curcumin and naringenin but not quercetin and fisetin in a 6-OHDA model of Parkinson’s disease. Free Radical Res. 2005; 39: 1119–25 https://doi.org/10.1080/10715760500233113
Wu P, Zhang Z, Wang F, Che J. Natural compounds from traditional medicinal herbs in the treatment of cerebral ischemia/reperfusion injury. Acta Pharmacologica Sinica. 2010; 31: 1523–1531 https://doi.org/10.1038/aps.2010.186
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest: The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Martínez Leo, E.E., Segura Campos, M.R. Systemic Oxidative Stress: A Key Point in Neurodegeneration — A Review. J Nutr Health Aging 23, 694–699 (2019). https://doi.org/10.1007/s12603-019-1240-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12603-019-1240-8