
Abstract
Blood–brain barrier (BBB) is comprised of brain microvascular endothelial cells (ECs), astrocytes, perivascular microglia, pericytes, neuronal processes, and the basal lamina. As a complex and dynamic interface between the blood and the central nervous system (CNS), BBB is responsible for transporting nutrients essential for the normal metabolism of brain cells and hinders many toxic compounds entering into the CNS. The loss of BBB integrity following stroke induces tissue damage, inflammation, edema, and neural dysfunction. Thus, BBB disruption is an important pathophysiological process of acute ischemic stroke. Understanding the mechanism underlying BBB disruption can uncover more promising biological targets for developing treatments for ischemic stroke. Ischemic stroke-induced activation of microglia and astrocytes leads to increased production of inflammatory mediators, containing chemokines, cytokines, matrix metalloproteinases (MMPs), etc., which are important factors in the pathological process of BBB breakdown. In this review, we discussed the current knowledges about the vital and dual roles of astrocytes and microglia on the BBB breakdown during ischemic stroke. Specifically, we provided an updated overview of phenotypic transformation of microglia and astrocytes, as well as uncovered the crosstalk among astrocyte, microglia, and oligodendrocyte in the BBB disruption following ischemic stroke.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The neurovascular unit (NVU) has molecular signaling and physical attributes and exerts a vital role in carrying out neurovascular coupling (NVC). NVC supports the high energy demand of the brain from the blood and modulates brain functions, such as modulating memory via synaptic plasticity [1, 2]. In addition to the vital roles of neurons, astrocytes, and vasculature in NVU [3], microglia and oligodendrocytes, once considered peripheral to the NVU, may indirectly contribute to NVC. Microglia and oligodendrocytes are conducive to NVU homeostasis in both health and disease conditions. NVC is a finely tuned process in contrast with auto-regulation. Auto-regulation ensures that blood flow is consistent with variation of systemic blood pressure [4] and is known as a more coarse method in regulating blood flow [5].
Ischemic stroke severely interrupts both the physical and molecular signaling aspects of the NVU, so that it is either entirely lost in some regions or merely dysfunctional in other regions. As a part of NVU, BBB is a dynamic regulatory boundary that regulates the exchange of ions and other molecules and prevents the uncontrolled exchange of bacteria, viruses, toxins, and cells between the blood and CNS. The loss of BBB integrity induces the injury of paracellular permeability following stroke, t, and leads to hemorrhagic transformation, vasogenic edema, and increased mortality. Moreover, BBB breakdown following ischemic stroke is the most important factor that limits the therapeutic time window of thrombolytic agent recombinant tissue plasminogen activator (rtPA) [6,7,8]. Microglia and astrocytes are activated after cerebral ischemia and release the chemokines, cytokines, matrix metalloproteinases (MMPs), etc. These inflammatory mediators from activated microglia and astrocytes are shown as important factors in the BBB breakdown [9].
In this review, we highlighted the current knowledge about the effects of ischemic stroke on the BBB breakdown. We particularly focused on the vital and dual roles of glial cells on the BBB breakdown after ischemic stroke and revealed the phenotypic transformation of microglia and astrocytes as well as the intercellular communication within astrocytes and microglia.
BBB Injury Following Ischemic Stroke
Structure and Function of BBB
BBB, a part of NVU, is formed by ECs, neuronal cells, glial cells, and pericytes. The pericytes are embedded in the basement membrane of capillary vessel. Innermost luminal side of the BBB is constituted by continuous non-fenestrated ECs, which are sealed by tight junctions (TJs) [10]. The close interaction among the astrocytes, microglia, ECs, pericytes, and neurons is indispensable for the integrity of BBB [11]. Cerebral ECs have unique characteristics distinguished from peripheral ECs, for instance, cerebral ECs form a continuous monolayer without fenestrations characterized by specified TJs and a low transcytosis rate [12, 13]. TJ and adherens junctions constitute a circumferential zipper-like seal between adjacent ECs; this action ensures they act as a gatekeeper for limiting paracellular permeability [14]. TJs contain three transmembrane proteins: occludin, claudins, and junction adhesion molecules (JAMs) [15, 16]. Adherens junctions encompass vascular endothelial (VE)-cadherin and transmembrane proteins, with extracellular segments homophilic interacting and cytoplasmic domains binding to the plaque proteins [17].
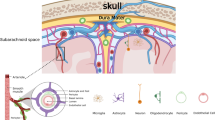
BBB is an anatomical and tightly regulated interface between the CNS and circulating blood [18] (Fig. 1). Only gaseous molecules (e.g., N2, CO2, and O2) and small lipophilic molecules (< 400 kD) can enter the cerebral parenchyma due to the low permeability of the BBB; the delivery of macromolecules from blood into the cerebral parenchyma is severely limited [18]. In order to meet the high-energy demands of neuronal activity, BBB is also tightly regulated and efficient transport barrier for enabling the delivery of essential nutrients to the CNS. Besides, there are much higher numbers of mitochondria, receptors, transporters, ion channels, and active efflux pumps in cerebral ECs than peripheral ECs [19], all of which ensure that cerebral ECs can selectively regulate molecular transport between the brain and blood [20]. In addition, BBB is an immunologic barrier for it can block various leukocytes entering from blood into the CNS and inhibit the infiltration of CNS-specific antigens into the peripheral immune system. As a result, BBB hinders many toxic compounds and pathogens entering into the CNS [21]. Therefore, BBB plays a key role in maintaining homeostasis in the neuronal microenvironment.

Diagram illustration of the BBB. BBB is formed by brain microvascular endothelial cells (ECs), pericytes, astrocytes, neurons, perivascular microglia, and basal lamina
Moreover, BBB is a continuous endothelial membrane within brain microvessels that have sealed cell‑to‑cell contacts and are sheathed by perivascular astrocyte end-feet and mural vascular cells [22]. Abluminal EC surface is almost surrounded by perivascular astrocytic endfeet [23]. Gap junctions, presented in the astrocyte endfeet that enwrap the blood vessel walls, mediate intercellular communication between astrocytes [24]. Astrocyte-released sonic hedgehog (Shh), the most widely studied molecule, acts on EC hedgehog (Hh) receptors and then regulates TJ formation and BBB permeability [25]. Other chemical mediators released from astrocytes, containing prostaglandins, nitric oxide (NO), glial cell-derived neurotrophic factor (GDNF), and arachidonic acid, likewise regulate TJs [26]. Thus, as the most abundant glial cells in the brain, astrocytes not only participate in regulating the cerebral blood flow but also adjusting the BBB permeability [27].
The extravasation of peripheral immune cells into CNS is dependent on adhesion molecules, including intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which are expressed in ECs and pericytes at extremely low levels [28]. ECs can inhibit the expression of pro-inflammatory genes and quiesce circulating leukocytes under normal physiological conditions [18]. Therefore, the BBB directly regulates immune reactions rather than acts as a neutral and passive barrier within the CNS, as well as can modulate the function and fate of infiltrating immune cells under physiological condition [29]. Under systemic inflammatory conditions, excessive immune responses injure TJs and ECs and then induce the BBB breakdown [30].
BBB Breakdown Following Ischemic Stroke
BBB breakdown following ischemic stroke results in infiltration and accumulation of molecules and peripheral immune cells entering into brain parenchyma. Hence, cerebral ischemia insults rapidly induce cerebral edema, containing cytotoxic edema and vasogenic edema. Cytotoxic edema is resulted by the excess accumulation of fluid in the intracellular space and occurs minutes after ischemia onset. The vasogenic edema appears after cytotoxic edema and is in particular related to BBB breakdown [31, 32]. In addition to brain edema, BBB disruption likewise induces tissue damage, neuronal inflammation, and dysfunction [33]. During the first 3 h after ischemic stroke, BBB disruption in patients can be identified by magnetic resonance imaging (MRI) due to the development of vasogenic edema [34]. Consistently, researchers have reported that cerebral edema forms in the first few hours after ischemia onset based on animal models [35].
BBB dysfunction likewise contributes to the tPA treatment-induced hemorrhagic transformation and the increased mortality after ischemic stroke. For instance, researchers have revealed that tPA-associated hemorrhagic transformation often occurred as a result of the catastrophic breakdown of the BBB [36]. Li et al. also demonstrated that BBB breakdown was correlated with intracerebral hemorrhage following tPA thrombolysis [37]. Furthermore, it is well accepted that BBB dysfunction contributes to the infiltration and accumulation of molecules and peripheral immune cells into brain parenchyma following stroke and then induces the injury progression [38, 39].
The potential mechanism and vital factors involved in BBB breakdown have attracted the attention of more and more researchers. Recent study has revealed that within the first few hours after ischemia, microglial cells are activated and then release pro-inflammatory cytokines, containing interleukin (IL)-1 and IL-6. Besides, the activated microglia likewise promote the expression of ICAM-1, P-selectin, and E-selectin. These molecules further promote the adherence and accumulation of leukocytes and enable the leukocytes to migrate across the blood vessels. This action induces inflammatory cascades and BBB breakdown [40] and further exaggerates the cerebral infarction [41].
BBB Breakdown and Basement Membrane
The basement membrane is a sheet-like extracellular matrix (ECM) complex beneath epithelium and endothelium and encircles the abluminal side of blood vessels at the BBB. Brain basement membrane contains collagen IV, laminin, nidogen, and heparan sulfate proteoglycans. Collagen IV is the most abundant component of the basement membrane [42]. Basement membrane displays substantial changes during the ischemic stroke onset. Loss and degradation of basement membrane have been found to occur soon after ischemia [43]. In addition, the extravasation of blood constituents to the brain tissues through BBB breakdown is highly correlated with loss of basement membrane [44]. Thus, maintaining basement membrane integrity is a key challenge to prevent brain damage and hemorrhagic complications following ischemic stroke [45]. High content of local matrix metalloproteinase-9 (MMP-9) is closely related to basal lamina collagen IV degradation and BBB breakdown, which results in neutrophil infiltration in the infarcted and hemorrhagic areas [46]. Furthermore, MMP-9 plays aggravated effect in tPA-associated BBB disruption [47].
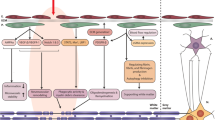
The Role of Glial Cells in BBB Injury Following Ischemic Stroke (Fig. 2)

The relationship among BBB disruption and glial cells after ischemic stroke. Activation of microglia and astrocytes after ischemia induces the increment of cytokines, chemokines, VCAM-1, and ICAM-1 in ischemic brain tissue. These inflammatory mediators result in the BBB breakdown, which leads to edema and neuroinflammation. In pathological condition, OPCs often fail to differentiate into mature oligodendrocytes
Astrocyte
Accumulating research have revealed the prominent position and role of astrocytes in the NVU, such as providing neurotrophic support and regulating synaptic activity [48]. Astrocytes provide a cellular link among blood vessels and the neuronal circuitry in the NVU [49, 50]. For instance, astrocytes extend their foot process to wrap around neuronal synapses [51]. Astrocytes take up the neurotransmitters at the cleft to temporally and spatially refine synaptic signaling at the synapses, thereby regulating the information transfer between neurons [52]. Besides, astrocytes likewise provide structural and nutritional support for neurons, modulate the cerebral blood flow, and regulate the function of BBB in response to the neuronal activity [53].
However, astrocytes become hyperactive and then induce the formation of glial scars under pathological conditions such as cerebral ischemia and mechanical injury. Astrocyte undergoes molecular, morphological, and functional remodeling in response to CNS injury and is named as reactive astrocytes, which is considered as the major contributor to the pathological process of ischemic stroke [54]. Accumulating studies have suggested that astrocytes played a vital role in immune responses after ischemic stroke. The activation of astrocytes during acute ischemic stroke can be identified by the upregulated expression of glial fibrillary acidic protein (GFAP), which is widespread and long-lasting. Astrocyte activation following ischemic stroke can be induced by various factors, including hypoxia, blood vessel disruption, neurotransmitters released from neighboring neurons, cell death, cytokines IL-1 and IL-6, ciliary neurotrophic factor (CNTF), transforming growth factor (TGF)-α, and kallikrein-related peptidase 6 (KLK6). Importantly, activation of astrocyte following ischemic stroke can further release various pro-inflammatory mediators, containing tumor necrosis factor (TNF)-α, IL-1α, IL-1β, IL-6, interferon-gamma (IFN-γ), and reactive oxygen species (ROS)/reactive nitrogen species (RNS) [55].
Increasing findings have demonstrated the dualistic effects of astrocytes following ischemic stroke. For example, transcriptomics has revealed a fundamental discovery: astrocytes are diverse and specialized in the healthy brain to perform specific roles in distinct CNS circuits [56]. Likewise, reactive astrocytes are also diverse, and it is proposed that reactive astrocytes have two polarization states: neurotoxic (pro-inflammatory) and neuroprotection (anti-inflammatory). The polarization of pro-inflammatory astrocytes is induced by pro-inflammatory factors, including TNF-α and IL-1α, and can be characterized by the expression of C3 and inducible nitric oxide synthase (iNOS). Anti-inflammatory astrocytes, known as neuroprotective subtype, can be identified by testing the expression of S100A10 and pentraxin-3 (PTX3) and the neurotrophic factors, containing IL-2, IL-10, and TGF-β [57].
On one hand, pro-inflammatory astrocytes directly exert detrimental effects on the BBB integrity via increasing the expression of vascular endothelial growth factor (VEGF), chemokines (CCL2 and CCL5), cytokines (TNF-α, IL-1β, IL-6, and IL-15), ROS, MMP, and lipocalin-2 (LCN-2). Astrocyte-produced VEGF reduces the TJs expression in ECs, which exacerbates the BBB damage and the neurological deficits [58]. Polymerase δ-interacting protein 2 (Poldip2) is likewise upregulated in astrocytes after stroke, which can cause the damaged BBB integrity by upregulating the expression of MCP-1, VEGF, TNF-α, IL-6, and MMP [59]. On the other hand, astrocytes also produce soluble factors that recruit peripheral immune cells via upregulating the expression of ICAM-1 and VCAM-1 in ECs and activate microglia, which in turn indirectly accelerates the inflammation-induced BBB disruption [60].
In contrast, anti-inflammatory astrocytes have been found to promote BBB repair via resolving inflammation. For example, anti-inflammatory (S100A-positive) astrocytes accelerate inflammation resolution via secreting IL-2, IL-10, and TGF-β. Astrocyte-released PTX3 attenuates IgG staining in ischemic brain tissue through restraining the expression of VEGF [61]. Insulin-like growth factor-1 (IGF-1) released from astrocyte protects BBB integrity and ameliorates neurological function following stoke via shifting immune cells toward an anti-inflammatory profile in the ischemic area [62]. Furthermore, astrocytes can restrain overactivation of microglia by promoting the expression of C-X3-C Motif Chemokine Ligand 1 (CX3CR1) receptor and interleukin 4 receptor-α (IL-4Rα) in microglia [63]. Importantly, neuroprotective astrocyte was found to surpass the transcripts of neurotoxic astrocytes 3 days after ischemic stroke. Expressed genes in neuroprotective astrocytes are associated with regulation of ECM integrity and scar formation, which is known as astrogliosis. Astrogliosis restrains the immune reaction within the infarct region and inhibits the migration of infiltrating immune cells [64].
Surprisingly, accumulating studies have revealed that, under pathological situations, reactive astrocytes obtain the ability to engulf injured cells and degrade cellular debris in the penumbra; this action assists with the resolution of inflammation. Besides, the researchers revealed that the phagocytotic activity of astrocytes starts 3 days after ischemic stroke onset [65]. Further study has demonstrated that neuroprotective (S100a10-positive) astrocytes possess the phagocytotic activity and protect against the brain injury through phagocytosis of apoptotic neurons [66]. Therefore, promoting the astrocytic polarization to the neuroprotective (S100a10-positive) phenotype significantly mitigates the BBB permeabilization and accelerates the stroke recovery [67] (Fig. 3).

The dual roles of microglia and astrocytes on the BBB injury following ischemic stroke
Microglia
Microglia are the primary immune cells and account for 5–15% of all cells in the human brain. Besides, microglia are important partners of the NVU [68, 69] and are derived from the yolk sac and seeded in the brain in the early development of CNS, which are known as the first glial cells. Both microglia and neurons develop concurrently into highly plastic cells with mobility [70,71,72]. Under physiological condition, microglia continuously survey their surrounding environment in the CNS. Therefore, microglia are always the “pioneers” in the NVU. Microglial cells wander more observantly and detect their environment via scattering throughout the brain as sentinels [73]. While in the pathological status, microglia always first respond to the brain insults [9, 74, 75].
As the first activated innate immune cells, microglia can be activated within minutes after tissue damage onset [76]. Activated microglia acquire the ability of phagocytosis and can secrete numerous inflammation factors, with the morphological changes from the ramification to an amoeboid shape [77]. The dichotomies of activated microglia, such as ‘‘M1 versus M2″ and “resting versus activated,” have been defined in accumulating studies. Intermediate phenotypes of microglia display with diverse combination of polarization markers ranging; these intermediate phenotypes represent the crossroads of various pro-and anti-inflammatory effects [78,79,80]. Thus, the supposed dichotomy, M1 and M2, hardly reflects a wide range of microglial phenotypes. However, this dichotomy of activated microglia facilitates understanding the state and function of microglia in various CNS disorders [81].
The neurotoxic (pro-inflammatory) microglia, known as “M1-like” phenotype previously, can be determined by testing the expression of CD32, CD16, CD11b, CD68, CD86, iNOS, etc. [82]. The characteristics of activated microglia and switch of phenotypes were determined by local extracellular and intracellular signals. Besides, the polarization of neurotoxic microglia is considered to be destructive to NVU [83] and BBB dysfunction [84] and can be characterized by increased inflammatory mediators, including TNF-α, IL-1β, and ROS [85]. Neuroprotective microglia, formerly known as “M2-like” microglia, are characterized by upregulation of anti-inflammatory mediators, containing TGF-β, IL-10, and glucocorticoids [85]. The transformation of neuroprotective microglia can be identified by testing the expression of arginase-1 (Arg-1) and CD206 [86].
After cerebral ischemia, the number of microglia in the infarct core decreases immediately. However, the number of microglia in the penumbra increases within hours, peaks at 48–72 h after ischemic stroke, and persists in this region for several weeks [87]. In addition, another research has reported that microglia were activated by damage-associated molecular pattern (DAMP) within minutes in the peri-infarct area, and these activated microglia became round- or amoeboid-like in morphology within 12–24 h after experimental cerebral ischemia [88].
Besides, microglia begin migrating to the infarct core from the penumbra 1 day after stroke onset, and the migration of microglia is mediated by the annexin-1/casein kinase II pathway [89]. After morphological changes, the microglial functions and signaling pathways in microglia are differentiated between CD16-positive microglia versus CD206-positive subtype. Within 48 h after ischemic stroke onset, resident CD206-positive microglia shift to the CD16-positive subtype in the peri-infarct region [90]. Neurotoxic (iNOS-positive) microglia activate the nuclear factor-kappa B (NF-κB) and promote the formation of NLRP3 inflammasome and then induce the elevated levels of pro-inflammatory cytokines [91]. Inhibiting the polarization of neuroprotective (iNOS-positive) microglia can attenuate the cerebral ischemia/reperfusion (I/R)-induced BBB injury [92].
The polarization of neuroprotective (CD206-positive) microglia induced by IL-4, IL10, and VEGF begins several days after ischemic stroke [93]. The CD206-positive microglia obtain the phagocytic ability, which is similar to the infiltrating macrophages [94]. The comparative analysis of monocyte-derived macrophage-specific and microglia-specific transcripts has revealed that microglia are more likely liable to transition to the neurotoxic (CD86/32/16-positive) subtype in response to stroke compared to infiltrating macrophages [95].
Both IL-4 and IL-10 from neuroprotective microglia [96] could restrain IL-1β, IFN-g, and TNF-α expression in the ischemic brain but elevate the levels of anti-inflammatory factors by inhibiting the NF-κB pathway [97]. In addition, neuroprotective microglia likewise can produce the TGF-β [94], which can reduce the levels of TNF-α and monocyte chemoattractant protein-1 (MCP-1) by affecting the ALK5-p-Smad2/3 signaling pathway [98]. IL-10 reduces the ICAM-1 and VCAM-1 expressions in ECs and limits the infiltration of immune cells into the brain [99]. Thus, IL-10 and TGF-β are vital factors in maintaining the functional and structural integrity of the BBB following ischemic stroke. Promoting the polarization of neuroprotective (CD206-positive) microglia could accelerate the resolution of inflammation, promote the BBB repair, and accelerate functional recovery (Fig. 3).
Oligodendrocyte
The component of the NVU likewise contains oligodendrocytes. The effect of oligodendrocyte on the NVU and BBB breakdown following brain injury came into focus [100,101,102]. Oligodendrocytes are supported by other cells in the NVU, and these cells collaborate in various processes, containing neurogenesis, angiogenesis, and oligodendrogenesis [103]. Oligodendrocytes are the myelin-producing cells in the CNS and are critical for function and survival of the axons [104]. The researchers have revealed the oligodendrocytes’ ability to preserve neurons during ischemic stroke with minimal glucose. Besides, as a member of NVU, oligodendrocyte lineage cell also monitors the EC processes and BBB function [105, 106].
Strikingly, ECs can also control the various aspects of oligodendrocytes when they regulate the BBB function. For instance, cerebral EC-produced fibroblast growth factor (FGF) and brain-derived neurotrophic factor (BDNF) enable oligodendrocyte precursor cells (OPCs) to survive and increase the number of OPCs [107]. In addition, VEGF-A secreted from cerebral endothelial cells has been found to promote OPC migration [108]. Studies have reported that BBB disruption is a key factor in the acute ischemic stroke-induced pathological damage in white matter [109, 110]. Of note, loss of white matter integrity and microvascular dysfunction after acute ischemic stroke predict poor outcomes [111]. NG2-glia cells, known as OPCs, constitute the fifth major cell population in the CNS and possess the ability to promote remyelination of axons after ischemic injury [112].
The effects of NG2-glia cells on BBB integrity are inconsistent under physiological and pathological conditions. During CNS development, NG2-glia cells improve the BBB tightness through upregulating the expression of occludin and claudins (TJs) through activation of TGF-β signaling in ECs. Besides, NG2-glia cells can attach to cerebral ECs found in neonatal mouse brains via the basal lamina [108]. In a white matter damage model of prolonged cerebral hypoperfusion stress, NG2-glia cells can respond rapidly before BBB dysfunction via secreting MMP-9, which results in neutrophil infiltration [113]. Moreover, NG2-glia cells are necessary for maintaining the microglial homeostasis [114] and possess the ability to differentiate into reactive astrocytes following permanent brain ischemia [115]. Therefore, we predict that NG2-glia cells may promote the repair of the BBB function via promoting the expression of occludin and claudins (TJs) in ECs during the recovery phase after ischemic stroke.
Crosstalk Among Glial Cells in BBB Injury (Fig. 4)

The crosstalk between the glial cells
Crosstalk Between Astrocyte and Oligodendrocyte
Oligodendrocytes are highly vulnerable to ischemia [116]. The restrained maturation of OPCs to oligodendrocytes after ischemic stroke results in remyelination failure and then hampers the neurological recovery [117]. BBB breakdown following ischemic stroke tends to promote the leakage of brain auto-antigens, such as myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MBP), and proteolipid protein (PLP), to the periphery. These auto-antigens in blood activate the immune system and promote the migration of activated immunocytes into the CNS and then exacerbate the brain injury [118].
Moreover, BBB disruption induces the changes of composition in the brain microenvironment and then results in the infiltration of blood proteins into the CNS. Under this condition, OPCs often fail to differentiate into mature oligodendrocytes, which hinders the remyelination and myelin repair [119]. Therefore, the maturation of OPCs is strongly associated with the perivascular condition, which becomes terrible for OPC maturation when increased BBB permeability allows blood proteins to enter the brain. Thus, the contribution of blood-derived signals to OPC maturation is detrimental, but the mechanism is unclear.
Both astrocytes and microglia could promote the proliferation and differentiation of OPCs through noncell autonomous means. Astrocytes can attach to oligodendrocytes through gap junctions and utilize Cx43 hemichannels to transfer ATP and other small molecules [120,121,122]. Glutamate transporters in astrocytes expel glutamate by activating N-methyl-D-aspartatic acid (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors on the oligodendrocyte lineage cells in an ischemic environment, thereby preventing the differentiation of OPCs [123,124,125,126].
Moreover, in addition to the adverse effect of astrocytes on the OPC differentiation, astrocyte can significantly facilitate OPC differentiation via precluding the release of glutamate through blocking the Cx43 hemichannel [127]. Therefore, modulation of Cx43 may be key to enabling the damaged axons to myelinate and promote the generation of new oligodendrocytes. Astrocytes likewise produce beneficial growth factors following brain damage that promote neuronal regenerative processes. For instance, astrocyte-derived BDNF could promote the quantity of oligodendrocytes in white matter in a mouse model of cerebral hypoperfusion [128]. Additionally, pluripotent stem cells derived from astrocytes boost OPC maturation and release tropic factors that assist with oligodendrogenesis [129].
Furthermore, astrocytes can become reactive and re-acquire immature stem cell like properties under inflammatory conditions or following CNS injury [130]. These reactive astrocytes are mainly derived from static astrocytes rather than endogenous glial precursor cells. Reactive astrocytes obtain the pluripotency, self-renewal, and display remarkable plasticity [131]. Accumulating studies have shown that these reactive astrocytes have stem cell properties and have multiple differentiation potentials [132]. Membrane-bound neuregulin-1 (Nrg1) is an essential factor for promoting differentiation and maturation of oligodendrocytes. Ding et al. have found that Nrg 1 could induce reactive astrocytes to express oligodendrocyte markers O4 and PDGFR-α at both mRNA and protein levels, suggesting that Nrg1 could directly convert reactive astrocytes toward oligodendrocyte lineage cells and then promote the remyelination following CNS injury [133].
Further studies have demonstrated the dual roles of reactive astrocyte on the oligodendrocyte contribute to the phenotypic transformation. For instance, C3-positive (neurotoxic) astrocytes have inhibitory effects on oligodendrogenesis and OPCs’ differentiation via releasing pro-inflammatory cytokines. In contrast, PTX3-positive (neuroprotective) astrocytes promote the oligodendrocytes maturation via releasing trophic factors, such as BDNF and VEGF, and produce the anti-inflammatory cytokines [134]. Similarly, transplantation of neuroprotective (S100a10-positive) astrocytes could improve motor recovery of spinal cord injury (SCI) via promoting the myelination. However, neurotoxic (C3-positive) astrocyte-transplanted SCI mice showed a higher extent of disorganized structures of neurons and a lower number of myelinated axons [135].
Crosstalk Between Microglia and Oligodendrocyte
Microglia might regulate the oligodendrocyte function and accelerate remyelination by eliminating defective myelin and apoptotic cells during diseased states [136]. Microglia can transform to the CD206/Arg-1-positive (neuroprotective) subtype from iNOS/IL-β-positive (neurotoxic) subtype during the remyelination process. CD206/Arg-1-positive microglia could promote the differentiation of oligodendrocytes both in vivo and in vitro [137]. For instance, the mechanism of ethyl pyruvate-mediated differentiation of OPCs is found to be related to increasing the CD206-positive microglia and decreasing CD16/32-positive microglia [138]. In addition, microglia can localize at the demyelinated region after axonal demyelination and utilize progenitor cells and neural stem cells to produce additional OPCs in the corpus callosum [139]. Of note, activated microglia induced by lipopolysaccharide (LPS) could decrease the OPC generation through TLR4 signaling pathways [140]. Therefore, promoting the polarization of neuroprotective microglia could initiate remyelination and heal the ischemic stroke-induced brain injury.
Crosstalk Between the Microglia and Astrocyte
Anti-inflammatory factors derived from neuroprotective microglia promote the transformation of astrocytes to the neuroprotective phenotype via reducing the expression of purinergic 2Y1 receptor (P2Y1R) in a brain trauma model [141], suggesting that neuroprotective astrocytes might also be induced by activated microglia in the acute ischemic stroke. The phenotypic transformation of astrocytes to neurotoxic (pro-inflammatory) or neuroprotective (anti-inflammatory) following ischemic stroke is parallel to the activated microglia. In addition, both activated microglia and reactive astrocytes play dual roles in BBB breakdown and brain injury after ischemic stroke. For instance, astrocytes accelerate BBB disruption by exacerbating inflammation injury and promoting the secretion of soluble factors. Transcriptome analysis has revealed that genes involved in leukocyte transendothelial migration, inflammation, and JAK/STAT3 signaling were up-regulated in reactive astrocytes [64].
The activation of toll-like receptor 4 (TLR4) and NF-κB pathway has been identified as the key molecules in mediating the activation of microglia and astrocytes [142]. Activation of NF-κB pathway initiates the neuroinflammation following ischemic stroke via promoting the expression and secretion of inflammation-related genes in astrocytes and microglia [143, 144]. Besides, the vital roles of TLR4 and NF-κB in the activation of microglia and astrocytes were further confirmed by Liu et al. [9] using rat middle cerebral artery occlusion (MCAO)/reperfusion model. Furthermore, accumulating studies have revealed that inhibiting the expression of TLR4 and NF-κB activation obviously reduces neuroinflammation and ameliorates subsequent cerebral ischemia injury [145, 146]. Another study verified that inhibiting the TLR4/NF-κB signaling pathway could protect against rat cerebral I/R injury [147].
In an astrocytes-microglia co-culture model of inflammation, the researchers have found that activation of microglial P2Y6 receptors induced the release of nitric oxide (NO), which caused astrocyte apoptosis [148]. Besides, the activated microglia and astrocytes were found to be adjacent to each other in the penumbra, indicating the potential interaction and “co-activation” between microglia and astrocytes. Importantly, the polarization of neurotoxic (pro-inflammatory) astrocyte is found to be induced by IL-1α, TNF-α, and C1q, which are released by activated microglia [149, 150]. Additionally, inhibiting the TLR4-mediated activation microglia could decrease the secretion of TNF-α and then restrain the NF-κB-induced activation of neurotoxic astrocyte and reduce the neuronal damage following cerebral ischemia [9].
As aforementioned, BBB disruption-induced brain edema, potentially leading to brain herniation and death, is a life-threatening consequence of stroke [151]. BBB integrity is dependent on ECs and astrocytes and cell–cell junctions, containing adherens junctions, tight junctions, vascular endothelial (VE)-cadherin, claudin-5, and claudin-1 [152,153,154]. VE-cadherin is expressed in ECs and promotes junction stability via interacting with the actin cytoskeleton [153]. Claudin-1 is strongly expressed in leaky microvessels in brain after stroke, but rarely expressed in the normal BBB [152]. Additionally, AQP4 is densely expressed in the astrocytic endfeet and mediates water accumulation, which results in the cytotoxic edema and is associated with the stroke onset [154].
Activated microglia are strongly associated with the degradation of the BBB after cerebral ischemia. Activated microglia engulf blood vessels in the penumbra through extending cellular protrusions toward vessels, which results in the extravasation of macrophages from blood. Besides, microglia in peripheral vessels engulf the ECs, which induces the dysfunction of endothelium and BBB disintegration [155]. Activated microglia that have detrimental effect on BBB disruption have been identified as CD68-positive (neurotoxic) subtype, and they can produce pro-inflammatory factors, such as TNF-a, IFN-g, CCL2, IL-1α, IL-1β, IL-6, VEGF, and MMP-9. The expressions of IL-1α and IL-1β are strongly upregulated in the ischemic brain tissues 6–24 h after stroke onset [156]. Removal of IL-1α or IL-1β could attenuate BBB disintegration and reduce brain injury in experimental stroke. IL-1α is a key mediator of the CXCL1 and IL-6 expression in the ECs. Besides, IL-1α can induce the expression of AQP4 in astrocytes, which is known as a deteriorated factor of BBB and brain edema [157].
Furthermore, IL-1β from neurotoxic microglia likewise increases the degradation and relocation of occludin and ZO-1 in ECs [158] and promotes the expression and release of VEGF from astrocytes [159]. Moreover, IL-1β could cause an incremental release of CCL2, CCL20, and CXCL2 and downregulate the expression and release of sonic hedgehog from astrocyte. Sonic hedgehog is known as an important signal in maintaining BBB integrity [160]. Furthermore, neurotoxic microglia-produced TNF-α induces downregulation of occluding and promotes the endothelial necroptosis via binding to TNF receptor 1 [83]. TNF-α could upregulate the expression of MMP-9 but decrease collagen IV expression in ECs; both of these actions obviously increase BBB permeability in vitro [161]. Moreover, IL-1β, IL-6, IFN-g, and TNF-α increase the expression of ICAM-1 and VCAM-1 in the ECs and then facilitate the infiltration of peripheral immune cells [162]. Therefore, inhibiting activation of microglia or blocking the release of detrimental cytokines after stroke may reduce the BBB breakdown.
Conclusion
It has become apparent that BBB is a mechanical and immunologic barrier between blood and CNS. BBB can block various leukocytes and proteins entering the CNS and inhibit the infiltration of CNS-specific antigens into the peripheral immune system. During the pathological process of ischemic stroke, brain edema, hemorrhagic transformation, and neuronal damage occur in close association with BBB breakdown. As reviewed in this article, microglia, astrocytes, and oligodendrocytes regulate BBB permeability under physiological state. After cerebral ischemia onset, the activation of astrocytes and microglia and the injured oligodendrocyte are deeply involved in BBB disruption. Importantly, both activated microglia and reactive astrocytes show the dual effects on BBB breakdown due to the phenotypic transformation. To better understand the roles of activated microglia, reactive astrocytes and oligodendrocyte on the BBB breakdown will provide more opportunities and insight to explore appropriate therapeutic interventions for ischemic brain injury. Moreover, in the pathological process of BBB disruption, there is a close relationship among microglia, astrocyte, and oligodendrocyte. Moving forward, more research will need to further explore the agents that can promote the phenotypic transformation of astrocytes and microglia to neuroprotective subtype.
Data Availability
Not applicable.
References
Zhou Z, Okamoto K, Onodera J et al (2021) Astrocytic cAMP modulates memory via synaptic plasticity. Proc Natl Acad Sci U S A 118:e2016584118
Vermeulen TD, Benbaruj J, Brown CV, Shafer BM, Floras JS, Foster GE (2020) Acute intermittent hypercapnic hypoxia and cerebral neurovascular coupling in males and females. Exp Neurol 334:113441
Heo C, Kwak HJ, Ngo LH, Woo RS, Lee SJ (2023) Implementation of the neuro-glia-vascular unit through co-culture of adult neural stem cells and vascular cells and transcriptomic analysis of diverse Abeta assembly types. J Neurosci Methods 402:110029
Wang CH, Chang WT, Huang CH, Tsai MS, Liu SH, Chen WJ (2020) Cerebral blood flow-guided manipulation of arterial blood pressure attenuates hippocampal apoptosis after asphyxia-induced cardiac arrest in rats. J Am Heart Assoc 9:e016513
Agrawal S, Placek MM, White D et al (2023) Studying Trends of Auto-Regulation in Severe Head Injury in Paediatrics (STARSHIP): protocol to study cerebral autoregulation in a prospective multicentre observational research database study. BMJ Open 13:e071800
Song S, Huang H, Guan X et al (2021) Activation of endothelial Wnt/beta-catenin signaling by protective astrocytes repairs BBB damage in ischemic stroke. Prog Neurobiol 199:101963
Nguyen QL, Okuno N, Hamashima T et al (2021) Vascular PDGFR-alpha protects against BBB dysfunction after stroke in mice. Angiogenesis 24:35–46
Wang Y, Wang X, Zhang X et al (2020) D1 receptor-mediated endogenous tPA upregulation contributes to blood-brain barrier injury after acute ischaemic stroke. J Cell Mol Med 24:9255–9266
Liu M, Xu Z, Wang L et al (2020) Cottonseed oil alleviates ischemic stroke injury by inhibiting the inflammatory activation of microglia and astrocyte. J Neuroinflammation 17:270
Cottarelli A, Shahriar S, Arac A et al (2023) Rab7a activation promotes degradation of select tight junction proteins at the blood-brain barrier after ischemic stroke. bioRxiv. Preprint
Taler M, Aronovich R, Henry Hornfeld S et al (2021) Regulatory effect of lithium on hippocampal blood-brain barrier integrity in a rat model of depressive-like behavior. Bipolar Disord 23:55–65
He J, Shen R, Liu Q et al (2022) RGD nanoarrays with nanospacing gradient selectively induce orientation and directed migration of endothelial and smooth muscle cells. ACS Appl Mater Interfaces 14:37436–37446
Hasannejad-Asl B, Pooresmaeil F, Choupani E et al (2023) Nanoparticles as powerful tools for crossing the blood-brain barrier. CNS Neurol Disord: Drug Targets 22:18–26
Orlando A, Linsalata M, Bianco G et al (2018) Lactobacillus rhamnosus GG protects the epithelial barrier of Wistar rats from the pepsin-trypsin-digested gliadin (PTG)-induced enteropathy. Nutrients 10:1698
Guo W, Wang P, Liu ZH, Ye P (2018) Analysis of differential expression of tight junction proteins in cultured oral epithelial cells altered by Porphyromonas gingivalis, Porphyromonas gingivalis lipopolysaccharide, and extracellular adenosine triphosphate. Int J Oral Sci 10:e8
Aydin S, Billur D, Kizil S et al (2020) Evaluation of blood-testis barrier integrity in terms of adhesion molecules in nonobstructive azoospermia. Andrologia 52:e13636
Chrifi I, Louzao-Martinez L, Brandt MM et al (2019) CMTM4 regulates angiogenesis by promoting cell surface recycling of VE-cadherin to endothelial adherens junctions. Angiogenesis 22:75–93
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV (2019) Blood-brain barrier: from physiology to disease and back. Physiol Rev 99:21–78
Xie Y, He L, Lugano R et al (2021) Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 6:e150861
Fisher D, Thomas KA, Abdul-Rasool S (2020) The synergistic and neuroprotective effects of alcohol-antioxidant treatment on blood-brain barrier endothelial cells. Alcohol Clin Exp Res 44:1997–2007
Kaddoumi A, Denney TS Jr, Deshpande G et al (2022) Extra-virgin olive oil enhances the blood-brain barrier function in mild cognitive impairment: a randomized controlled trial. Nutrients 14:5102
Zhang L, Zhou L, Bao L et al (2021) SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther 6:337
Ayloo S, Lazo CG, Sun S, Zhang W, Cui B, Gu C (2022) Pericyte-to-endothelial cell signaling via vitronectin-integrin regulates blood-CNS barrier. Neuron 110(1641–1655):e1646
Kenny A, Plank MJ, David T (2018) Macro scale modelling of cortical spreading depression and the role of astrocytic gap junctions. J Theor Biol 458:78–91
Liu S, Chang L, Wei C (2019) The sonic hedgehog pathway mediates Tongxinluo capsule-induced protection against blood-brain barrier disruption after ischaemic stroke in mice. Basic Clin Pharmacol Toxicol 124:660–669
Yue Q, Xu Y, Lin L, Hoi MPM (2022) Canthin-6-one (CO) from Picrasma quassioides (D.Don) Benn. ameliorates lipopolysaccharide (LPS)-induced astrocyte activation and associated brain endothelial disruption. Phytomedicine 101:154108
Zhang Q, Liu C, Shi R et al (2022) Blocking C3d(+)/GFAP(+) A1 astrocyte conversion with semaglutide attenuates blood-brain barrier disruption in mice after ischemic stroke. Aging Dis 13:943–959
Li G, Jiang X, Liang X et al (2023) BAP31 regulates the expression of ICAM-1/VCAM-1 via MyD88/NF-kappaB pathway in acute lung injury mice model. Life Sci 313:121310
Koch K, Lindner M, Fleck AK et al (2022) CNS pericytes modulate local T cell infiltration in EAE. Int J Mol Sci 23:13081
Khan A, Ni W, Lopez-Giraldez F, Kluger MS, Pober JS, Pierce RW (2021) Tumor necrosis factor-induced ArhGEF10 selectively activates RhoB contributing to human microvascular endothelial cell tight junction disruption. FASEB J 35:e21627
Jang M, Han S, Cho H (2023) Correspondence between development of cytotoxic edema and cerebrospinal fluid volume and flow in the third ventricle after ischemic stroke. J Stroke Cerebrovasc Dis 32:107200
Shi ZF, Fang Q, Chen Y et al (2021) Methylene blue ameliorates brain edema in rats with experimental ischemic stroke via inhibiting aquaporin 4 expression. Acta Pharmacol Sin 42:382–392
Momenabadi S, Vafaei AA, Bandegi AR, Zahedi-Khorasani M, Mazaheri Z, Vakili A (2020) Oxytocin reduces brain injury and maintains blood-brain barrier integrity after ischemic stroke in mice. NeuroMol Med 22:557–571
Lee K, Yoo RE, Cho WS et al (2023) Blood-brain barrier disruption imaging in postoperative cerebral hyperperfusion syndrome using DCE-MRI. J Cereb Blood Flow Metab 44:271678X231212173
Gono R, Sugimoto K, Yang C et al (2023) Molecular mechanism of cerebral edema improvement via IL-1RA released from the stroke-unaffected hindlimb by treadmill exercise after cerebral infarction in rats. J Cereb Blood Flow Metab 43:812–827
Wang R, Zhu Y, Liu Z et al (2021) Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood 138:91–103
Li M, Chen S, Shi X et al (2018) Cell permeable HMGB1-binding heptamer peptide ameliorates neurovascular complications associated with thrombolytic therapy in rats with transient ischemic stroke. J Neuroinflammation 15:237
Yu X, Bai Y, Han B et al (2022) Extracellular vesicle-mediated delivery of circDYM alleviates CUS-induced depressive-like behaviours. J Extracell Vesicles 11:e12185
Logsdon AF, Schindler AG, Meabon JS et al (2020) Nitric oxide synthase mediates cerebellar dysfunction in mice exposed to repetitive blast-induced mild traumatic brain injury. Sci Rep 10:9420
Bellut M, Papp L, Bieber M, Kraft P, Stoll G, Schuhmann MK (2021) NLPR3 inflammasome inhibition alleviates hypoxic endothelial cell death in vitro and protects blood-brain barrier integrity in murine stroke. Cell Death Dis 13:20
Wei P, Wang K, Luo C et al (2021) Cordycepin confers long-term neuroprotection via inhibiting neutrophil infiltration and neuroinflammation after traumatic brain injury. J Neuroinflammation 18:137
Howe MD, Furr JW, Munshi Y et al (2019) Transforming growth factor-beta promotes basement membrane fibrosis, alters perivascular cerebrospinal fluid distribution, and worsens neurological recovery in the aged brain after stroke. Geroscience 41:543–559
Yang ZH, Liu YJ, Ban WK et al (2023) Pterostilbene alleviated cerebral ischemia/reperfusion-induced blood-brain barrier dysfunction via inhibiting early endothelial cytoskeleton reorganization and late basement membrane degradation. Food Funct 14:8291–8308
Skuja S, Jain N, Smirnovs M, Murovska M (2022) Alcohol-induced alterations in the vascular basement membrane in the substantia nigra of the adult human brain. Biomedicines 10:830
Yao Y (2019) Basement membrane and stroke. J Cereb Blood Flow Metab 39:3–19
Yu H, Luo H, Chang L et al (2022) The NEDD8-activating enzyme inhibitor MLN4924 reduces ischemic brain injury in mice. Proc Natl Acad Sci U S A 119:e2111896119
Yao SQ, Ye Y, Li Q et al (2024) YangXueQingNaoWan attenuated blood brain barrier disruption after thrombolysis with tissue plasminogen activator in ischemia stroke. J Ethnopharmacol 318:117024
Wang HJ, Ran HF, Yin Y et al (2022) Catalpol improves impaired neurovascular unit in ischemic stroke rats via enhancing VEGF-PI3K/AKT and VEGF-MEK1/2/ERK1/2 signaling. Acta Pharmacol Sin 43:1670–1685
Spitzer D, Guerit S, Puetz T et al (2022) Profiling the neurovascular unit unveils detrimental effects of osteopontin on the blood-brain barrier in acute ischemic stroke. Acta Neuropathol 144:305–337
Kameyama T, Miyata M, Shiotani H et al (2023) Heterogeneity of perivascular astrocyte endfeet depending on vascular regions in the mouse brain. iScience 26:108010
Thomas CI, Ryan MA, Mcnabb MC, Kamasawa N, Scholl B (2023) Astrocyte coverage of excitatory synapses correlates to measures of synapse structure and function in primary visual cortex. bioRxiv: Preprint
Darvishmolla M, Heysieattalab S, Saeedi N, Hosseinmardi N, Janahmadi M (2022) Involvement of hippocampal astrocytic connexin-43 in morphine dependence. Physiol Behav 247:113710
Diaz-Castro B, Robel S, Mishra A (2023) Astrocyte endfeet in brain function and pathology: open questions. Annu Rev Neurosci 46:101–121
Feng XF, Li MC, Lin ZY et al (2023) Tetramethylpyrazine promotes stroke recovery by inducing the restoration of neurovascular unit and transformation of A1/A2 reactive astrocytes. Front Cell Neurosci 17:1125412
Li Y, Xie Y, Liu R et al (2023) Knockout of microglial Hv1 proton channel reduces neurotoxic A1 astrocytes and neuronal damage via the ROS/STAT3 pathway after spinal cord injury. Glia 71:2418–2436
Acioglu C, Li L, Elkabes S (2021) Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res 1758:147291
Ma M, Li H, Wu J et al (2020) Roles of prokineticin 2 in subarachnoid hemorrhage-induced early brain injury via regulation of phenotype polarization in astrocytes. Mol Neurobiol 57:3744–3758
Kim Y, Lee S, Zhang H et al (2020) CLEC14A deficiency exacerbates neuronal loss by increasing blood-brain barrier permeability and inflammation. J Neuroinflammation 17:48
Hernandes MS, Lassegue B, Hilenski LL et al (2018) Polymerase delta-interacting protein 2 deficiency protects against blood-brain barrier permeability in the ischemic brain. J Neuroinflammation 15:45
Wang N, Guo W, Liu T, Chen X, Lin M (2023) Toll-like receptors (TLR2 and TLR4) antagonist mitigates the onset of cerebral small vessel disease through PI3K/Akt/GSK3beta pathway in stroke-prone renovascular hypertensive rats. Biotechnol Genet Eng Rev 39:1–21
Tan S, Shan Y, Lin Y et al (2019) Neutralization of interleukin-9 ameliorates experimental stroke by repairing the blood-brain barrier via down-regulation of astrocyte-derived vascular endothelial growth factor-A. FASEB J 33:4376–4387
Tunc BS, Toprak F, Toprak SF, Sozer S (2021) In vitro investigation of growth factors including MGF and IGF-1 in neural stem cell activation, proliferation, and migration. Brain Res 1759:147366
Guo X, Kimura A, Namekata K et al (2022) ASK1 signaling regulates phase-specific glial interactions during neuroinflammation. Proc Natl Acad Sci U S A 119:e2103812119
Rakers C, Schleif M, Blank N et al (2019) Stroke target identification guided by astrocyte transcriptome analysis. Glia 67:619–633
Schroder LJ, Mulenge F, Pavlou A et al (2023) Dynamics of reactive astrocytes fosters tissue regeneration after cuprizone-induced demyelination. Glia 71:2573–2590
Gao Y, Liu J, Wang J et al (2022) Proteomic analysis of human hippocampal subfields provides new insights into the pathogenesis of Alzheimer’s disease and the role of glial cells. Brain Pathol 32:e13047
Zong X, Li Y, Liu C et al (2020) Theta-burst transcranial magnetic stimulation promotes stroke recovery by vascular protection and neovascularization. Theranostics 10:12090–12110
Liu LR, Liu JC, Bao JS, Bai QQ, Wang GQ (2020) Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol 11:1024
Fattorelli N, Martinez-Muriana A, Wolfs L, Geric I, De Strooper B, Mancuso R (2021) Stem-cell-derived human microglia transplanted into mouse brain to study human disease. Nat Protoc 16:1013–1033
Sobierajski E, Lauer G, Aktas M et al (2022) Development of microglia in fetal and postnatal neocortex of the pig, the European wild boar (Sus scrofa). J Comp Neurol 530:1341–1362
Belayev L, Hong SH, Freitas RS et al (2020) DHA modulates MANF and TREM2 abundance, enhances neurogenesis, reduces infarct size, and improves neurological function after experimental ischemic stroke. CNS Neurosci Ther 26:1155–1167
Utz SG, See P, Mildenberger W et al (2020) Early fate defines microglia and non-parenchymal brain macrophage development. Cell 181(557–573):e518
Xingi E, Koutsoudaki PN, Thanou I et al (2023) LPS-induced systemic inflammation affects the dynamic interactions of astrocytes and microglia with the vasculature of the mouse brain cortex. Cells 12:1418
Zhu Y, Yu J, Gong J et al (2021) PTP1B inhibitor alleviates deleterious microglial activation and neuronal injury after ischemic stroke by modulating the ER stress-autophagy axis via PERK signaling in microglia. Aging (Albany NY) 13:3405–3427
Kim S, Lee W, Jo H et al (2022) The antioxidant enzyme peroxiredoxin-1 controls stroke-associated microglia against acute ischemic stroke. Redox Biol 54:102347
Han B, Jiang W, Cui P et al (2021) Microglial PGC-1alpha protects against ischemic brain injury by suppressing neuroinflammation. Genome Medicine 13:47
Takagi S, Furube E, Nakano Y, Morita M, Miyata S (2019) Microglia are continuously activated in the circumventricular organs of mouse brain. J Neuroimmunol 331:74–86
Liu X, Zhang M, Liu H et al (2021) Bone marrow mesenchymal stem cell-derived exosomes attenuate cerebral ischemia-reperfusion injury-induced neuroinflammation and pyroptosis by modulating microglia M1/M2 phenotypes. Exp Neurol 341:113700
Deng W, Mandeville E, Terasaki Y et al (2020) Transcriptomic characterization of microglia activation in a rat model of ischemic stroke. J Cereb Blood Flow Metab 40:S34–S48
O’neil SM, Witcher KG, Mckim DB, Godbout JP (2018) Forced turnover of aged microglia induces an intermediate phenotype but does not rebalance CNS environmental cues driving priming to immune challenge. Acta Neuropathol Commun 6:129
Zhong Y, Gu L, Ye Y et al (2022) JAK2/STAT3 axis intermediates microglia/macrophage polarization during cerebral ischemia/reperfusion injury. Neuroscience 496:119–128
Atta AA, Ibrahim WW, Mohamed AF, Abdelkader NF (2023) Targeting alpha7-nAChR by galantamine mitigates reserpine-induced fibromyalgia-like symptoms in rats: Involvement of cAMP/PKA, PI3K/AKT, and M1/M2 microglia polarization. Eur J Pharmacol 952:175810
Chen AQ, Fang Z, Chen XL et al (2019) Microglia-derived TNF-alpha mediates endothelial necroptosis aggravating blood brain-barrier disruption after ischemic stroke. Cell Death Dis 10:487
Zhou C, Su M, Sun P, Tang X, Yin KJ (2021) Nitro-oleic acid-mediated blood-brain barrier protection reduces ischemic brain injury. Exp Neurol 346:113861
Radpour M, Khoshkroodian B, Asgari T, Pourbadie HG, Sayyah M (2023) Interleukin 4 reduces brain hyperexcitability after traumatic injury by downregulating TNF-alpha, upregulating IL-10/TGF-beta, and potential directing macrophage/microglia to the M2 anti-inflammatory phenotype. Inflammation 46:1810–1831
Hsu CH, Pan YJ, Zheng YT, Lo RY, Yang FY (2023) Ultrasound reduces inflammation by modulating M1/M2 polarization of microglia through STAT1/STAT6/PPARgamma signaling pathways. CNS Neurosci Ther 29:4113–4123
Santos-Galdiano M, Perez-Rodriguez D, Anuncibay-Soto B et al (2018) Celecoxib treatment improves neurologic deficit and reduces selective neuronal loss and glial response in rats after transient middle cerebral artery occlusion. J Pharmacol Exp Ther 367:528–542
Sun Y, Zhu X, Zhu K, Yu J, Cheng L, Hei M (2022) High-mobility group box 1 contributes to hypoxic-ischemic brain damage by facilitating imbalance of microglial polarization through RAGE-PI3K/Akt pathway in neonatal rats. Int J Med Sci 19:2093–2103
Liu J, Tian J, Xie R, Chen L (2023) CK2 inhibitor DMAT ameliorates spinal cord injury by increasing autophagy and inducing anti-inflammatory microglial polarization. Neurosci Lett 805:137222
Ji XC, Shi YJ, Zhang Y, Chang MZ, Zhao G (2020) Reducing suppressors of cytokine signaling-3 (SOCS3) expression promotes M2 macrophage polarization and functional recovery after intracerebral hemorrhage. Front Neurol 11:586905
Tian Y, Liu B, Li Y et al (2022) Activation of RARalpha receptor attenuates neuroinflammation after SAH via promoting M1-to-M2 phenotypic polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-kappaB pathway. Front Immunol 13:839796
Qu Y, Wang L, Mao Y (2022) Gallic acid attenuates cerebral ischemia/re-perfusion-induced blood-brain barrier injury by modifying polarization of microglia. J Immunotoxicol 19:17–26
Esposito E, Hayakawa K, Ahn BJ et al (2018) Effects of ischemic post-conditioning on neuronal VEGF regulation and microglial polarization in a rat model of focal cerebral ischemia. J Neurochem 146:160–172
He Y, Gao Y, Zhang Q, Zhou G, Cao F, Yao S (2020) IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience 437:161–171
Kronenberg G, Uhlemann R, Richter N et al (2018) Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol 135:551–568
Wang T, Zhao N, Peng L et al (2020) DJ-1 regulates microglial polarization through P62-mediated TRAF6/IRF5 signaling in cerebral ischemia-reperfusion. Front Cell Dev Biol 8:593890
Li R, Zhou Y, Zhang S, Li J, Zheng Y, Fan X (2022) The natural (poly)phenols as modulators of microglia polarization via TLR4/NF-kappaB pathway exert anti-inflammatory activity in ischemic stroke. Eur J Pharmacol 914:174660
Yang E, Cai Y, Yao X et al (2019) Tissue plasminogen activator disrupts the blood-brain barrier through increasing the inflammatory response mediated by pericytes after cerebral ischemia. Aging (Albany NY) 11:10167–10182
Matsuda M, Inaba M, Hamaguchi J et al (2022) Local IL-10 replacement therapy was effective for steroid-insensitive asthma in mice. Int Immunopharmacol 110:109037
Michalski D, Keck AL, Grosche J, Martens H, Hartig W (2018) Immunosignals of oligodendrocyte markers and myelin-associated proteins are critically affected after experimental stroke in wild-type and Alzheimer modeling mice of different ages. Front Cell Neurosci 12:23
Niu J, Tsai HH, Hoi KK et al (2019) Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat Neurosci 22:709–718
Zhang J, Buller BA, Zhang ZG et al (2022) Exosomes derived from bone marrow mesenchymal stromal cells promote remyelination and reduce neuroinflammation in the demyelinating central nervous system. Exp Neurol 347:113895
Zhang W, Pu H, Hu X et al (2023) Poststroke intravenous transplantation of human mesenchymal stem cells improves brain repair dynamics and functional outcomes in aged mice. Stroke 54:1088–1098
Meyer N, Richter N, Fan Z et al (2018) Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep 22:2383–2394
Zhao Y, Zhu W, Wan T et al (2022) Vascular endothelium deploys caveolin-1 to regulate oligodendrogenesis after chronic cerebral ischemia in mice. Nat Commun 13:6813
Takahashi S (2022) Metabolic contribution and cerebral blood flow regulation by astrocytes in the neurovascular unit. Cells 11:813
Miyamoto N, Magami S, Inaba T et al (2020) The effects of A1/A2 astrocytes on oligodendrocyte linage cells against white matter injury under prolonged cerebral hypoperfusion. Glia 68:1910–1924
Girolamo F, Errede M, Longo G et al (2019) Defining the role of NG2-expressing cells in experimental models of multiple sclerosis. A biofunctional analysis of the neurovascular unit in wild type and NG2 null mice. PLoS One 14:e0213508
Yang Y, Kimura-Ohba S, Thompson JF et al (2018) Vascular tight junction disruption and angiogenesis in spontaneously hypertensive rat with neuroinflammatory white matter injury. Neurobiol Dis 114:95–110
Yang X, Chang L, Liu Z et al (2024) Neddylation in the chronically hypoperfused corpus callosum: MLN4924 reduces blood-brain barrier injury via ERK5/KLF2 signaling. Exp Neurol 371:114587
Rost NS, Cougo P, Lorenzano S et al (2018) Diffuse microvascular dysfunction and loss of white matter integrity predict poor outcomes in patients with acute ischemic stroke. J Cereb Blood Flow Metab 38:75–86
Xu J, Wang R, Luo W et al (2023) Oligodendrocyte progenitor cell-specific delivery of lipid nanoparticles loaded with Olig2 synthetically modified messenger RNA for ischemic stroke therapy. Acta Biomater 174:297–313
Zhang Y, Liu Y, Zhang X, Yong VW, Xue M (2023) Omarigliptin protects the integrity of the blood-brain barrier after intracerebral hemorrhage in mice. J Inflamm Res 16:2535–2548
Liu Y, Aguzzi A (2020) NG2 glia are required for maintaining microglia homeostatic state. Glia 68:345–355
Kirdajova D, Valihrach L, Valny M et al (2021) Transient astrocyte-like NG2 glia subpopulation emerges solely following permanent brain ischemia. Glia 69:2658–2681
Lin Q, Lin L, Li L, Zheng YF, Hu DW, Zhang G (2023) Dynamic changes of oligodendrogenesis in neonatal rats with hypoxic-ischemic white matter injury. Brain Res 1817:148495
Liu C, Han S, Zheng J, Wang H, Li S, Li J (2022) EphA4 regulates white matter remyelination after ischemic stroke through Ephexin-1/RhoA/ROCK signaling pathway. Glia 70:1971–1991
Piatek P, Lewkowicz N, Michlewska S et al (2022) Natural fish oil improves the differentiation and maturation of oligodendrocyte precursor cells to oligodendrocytes in vitro after interaction with the blood-brain barrier. Front Immunol 13:932383
Wang J, He X, Meng H et al (2020) Robust myelination of regenerated axons induced by combined manipulations of GPR17 and microglia. Neuron 108(876–886):e874
Hattori T, Kaji M, Ishii H et al (2017) CD38 positively regulates postnatal development of astrocytes cell-autonomously and oligodendrocytes non-cell-autonomously. Glia 65:974–989
Li T, Niu J, Yu G et al (2020) Connexin 43 deletion in astrocytes promotes CNS remyelination by modulating local inflammation. Glia 68:1201–1212
Liang Z, Wang X, Hao Y et al (2020) The multifaceted role of astrocyte connexin 43 in ischemic stroke through forming hemichannels and gap junctions. Front Neurol 11:703
Niu J, Li T, Yi C et al (2016) Connexin-based channels contribute to metabolic pathways in the oligodendroglial lineage. J Cell Sci 129:1902–1914
Baldassarro VA, Marchesini A, Giardino L, Calza L (2020) Differential effects of glucose deprivation on the survival of fetal versus adult neural stem cells-derived oligodendrocyte precursor cells. Glia 68:898–917
Xu Y, Tian Y, Wang Y et al (2021) Exosomes derived from astrocytes after oxygen-glucose deprivation promote differentiation and migration of oligodendrocyte precursor cells in vitro. Mol Biol Rep 48:5473–5484
An J, He Y, Yin JJ et al (2021) Temporal and spatial evolution of various functional neurons during demyelination induced by cuprizone. J Neurophysiol 126:1756–1771
Wang Q, Wang Z, Tian Y et al (2018) Inhibition of astrocyte connexin 43 channels facilitates the differentiation of oligodendrocyte precursor cells under hypoxic conditions in vitro. J Mol Neurosci 64:591–600
Li M, Xia M, Chen W et al (2020) Lithium treatment mitigates white matter injury after intracerebral hemorrhage through brain-derived neurotrophic factor signaling in mice. Transl Res 217:61–74
Mattingly Z, Chetty S (2023) Generation of oligodendrocytes from human pluripotent and embryonic stem cells. Methods Mol Biol 2683:89–101
Roll L, Eysel UT, Faissner A (2020) Laser lesion in the mouse visual cortex induces a stem cell niche-like extracellular matrix, produced by immature astrocytes. Front Cell Neurosci 14:102
Chen M, Ingle L, Plautz EJ et al (2022) LZK-dependent stimulation of astrocyte reactivity promotes corticospinal axon sprouting. Front Cell Neurosci 16:969261
Wang LL, Serrano C, Zhong X, Ma S, Zou Y, Zhang CL (2021) Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 184(5465–5481):e5416
Ding Z, Dai C, Zhong L et al (2021) Neuregulin-1 converts reactive astrocytes toward oligodendrocyte lineage cells via upregulating the PI3K-AKT-mTOR pathway to repair spinal cord injury. Biomed Pharmacother 134:111168
Kaminski N, Koster C, Mouloud Y et al (2020) Mesenchymal stromal cell-derived extracellular vesicles reduce neuroinflammation, promote neural cell proliferation and improve oligodendrocyte maturation in neonatal hypoxic-ischemic brain injury. Front Cell Neurosci 14:601176
Chang J, Qian Z, Wang B et al (2023) Transplantation of A2 type astrocytes promotes neural repair and remyelination after spinal cord injury. Cell Commun Signal 21:37
Au NPB, Ma CHE (2022) Neuroinflammation, microglia and implications for retinal ganglion cell survival and axon regeneration in traumatic optic neuropathy. Front Immunol 13:860070
Li Y, Liu Z, Song Y et al (2022) M2 microglia-derived extracellular vesicles promote white matter repair and functional recovery via miR-23a-5p after cerebral ischemia in mice. Theranostics 12:3553–3573
He Y, An J, Yin JJ et al (2019) Ethyl pyruvate enhances spontaneous remyelination by targeting microglia phagocytosis. Int Immunopharmacol 77:105929
Naruse M, Shibasaki K, Shimauchi-Ohtaki H, Ishizaki Y (2018) Microglial activation induces generation of oligodendrocyte progenitor cells from the subventricular zone after focal demyelination in the corpus callosum. Dev Neurosci 40:54–63
Chai Z, Ma T, Li Y et al (2023) Inhibition of inflammatory factor TNF-alpha by ferrostatin-1 in microglia regulates necroptosis of oligodendrocyte precursor cells. NeuroReport 34:583–591
Shinozaki Y, Shibata K, Yoshida K et al (2017) Transformation of astrocytes to a neuroprotective phenotype by microglia via P2Y(1) receptor downregulation. Cell Rep 19:1151–1164
Wang L, Yang JW, Lin LT et al (2020) Acupuncture attenuates inflammation in microglia of vascular dementia rats by inhibiting miR-93-mediated TLR4/MyD88/NF-kappaB signaling pathway. Oxid Med Cell Longev 2020:8253904
Liu Z, Yao X, Jiang W et al (2020) Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-kappaB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation 17:90
Nong X, Lan Y (2018) Picroside II attenuates CCI-induced neuropathic pain in rats by inhibiting spinal reactive astrocyte-mediated neuroinflammation through the NF-kappaB pathway. Neurochem Res 43:1058–1066
Zhang Z, Qin P, Deng Y et al (2018) The novel estrogenic receptor GPR30 alleviates ischemic injury by inhibiting TLR4-mediated microglial inflammation. J Neuroinflammation 15:206
Deng YL, Ma YL, Zhang ZL et al (2018) Astrocytic N-Myc downstream-regulated gene-2 is involved in nuclear transcription factor kappaB-mediated inflammation induced by global cerebral ischemia. Anesthesiology 128:574–586
Zhao H, Chen Z, Xie LJ, Liu GF (2018) Suppression of TLR4/NF-kappaB signaling pathway improves cerebral ischemia-reperfusion injury in rats. Mol Neurobiol 55:4311–4319
Von Kugelgen I (2023) Pharmacological characterization of P2Y receptor subtypes - an update. Purinergic Signal: Early Access
Fang Y, Ding X, Zhang Y et al (2022) Fluoxetine inhibited the activation of A1 reactive astrocyte in a mouse model of major depressive disorder through astrocytic 5-HT(2B)R/beta-arrestin2 pathway. J Neuroinflammation 19:23
Sun M, You H, Hu X et al (2023) Microglia-astrocyte interaction in neural development and neural pathogenesis. Cells 12:1942
Liu CW, Wang EY, Wang HL et al (2022) Blood-brain barrier disruption in preclinical mouse models of stroke can be an experimental artifact caused by craniectomy. eNeuro 9:1–12
Sladojevic N, Stamatovic SM, Johnson AM et al (2019) Claudin-1-dependent destabilization of the blood-brain barrier in chronic stroke. J Neurosci 39:743–757
Pulous FE, Grimsley-Myers CM, Kansal S, Kowalczyk AP, Petrich BG (2019) Talin-dependent integrin activation regulates VE-cadherin localization and endothelial cell barrier function. Circ Res 124:891–903
Sucha P, Hermanova Z, Chmelova M et al (2022) The absence of AQP4/TRPV4 complex substantially reduces acute cytotoxic edema following ischemic injury. Front Cell Neurosci 16:1054919
Swissa E, Serlin Y, Vazana U, Prager O, Friedman A (2019) Blood-brain barrier dysfunction in status epileptics: mechanisms and role in epileptogenesis. Epilepsy Behav 101:106285
Clausen BH, Wirenfeldt M, Hogedal SS et al (2020) Characterization of the TNF and IL-1 systems in human brain and blood after ischemic stroke. Acta Neuropathol Commun 8:81
Murata Y, Sugimoto K, Yang C et al (2020) Activated microglia-derived macrophage-like cells exacerbate brain edema after ischemic stroke correlate with astrocytic expression of aquaporin-4 and interleukin-1 alpha release. Neurochem Int 140:104848
Kangwantas K, Pinteaux E, Penny J (2016) The extracellular matrix protein laminin-10 promotes blood-brain barrier repair after hypoxia and inflammation in vitro. J Neuroinflammation 13:25
Yu X, Wang Y, Qiu H et al (2018) AEG-1 contributes to metastasis in hypoxia-related ovarian cancer by modulating the HIF-1alpha/NF-kappaB/VEGF pathway. Biomed Res Int 2018:3145689
Xing G, Zhao T, Zhang X et al (2020) Astrocytic sonic hedgehog alleviates intracerebral hemorrhagic brain injury via modulation of blood-brain barrier integrity. Front Cell Neurosci 14:575690
Ding XW, Sun X, Shen XF et al (2019) Propofol attenuates TNF-alpha-induced MMP-9 expression in human cerebral microvascular endothelial cells by inhibiting Ca(2+)/CAMK II/ERK/NF-kappaB signaling pathway. Acta Pharmacol Sin 40:1303–1313
Bai M, Sun R, Cao B, Feng J, Wang J (2023) Monocyte-related cytokines/chemokines in cerebral ischemic stroke. CNS Neurosci Ther 29:3693–3712
Funding
The authors acknowledge operating grant support from Natural Science Foundation of Colleges and Universities in Anhui Province in 2023 (Nos. 2023AH052541 and 2023AH050672).
Author information
Authors and Affiliations
Contributions
WL prepared figures and drafted the manuscript; JW edited and revised the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Research Involving Human Participants and/or Animals
Not applicable.
Informed Consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lu, W., Wen, J. Crosstalk Among Glial Cells in the Blood–Brain Barrier Injury After Ischemic Stroke. Mol Neurobiol 61, 6161–6174 (2024). https://doi.org/10.1007/s12035-024-03939-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-024-03939-6