
Abstract—
Macrophage/microglia are activated after Traumatic brain injury (TBI), transform to inflammatory phenotype (M1) and trigger neuroinflammation, which provokes epileptogenesis. Interleukin-4 (IL-4) is a well-known drive of macrophage/microglia to the anti-inflammatory phenotype (M2). We tested effect of IL-4 on speed of epileptogenesis, brain expression of inflammatory and anti-inflammatory cytokines, and lesion size in TBI-injured male rats. Rats underwent TBI by Controlled Cortical Impact. Then 100 ng IL-4 was injected into cerebral ventricles. One day after TBI, pentylenetetrazole (PTZ) kindling started and development of generalized seizures was recorded. The lesion size, cell survival rate, TNF-α, TGF-β, IL-10, and Arginase1 (Arg1) was measured in the brain 6 h, 12 h, 24 h, 48 h, and 5 days after TBI. Astrocytes and macrophage/microglia activation/polarization was assessed by GFAP/Arg1 and Iba1/Arg1 immunostaining. TBI-injured rats were kindled by 50% less PTZ injections than control and sham-operated rats. IL-4 did not change kindling rate in sham-operated rats but inhibited acceleration of kindling rate in the TBI-injured rats. IL-4 decreased damage volume and number of destroyed neurons. IL-4 stopped TNF-α whereas upregulated TGF-β, IL-10, and Arg1 expressions. Iba1/Arg1 positive macrophage/microglia was notably increased 48 h after IL-4 administration. IL-4 suppresses TBI-induced acceleration of epileptogenesis in rats by directing TBI neuroinflammation toward an anti-inflammatory tone and inhibition of cell death.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Traumatic brain injury (TBI) is an injury to the brain caused by an external force such as mechanical forces and supersonic blast waves. The global incidence of all-cause, all-severity TBI is estimated to be about 1%; of which 0.74% is related to mild TBI, and 0.07% associated with severe TBI [1]. Sixty-nine million people are estimated to suffer TBI from all causes each year, with the Southeast Asian and Western Pacific regions experiencing the greatest overall burden of disease [1]. TBI prompts an array of cellular processes that result in neuroinflammation [2,3,4,5]. Neuroinflammation, in turn, triggers a series of pathological cascades and underlies neurological diseases such as Alzheimer's disease, Parkinson's disease, stroke, chronic traumatic encephalopathy, and epilepsy [5,6,7].
Epilepsy is a neurologic disease that has around 0.76% international prevalence [8]. Post-traumatic epilepsy (PTE) is a form of acquired epilepsy [9]. TBI is responsible of 5% of all epilepsy cases; and over than 20% of cases of acquired epilepsy [10]. No strategy is yet available to avoid PTE. Neuroinflammation has a central role in PTE [11,12,13].
Interleukin-4 (IL-4) is a cytokine serves as a potent regulator of immunity [14]. It often acts as an anti-inflammatory agent by down-regulation of the inflammatory cytokines such as TNF-α [15] and upregulation of anti-inflammatory cytokines such as IL-10 [16, 17]. IL-4 knockout mice display increased brain injury and worsened neurological outcome after transient focal cerebral ischemia [18] and traumatic spinal cord injury (SCI) [19]. IL-4 is able to polarize macrophage/microglia to the anti-inflammatory phenotype [20]. It is found that IL-4 is produced in neurons after stroke [21] and focal cerebral ischemia [22], and modulates macrophage/microglia function by polarizing them to the M2 “healing” phenotype [21, 22]. In vivo alternative activating M2 macrophage/microglia by IL-4 has been of interest as a therapeutic strategy for treatment of the injury-associated neurologic diseases [16, 23]. In this regard, administration of IL-4 to animals with TBI and SCI has been accompanied by beneficial behavioral improvements [24,25,26]. Meanwhile, IL-4 has been shown antiepileptic activity in the animal study [27]. It is found that early treatment of mice by IL-4 inhibits rate of recurrent seizures in pilocarpine model of epilepsy by inhibiting early increase of M1 macrophage/microglia, and promoting the increase of M2 phenotype [27].
Nevertheless, the possible inhibitory effect of IL-4 on development of PTE has not yet been examined. It is shown that TBI accelerates development of epileptogenesis in animal models [28, 29]. Accordingly, we aimed to examine impact of IL-4 on the accelerated rate of epileptogenesis in the TBI-injured rats. We injected IL-4 to brain of rats shortly after TBI. Then, development of generalized seizures was assessed using kindling model of epileptogenesis. Given the essential role of macrophage/microglia in TBI pathogenesis, and IL-4 as the transformer of macrophage/microglia to the M2 phenotype, the lesion size, cell survival, Inflammatory/anti-inflammatory cytokines, and astrocytes as well as macrophage/microglia activation and polarization were assessed in the TBI-injured region.
MATERIALS AND METHODS
Animals
Adult male Wistar rats (9-weeks old and 240-270 g weight, n = 228) were obtained from Pasteur Institute of Iran (Stock number 0094-0098). Due to estrus cycle-dependent variations in seizure and epilepsy, we used male rats to exclude effect of this factor on the results. The animals were housed in standard polypropylene cages in a room with controlled temperature (23 ± 2.0 °C) and 12 h light/dark cycle (08:00–20:00). They were fed ad libitum with rodent’s chow and free access to drinking water. All experiments were performed between 09:00–18:00. Animals were allocated to different experimental groups by simple randomization. All animal experiments were carried out in accordance with the Review Board and Ethics Committee of Pasteur Institute of Iran, the National Institute for Medical Research Development (Authorization code 943697), and Council Directive 2010/63EU of the European Parliament, and the Council of 22 September 2010 on the protection of animals used for scientific purposes. Animal procedures were performed by skilled study personnel to minimize pain or distress during experimentation who do not aware of the nature of the experimental groups. After the operation, animals were kept warm until awakening. Food and water intake, body weight, and general behavior of the rats were closely monitored daily. Rats with > 15% loss of body weight and not used for behavioral and biological analysis were euthanized by CO2 asphyxiation.
Induction of TBI
Rats were anaesthetized with intraperitoneal (i.p.) injection of ketamine (60 mg/kg, Alfasan, Netherlands, Cat. No. 1508249-05 (year 2017)), and xylazine (10 mg/kg, Alfasan, Netherlands, Cat. No. 1402044-01 (year 2016)). The combination of ketamine–xylazine provides a relatively safe and rapid anesthesia with adequate analgesia, muscle relaxation, and sedation that can be administered without the need for specialized equipment. A 5-mm burr hole was drilled at left parieto-temporal cortex at coordinates; AP, -4 mm from bregma; ML, -4 mm from bregma; according to atlas of rat brain [30]. The bone was removed. TBI was induced by a Controlled Cortical Impact (CCI) device (AmScien Instruments, USA, Cat. No. AMS-201-A1 (year 2011)) with 5 mm round tip, 4.5 m/s velocity, 150 ms duration. Then, the dissected bone was brought back to its position on the skull, and fixed with dental acrylic. The skin was then closed. Sham-operated animals underwent the whole procedure except that they did not received CCI injury.
Intracerebroventricular Injection of IL-4
IL-4 (BIOBASIC, CANADA, Cat.No.RC232-15 (year 2018)) was dissolved in phosphate-buffered saline (PBS). It is injected (100 ng/5 µl/rat) into the right lateral ventricle (i.c.v.; AP, -0.9 mm from bregma; ML, 1.7 mm from bregma; V, 3.5 mm from dura) of the anesthetized rats through a 27-gauge needle that was fitted to a Hamilton microsyringe. The dose of IL-4 was selected based on the effective doses used in stroke and trauma studies [16, 31].
Pentylenetetrazole Kindling
Twenty-four h after TBI or sham operation, rats received 35 mg/kg pentylenetetrazole, (PTZ, Sigma-Aldrich, Germany, Lot.No.79H0703 (year 2011)) though i.p. route. PTZ injections were repeated every 48 h until generalized seizures were developed in rats in 3 sequential PTZ injections. The seizure behaviors were scored according to Racine’s classification [32]. Forelimbs clonic seizures associated with rearing (score 4), and generalized convulsions with loss of balance (score 5) were considered as generalized seizures.
Histology
2, 3, 5-Triphenyl Tetrazolium Chloride Staining
The depth and volume of brain contusion was evaluated after CCI by 2, 3, 5-Triphenyl tetrazolium chloride (TTC; Sigma-Aldrich, Austria, Lot.No.101152321 (year 2011)) staining according to previously described method [33]. The animals were euthanized by CO2 asphyxiation. Brains were quickly removed and cut freshly into 2 mm coronal sections by a rat brain matrix. The slices were instantly immersed in TTC 2% (w/v in 0.9% NaCl) at 37 ºC for 10 min. The area with no staining was determined as the damaged area. The non-stained area as well as the whole area of each section was scanned by ImageJ software version 1.8 (RRID: SCR_003070). The depth of lesion was determined by micrometer. Volume of section and volume of lesion area of that section were calculated by multiplying their area by the thickness (2 mm). The whole lesion volume was determined by summing lesion volume of the sections with traumatic injury. Data are presented as mean ratio of whole lesion volume to the whole volume of the relevant sections.
Fluorescent Staining
The viable cells in the CCI-injured area were assessed by hematoxylin (Sigma-Aldrich, Germany, Lot.No.FN1218002704 (year 2019)), & eosin (Sigma-Aldrich, Germany, Lot.No.FN4857435117 (year 2019)), H&E, and propidium iodide (PI; Sigma-Aldrich, Germany, Lot.No.MKBR1007V (year 2011)) staining. The animals were euthanized by CO2 asphyxiation and immediately perfused transcardially with 4% paraformaldehyde (Merck, Germany, Lot.No.S5110715821 (year 2019)) in 0.1 M PBS. The brains were harvested, kept in 4% paraformaldehyde in 0.1 M PBS and processed for embedding in paraffin blocks. The brains were then cut horizontally to 8 µm thickness. The sections were deparaffinized, rehydrated in a descending alcohol series, incubated with Triton x-100 (Sigma-Aldrich, Germany, Lot.No.031M0301V (year 2011)) 0.1%, for 20 min, stained with PI (1:1000 in Tris-buffered saline) for 30 s, or H&E, and washed with distilled water for 50 min. The sections were cover-slipped with 90% glycerol mounting buffer. The H&E-stained sections were visualized with light microscope (Nikon, Japan). The PI-stained sections were visualized in the dark place with fluorescent microscope (Nikon, Japan) equipped with specific filter cube for fluorescence channels, and connected to a digital camera. Digital photographs were taken using 4x, 10x, and 20 × objective lenses. The number of PI-stained cells were counted in the CCI-injured area of every 10 sections (5 mm width and 2.02 mm depth from cerebral cortex) by ImageJ. Data are presented as mean number of viable cells per section.
Double Immunostaining
In the CCI-injured area, one 8 µm section was selected from every 9 sections. The sections were deparaffinized, rehydrated in a descending alcohol series, and incubated with Triton x-100, 0.1%, for 20 min. They were incubated for 2 h at room temperature in PBS containing 1% BSA (BIO BASIC, Canada, Lot.No.1203GEC59E09D (year 2019)) and 1.5% normal goat serum (Sigma-Aldrich, USA, Lot.No.SLBH2670V (year 2015)) to block the background, followed by incubation overnight at 4 °C with mouse anti-Iba1-HRP monoclonal antibody (1:50 dilution; Santa Cruz Biotechnology, USA, Lot.No.B1319 (year 2021)) and rabbit anti-Arginase1 (Arg1) polyclonal antibody (1:50 dilution; Santa Cruz Biotechnology, USA, Lot.No.F0816 (year 2016)), or mouse anti-glial fibrillary acidic protein (GFAP)-HRP monoclonal antibody (1:50 dilution; Santa Cruz Biotechnology, USA, Lot.No.L1416 (year 2021)) and rabbit anti-Arg1 polyclonal antibody. Sections were then washed in PBS and incubated for 2 h at room temperature with Texas Red–conjugated goat anti-mouse IgG antibody (1:100 dilution; Life Technologies, USA, Lot.No.1782466 (year 2013)) and FITC-conjugated goat anti-rabbit IgG antibody (1:100 dilution; Millipore, USA, Lot.No.2723039 (year 2013)). Then sections were washed for 5 min with PBS. Finally, the sections were cover slipped with 90% glycerol mounting buffer and visualized in a dark place with a fluorescence microscope (Nikon), equipped with specific filter cube for fluorescein Texas Red‐X and FITC fluorescence channels, and connected to a digital camera. Digital photographs were taken using 4 × and 20 × objective lenses. The Iba1, Arg1, GFAP, Iba1/Arg1, and GFAP/Arg1 positive cells, were assessed in TBI region of every five sections.
Western Blotting
A 5-mm3 piece of the left parieto-temporal cortex at the site of trauma injury was cut up and protein concentration of the tissue homogenate was determined by Bradford assay [34]. Twenty µg of total protein of each was electrophoresed in 15% SDS-PAGE gel, then transferred to polyvinylidene difluoride membrane (Amersham Bioscience, UK, Lot.No.NK0544 (year 2015)) and probed with mouse monoclonal antibodies of anti-TNF-α (1:1000 dilution; Santa Cruz Biotechnology, USA, Lot.No.E0115 (year 2016)), anti-IL-10 (1:1000 dilution; Santa Cruz Biotechnology, USA, Lot.No.A0512 (year 2015)), anti-TGF-β (1:1000 dilution; Sigma-Aldrich, China, Lot.No.QC11534 (year 2016)), and anti-β-actin (1:2000 dilution; Invitrogen, USA, Lot.No.UB2715987 (year 2015)) as internal control, and rabbit polyclonal antibody of anti-Arg1. After washing, the membrane was incubated with peroxidase conjugated anti-mouse IgG (1:50000; Santa Cruz Biotechnology, USA, Lot.No.F2718 (year 2016)). Immunoreactive polypeptides were detected by chemiluminescence using electrochemiluminescence reagents (Amersham Bioscience, UK, Lot.No.16820255 (year 2017)) and subsequent autoradiography. Quantification of results was performed by densitometry scan of the films using ImageJ. The relative level of the assessed polypeptide was expressed as ratio of the polypeptide blot density to the β-actin blot density.
Experimental Design
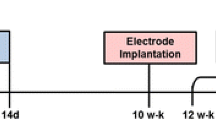
All experiments complied with the Animal Research: Reporting In Vivo Experiments (ARRIVE) guidelines. The experimental timeline is presented in Fig. 1. Five experimental groups with 9 rats in each group were assigned for kindling procedure. Group 1 (as control) included intact rats which underwent PTZ kindling. Group 2 (as sham) consisted of rats which underwent craniotomy with no TBI induction. Then they received PBS (5 µl/rat, i.c.v., up to 30 min after craniotomy), and chemical kindling started 24 h thereafter. In group 3 (IL-4) rats received IL-4 (100 ng/5 µl/rat, i.c.v.), and after 24 h kindling procedure started. Group 4 (Trauma) included rats which underwent TBI, and after 24 h kindling procedure started. In group 5 (IL-4 + Trauma) rats underwent TBI. Then they received IL-4 (100 ng/5 µl/rat, i.c.v., up to 30 min after TBI), and chemical kindling started 24 h thereafter. During kindling experiment 3 rats from the trauma group were excluded due to death after TBI. The data of one rat from control and one rat from sham groups were excluded from results because they were outliers and showed generalized seizures, score 5, immediately after first injection of PTZ.

Time line of the study. H&E: hematoxylin & eosin, PI: propidium iodide, PTZ: pentylenetetrazole, TBI: traumatic brain injury, TTC: 2, 3, 5-Triphenyltetrazolium chloride.
In order to determine the time course of the polypeptides expression after TBI, 13 experimental groups with 7 rats in each group were allocated for western blotting. These groups consisted of control (intact rats), “Sham+PBS” (including 6 subgroups, craniotomy with no TBI induction and injection of PBS 5 µl/rat, i.c.v., up to 30 min after craniotomy), and “Trauma+PBS” (including 6 subgroups, TBI induction and injection of PBS 5 µl/rat, i.c.v., up to 30 min thereafter). The brains were harvested at 3, 6, 12, 24, and 48 h as well as 5 days after sham operation or induction of TBI.
To determine effect of IL-4 on polypeptides expression, 6 experimental groups with 7 rats in each group were assigned. There were 3 “Sham + IL-4” groups in which animals underwent craniotomy with no trauma induction. Then, they received IL-4 (100 ng/5 µl/rat, i.c.v.) after craniotomy. The brain of rats was harvested at 6, 24, and 48 h after craniotomy. The 3 other groups were “Trauma + IL-4” groups in which rats underwent TBI. Then they received IL-4 (100 ng/5 µl /rat, i.c.v., after trauma induction), and brain was harvested 6, 24, and 48 h thereafter.
In order to determine lesion development after TBI and effect of IL-4 on lesion volume, 6 experimental groups with 7 rats in each group were allocated. Two groups were considered as “Sham+PBS”, and 2 groups were assigned as “Trauma+PBS”. Brain of rats was harvested at 24, and 48 h after sham operation or induction of TBI. In the last two groups effect of IL-4 on lesion volume was assessed. Rats underwent TBI. Then they received IL-4 (100 ng/5 µl /rat, i.c.v., after trauma induction), and brain was harvested 24, and 48 h thereafter. All the brains were then stained by TTC and injury volume was measured. Two rats from “Trauma+PBS 24 h”, and 1 rat from “Trauma+IL-4 48 h” groups died after TBI, and therefore, they were excluded from study.
In order to determine the effect of IL-4 on cell survival, and astrocytes as well as macrophage/microglia activation after trauma, 9 experimental groups with 3 rats in each group were allocated. Three groups were considered as “Sham+PBS”, and 3 groups were assigned as “Trauma” + PBS. The brain was harvested at 6, 24, and 48 h after sham operation or induction of TBI. In the remaining 3 groups, effect of IL-4 on cell viability and astrocytes as well as macrophage/microglia was assessed. Rats underwent TBI. Then they received IL-4 (100 ng/5 µl /rat, i.c.v., after trauma induction), and the brain was harvested 6, 24, and 48 h thereafter. The brains were then stained by H&E and PI for detecting number of viable cells. Moreover, they were double immunostained to detect Iba1, GFAP, Arg1, Iba1/Arg1 and GFAP/Arg1 positive cells.
Statistical Analysis
All statistical analyses were performed using Graph Pad Prism 8 Software (RRID: SCR_000306). Kolmogorov–Smirnov test was used to check the normal distribution of the data before analysis. Kaplan–Meier analysis was performed for PTZ kindling experiments. One-way ANOVA with Tukey–Kramer multiple comparisons test was used to analyze number of PTZ injections required to kindle rats. In order to determine significant interaction between treatment and time factors in the TBI volume, PI staining, and Western blot data, two-way ANOVA and multiple comparisons test were used. The results were expressed as mean ± SEM (standard error of mean) and the differences with p values less than 0.05 were considered statistically significant.
RESULTS
IL-4 Inhibited the Accelerated Rate of Kindling in the CCI-injured Rats
The data of one rat from control and one rat from sham groups were excluded from results because they were outliers and showed generalized seizures, score 5, immediately after first injection of PTZ. A Kaplan–Meier analysis showed that the CCI-injured rats became kindled significantly (p < 0.05; Log-rank Mantel-Cox test) earlier than the sham or control rats (Fig. 2a). Treatment of the CCI-injured rats with IL-4 reversed the accelerated rate of kindling epileptogenesis. Animals in control and sham groups became kindled with the similar rate with the mean number of 11 PTZ injections (Fig. 2b). Rats that received IL-4 were kindled after around 9 PTZ injections. However, there was no significant difference between IL-4, control and sham groups in number of injections. TBI significantly accelerated development of kindling as the CCI-injured rats were kindled after just 5 injections (p < 0.001 compared to control and sham groups). Rats which received IL-4 after trauma became kindled after mean number of 13 PTZ injections, which was more than that of sham and control groups. However, it was not significant compared to sham and control groups.

Effect of interleukin 4 on development of generalized seizures in rats with traumatic brain injury. a Kaplan–Meier analysis for development of PTZ kindling in rats with traumatic brain injury. The injured rats became kindled significantly earlier than the sham-operated rats. IL-4 restored the accelerated rate of kindling in the injured rats to the control level. *: p < 0.05; Log-rank Mantel-Cox test. b Number of PTZ injections needed for acquisition of generalized seizures. Each plot represents Mean ± SEM. Data were analyzed by one way ANOVA with Tukey’s post-hoc multiple comparisons test. n = 9 rats for each group. Three rats from the trauma group died after traumatic brain injury and were excluded from the experiment. The data of one rat from control and one rat from sham groups were excluded from results due to out of range values. ***: p < 0.001 compared to control and sham groups, ###: p < 0.001 compared to trauma group.
Effect of IL-4 on Brain Lesion Volume and Cell Viability in the CCI-injured Rats
With the severity applied in our study, CCI induced 2 ± 0.05 mm depth of deformation from dura 24 h and 48 h after CCI (Fig. 3). A lesion with 0.21 mm3 mean volume developed 24 h after CCI and preserved till 48 h. Two-way ANOVA revealed a significant effect of treatment [F (2, 33) = 121.4, p < 0.0001] but not time [F (1, 33) = 1.42, p = 0.24] factors. The mean lesion volume significantly increased 24 and 48 h after trauma (p < 0.0001). Treatment of the TBI-injured rats with IL-4 decreased injury volume only 48 h after TBI (p < 0.05). However, no significant interaction was found in treatment × time factor among groups [F (2, 33) = 1.43, p = 0.25].

Effect of intracerebroventricular administration of interleukin 4 on size of temporoparietal cortical tissue injury induced by controlled cortical impact in rats. The viable tissue (red stained) was distinguished from the dead tissue (white unstained) by 2, 3, 5-Triphenyltetrazolium staining. Data are presented as mean ± SEM, and analyzed by two way ANOVA with Tukey’s post-hoc multiple comparisons test. n = 7 rats for each group. Two rats from “Trauma+PBS 24 h”, and 1 rat from “Trauma + IL-4 48 h” groups died after TBI, and were excluded from study. ***: p < 0.0001 compared to corresponding “Sham+PBS group”. #: p < 0.05 compared to “Trauma+PBS 48 h” group.
Effect of TBI and IL-4 on the number of PI-positive cells in the CCI-injured region is demonstrated in Fig. 4. PI positive cells were significantly decreased 24 h (p < 0.01) and 48 h (p < 0.001) after TBI. Two-way ANOVA revealed a significant interaction [F (3, 16) = 3.88, p < 0.05] between treatment [F (3, 16) = 33.86, p < 0.0001] and time [F (1, 16) = 7.67, p = 0.01] factors among groups. Administration of IL-4 to the CCI-injured rats did not significantly change the number of PI-positive cells at 24 h after TBI. However, treatment of TBI-injured rats with IL-4 was accompanied by significant increase in number of PI-positive cells at 48 h post-TBI period, (p < 0.05).

Effect of interleukin 4 on the cell viability in the traumatic brain region of rats. The number of propidium iodide (PI)-stained cells were counted in the trauma area (5 mm width, and 2.02 mm depth from cerebral cortex) of every 10 sections (8 µm thickness of each section). Data are presented as mean ± SEM number of PI-positive cells per section. a a sample of brain slices with different magnifications from experimental groups. b development of eosinophilic and pyknotic changes in cells. c number of PI-positive cells in TBI region in the experimental groups.The cells with intact nucleus were considered as saved cells and counted in the TBI-injured area. Data were analyzed by two way ANOVA with Tukey’s post-hoc multiple comparisons test. n = 3 rats for each group. **: p < 0.01, and ***: p < 0.001 compared to corresponding sham group. #: p < 0.05 compared to “Trauma+PBS 48 h” group. &: p < 0.05 compared to “Trauma + IL-4 24 h” group.
IL-4 Prevented Overexpression of TNF-α 6 h after TBI
Figure 5a demonstrates time course of TNF-α protein expression from 3 h to 5 days after TBI. TNF-α significantly increased at early times (3 h and 6 h) after TBI, and returned to control level 24 h thereafter. Two-way ANOVA revealed a significant interaction [F (3, 40) = 35.84, p < 0.0001] between treatment [F (1, 40) = 72.87, p < 0.0001] and time [F (3, 40) = 45.12, p < 0.0001] factors among groups. The 6 h post-TBI period was selected to evaluate the effect of IL-4 on TNF-α expression.

Effect of interleukin 4 on TNF-α up-regulation after traumatic brain injury in rats. a Time course of TNF-α protein expression after Controlled Cortical Impact injury. b Effect of IL-4 on TNF-α overexpression 6 h after traumatic brain injury. Upper part: The representative blots of brain TNF-α and ß-actin (internal control) obtained by Western blot technique. Lower part: The mean ratio of TNF-α to ß-actin. Data are represented as Mean ± SEM for 7 rats in each group. Dara are analyzed by two-way ANOVA with multiple comparisons test (a) and one way ANOVA with Tukey’s post-hoc multiple comparisons test (b). **: p < 0.01, and ***: p < 0.0001 compared to corresponding sham group. # # #: p < 0.0001 compared to “Trauma+PBS 3 h” and “Trauma+PBS 6 h” groups (a), or corresponding “Trauma+PBS 6 h” group (b).
As shown in Fig. 5b, One way ANOVA revealed a significant difference between groups [F (3, 20) = 182.7, p < 0.0001]. Administration of IL-4 to sham-operated rats (“Sham + IL-4” group) increased TNF-α expression after 6 h (p < 0.01, compared to “Sham+PBS 6h” group). However, administration of IL-4 to the CCI-injured rats inhibited overexpression of TNF-α at this time point (p < 0.0001).
IL-4 Augmented Overexpression of IL-10 after TBI
The time course of IL-10 protein expression after TBI is demonstrated in Fig. 6a. IL-10 significantly increased 6 h after CCI. This increase lasted up to 24 h after TBI. Two-way ANOVA revealed a significant interaction [F (6, 60) = 16.37, p < 0.0001] between treatment [F (2, 60) = 230.60, p < 0.0001] and time [F (3, 60) = 20.70, p < 0.0001] factors among groups.

Effect of interleukin 4 on Interleukin 10 up-regulation after traumatic brain injury in rats. a Time course of IL-10 protein expression after Controlled Cortical Impact injury. b Effect of IL-4 on IL-10 overexpression 24 and 48 h after traumatic brain injury. Upper part: The representative blots of brain IL-10 and ß-actin (internal control) obtained by Western blot technique. Lower part: The mean ratio of IL-10 to ß-actin. Data are represented as Mean ± SEM for 7 rats in each group. Dara are analyzed by two-way ANOVA with multiple comparisons test. ***: p < 0.0001 compared to corresponding sham group. # # #: p < 0.0001 compared to other Trauma groups (a) or corresponding Trauma group (b). @@@: p < 0.0001 compared to “Sham + IL-4 24 h” group. &&&: p < 0.0001 compared to “Trauma + IL-4 24 h” group.
The 24 h and 48 h periods after TBI were selected to assess the effect of IL-4 on IL-10 expression. Two-way ANOVA revealed a significant interaction [F (3, 40) = 111.30, p < 0.0001] between treatment [F (3, 40) = 168.30, p < 0.0001] and time [F (1, 40) = 28.12, p < 0.0001] factors among groups (Fig. 6b). Administration of IL-4 to sham-operated rats (sham + IL-4 group) significantly increased IL-10 expression after 48 h (p < 0.0001 compared to the corresponding sham group). The extent of IL-10 expression at 48 h was significantly higher than that at 24 h after sham operation (p < 0.0001). Administration of IL-4 to the CCI-injured rats significantly amplified overexpression of IL-10 at both 24 h and 48 h after TBI (p < 0.0001 compared to the corresponding Trauma group).
IL-4 Amplified Overexpression of TGF-β after TBI
The time course of TGF-β protein expression after TBI is demonstrated in Fig. 7a. TGF-β significantly increased 6 h after CCI, persisted for 24 h and then decreased to a level less than control at 48 h after TBI (p < 0.0001). Two-way ANOVA revealed a significant interaction [F (4, 45) = 135.70, p < 0.0001] between treatment [F (2, 45) = 128.20, p < 0.0001] and time [F (2, 45) = 162.60, p < 0.0001] factors among groups.

Effect of interleukin 4 on Transforming growth factor-β expression after traumatic brain injury in rats. a Time course of TGF-β protein expression after Controlled Cortical Impact injury. b Effect of IL-4 on TGF-β overexpression 24 and 48 h after traumatic brain injury. Upper part: The representative blots of brain TGF-β and ß-actin (internal control) obtained by Western blot technique. Lower part: The mean ratio of IL-10 to ß-actin. Data are represented as Mean ± SEM for 7 rats in each group. Data are analyzed by two-way ANOVA with multiple comparisons test. ***: p < 0.0001 compared to corresponding sham group. # # #: p < 0.0001 compared to “Trauma 6 h” and “Trauma 24 h” groups (a), or corresponding Trauma group (b). @@@: p < 0.0001 compared to corresponding “Sham + IL-4″ group”.
The 24 h and 48 h periods after TBI were selected to evaluate effect of IL-4 on TGF-β expression. Two-way ANOVA revealed a significant interaction [F (3, 40) = 32.41, p < 0.0001] between treatment [F (3, 40) = 49.11, p < 0.0001] and time [F (1, 40) = 35.38, p < 0.001] factors among groups (Fig. 7b). Administration of IL-4 to sham-operated rats (“Sham + IL-4” group) did not change TGF-β expression after both 24 h and 48 h. Administration of IL-4 to the CCI-injured rats significantly amplified overexpression of TGF-β at 48 h after TBI (p < 0.0001). There was no significant difference between the 24 h and 48 h trauma groups receiving IL-4.
IL-4 Increased Expression of Arg1 in the CCI-injured Brain
As seen in Fig. 8a, expression of Arg1 significantly increased 6 h (p < 0.0001), and 24 h (p < 0.0001), but not 48 h after TBI. Two-way ANOVA revealed a significant interaction [F (4, 45) = 27.70, p < 0.0001] between treatment [F (2, 45) = 242.90, p < 0.0001] and time [F (2, 45) = 14.84, p < 0.0001] factors among groups.

Effect of IL-4 on brain expression of Arginase-1 in rats with traumatic brain injury. a Time course of Arginase-1 protein expression after Controlled Cortical Impact injury. b Effect of IL-4 on Arginase-1 expression 24 and 48 h after traumatic brain injury. Upper part: The representative Western blots of brain Arginase-1and ß-actin (internal control) obtained after Controlled Cortical Impact injury. Lower part: The mean ratio of Arginase-1 to ß-actin. Data are represented as Mean ± SEM for 7 rats in each group. Data are analyzed by two-way ANOVA with multiple comparisons test. ***: p < 0.0001 compared to corresponding sham group. # # #: p < 0.0001 compared to corresponding Trauma group. @@@: p < 0.0001 compared to corresponding “Sham + IL-4” group. &&: p < 0.01 compared to “Trauma + IL-4 24 h” group.
The 24 h and 48 h periods after TBI were selected to evaluate effect of IL-4 on Arg1 expression. Two-way ANOVA revealed a significant interaction [F (3, 40) = 32.44, p < 0.0001] between treatment [F (3, 40) = 145.20, p < 0.0001] and time [F (1, 40) = 77.81, p < 0.0001] factors among groups (Fig. 8b). Administration of IL-4 to sham-operated rats increased Arg1 expression after 24 h (p < 0.0001) but not 48 h. Administration of IL-4 to the CCI-injured rats significantly amplified overexpression of Arg1 at 48 h after TBI (p < 0.0001). There was significant difference in Arg1 expression between the 24 h and 48 h trauma groups receiving IL-4 (p < 0.01).
Effect of IL-4 on Astrocytes and Macrophage/Microglia State in the CCI-injured Rats
The Iba1 and Arg1 expression in the CCI-injured region is demonstrated in Fig. 9. Iba1 was considerably expressed 6, 24, and 48 h after TBI. Arg1 expression is also observed in the Iba1-positive cells at all three times but it was less prominent at 48 h after TBI. A significant increase in Iba1/Arg1 positive cells was observed in the IL-4-treated CCI-injured brain sections at 48 h post-TBI period. More number of Iba1/Arg1 positive cells was detected in the CCI-injured brain areas of rats treated with IL-4 at 48 h post-TBI period.

Effect of interleukin 4 on Iba1 and Arginase 1 co-expression in the trauma-injured brain area of rats. a an illustrative brain section in which Immunofluorescent images are taken from inside the dotted square. Immunofluorescent images represent Iba1, Arg1, and Iba1/Arg1 positive cells at 6 (b), 24 (c), and 48 (d) hours after traumatic brain injury induced by controlled cortical impact device. Arrows indicate cells labeled with antibodies of anti-Iba1, anti-Arg1, and coloacalization of both. Neural cells are seen as a black hole which might be surrounded by microglial cells. Scale bar represents 30 µm.
The GFAP and Arg1 expression in the CCI-injured brain sections are demonstrated in Fig. 10. As seen in the figure, GFAP was not significantly expressed 6 h after TBI. However, then at 24, and 48 h post-TBI periods, a remarkable GFAP positive cells are detected. Although Arg1-positive cells are seen, Arg1 expression was not detected in the GFAP-positive cells. Brain sections from the CCI-injured areas of the IL-4-treated rats show GFAP-positive cells without Arg1 co-expression. The CCI-injured brain areas of rats treated with IL-4 showed a considerable decrease in the GFAP-positive cells especially at 48 h post-TBI period.

Effect of interleukin 4 on GFAP and Arginase 1 co-expression in the trauma-injured brain area of rats. a an illustrative brain section in which Immunofluorescent images are taken from inside the dotted square. Immunofluorescent images represent GFAP, Arg1, and GFAP/Arg1 positive cells at 6 (b), 24 (c), and 48 (d) hours after traumatic brain injury induced by controlled cortical impact device. Arrows indicate cells labeled with anti-GFAP antibody. No colocalization of GFAP and Arg1 is detected. Neural cells are seen as a black hole which might be surrounded by astrocytes. Scale bar represents 30 µm.
DISCUSSION
The present study implies that the accelerating effect of TBI on development of kindled seizures is inhibited by single i.c.v. injection of IL-4 shortly after TBI. IL-4 could reduce lesion size, and significantly rescue neurons from death in the TBI-injured rats. IL-4 also suppressed expression of the M1 cytokine TNF-α, whereas amplified overexpression of the M2 cytokines IL-10, and TGF-β, and the M2 marker Arg1 in the damaged area. In addition, IL-4 decreased astrogliosis and increased macrophage/microglia activation and polarization to the anti-inflammatory phenotype. Therefore, directing TBI neuroinflammation toward an anti-inflammatory tone seems to be mainly involved in the blunting effect of IL-4 on the accelerated epileptogenesis in the TBI context.
It is reported that intraperitoneal administration of IL-4 to mice 5 h before and 4 days after induction of epilepsy by pilocarpine could decrease frequency of seizures and inhibits development of epilepsy [27]. Therefore, we first assessed the ineffective dose of IL-4 on kindling rate in non-traumatic rats. In addition, we intended to exclude potential effect of IL-4 on peripheral immune system. Therefore, the i.c.v. route of administration was selected. Given that antiepileptogenic interventions are necessary after the presumed epileptogenic insult, in order to translate the drug-therapy protocol into practical clinical application, we administered IL-4 shortly after TBI. IL-4, 100 ng/rat, slightly decreased the rate of kindling epileptogenesis in naïve rats. However, this effect was not statistically significant. Yet, this dose could successfully inhibit acceleration of kindling development in TBI state. The present study is the first report indicating that IL-4 if is administered for a while after trauma is able to prevent acceleration of epileptogenesis in the CCI-injured rats. There are indirect studies that support finding of the present study. It is reported that the bacterial endotoxin LPS and its derivative monophosphoryl lipid A inhibit the increased potential for acquisition of seizures in the TBI-injured rats [28, 29]. LPS can stimulate de novo IL-4 gene expression in murine microglia both in vitro and in vivo [35, 36]. Moreover, LPS is able to induce M2 polarization through a time-dependent [37] and species-dependent manner [38]. In this regard, short term exposure to LPS motivates inhibition of M1 polarization and directing the M1/M2 balance to M2 whereas long term exposure to LPS instigates M1 polarization and decreasing M2 polarization [37]. However, LPS is too toxic to be introduced in human clinical trials. In contrast to LPS, clinical studies suggest that IL-4 has acceptable safety. Clinical trials state recombinant IL-4 is safe and well tolerated in humans at the doses of 0.25–5 μg/kg/day and up to 10 μg/kg once administered 3 times/week [39, 40]. Therefore, IL-4 may have significant clinical relevance for prevention of PTE.
There is strong evidence on the beneficial impacts of IL-4 on tissue repair following brain injury [18, 41, 42]. It is shown that administration of IL-4 48 h after SCI markedly improves functional outcomes and reduces tissue damage after contusion injury [24]. Therefore, we first examined the possible impact of IL-4 on TBI size as the primary mechanism of anti-epileptogenic effect of IL-4 in the TBI state. In our study, TTC staining showed a remarkable cortical injury 24 h after CCI. The size of the injured tissue did not expand at 48 h post-TBI period. Then, 5 days after TBI, the injured area showed a size reduction. Our results are in agreement with the study by Başkaya et al. [43]. They showed that CCI with moderate severity (2 mm deformation depth) induces maximum cortical injury in rats after 1 and 2 days. Similar to our results, they found a gradual decrease in the lesion volume starting from day 4 and then a constant injury volume was observed from day 5 till 7. In our study, treatment of the CCI-injured rats with IL-4 caused significant reduction in injured size at 48 h post-TBI period. This finding is in line with the beneficial preventive effects of IL-4 on lesion development after SCI [24]. Some research groups suggested that TTC staining has low power and cannot detect the tissue viability below a limit because the cells that are between death and survival cannot be detected by this method [44]. Moreover, this technique is only able to detect irreversible brain damage [45]. At the same time, it is reported that despite protective effect on both neuronal and oligodendrocyte populations, IL-4 could not reduce the size of the injured tissue in SCI [25]. In order to further verify the potential protective effect of IL-4 against the TBI-induced tissue injury, we examined cell viability in the trauma area by PI staining. PI is a useful marker for loss of cell membrane integrity. Cell membrane permeability to PI occurs early after traumatic brain injury. Previous studies using CCI model of TBI suggested that some PI permeable neurons regain cell membrane integrity, once again some time after the injury [46]. Therefore, there exists an extended therapeutic window after CCI, to rescue PI-positive cells using neuroprotective therapies. Meanwhile, robust cell death is identified by cytoplasmic hypereosinophilia and nuclear shrinkage. In our study, before PI staining, the brain slices were first processed with Triton X-100 0.1%, to break membrane integrity of both intact and injured cells (in order to enable PI to enter the cell). Thereafter, the PI staining of these pre-Triton-treated cells helps to detect only cells with disrupted membranes but with intact nucleus (healthy cells as well as the cells which are in transition state from apoptotic to death), which have the potential to come back to life by the neuroprotective drug. Therefore, we took advantage of this approach to distinguish fully-dead cells (not stained with PI), and count only alive cells (cells with intact nucleus) regardless of whether they have been damaged by trauma or not. Thus, in the conditions of our study, PI-positive cells are indicator of live cells. Administration of IL-4 (100 ng/rat, i.c.v.) after TBI significantly prevented development of neuronal death 48 h after TBI. This finding is correlated with reduction of the injured size by IL-4 at 48 h post-TBI period. In contrast to our results, intrahippocampal injection of mesenchymal stromal cells transiently expressing 100 ng IL-4, 5 days after TBI could not reduce hippocampal and cortical neurodegeneration and improve functional outcomes in mice [47]. The early times after TBI are critical in subsiding development of TBI pathogenesis. Regardless of the tool used to deliver IL-4 into the CNS, we injected IL-4 acutely, up to 30 min, after TBI. This period is quite early compared to the 5 days post-TBI period of IL-4 injection used by Enam et al. [47]. This might be the main reason of positive results obtained in our study. It is known that IL-4 inhibits cell death and apoptosis [48]. IL-4 also enhances survival of murine basophils [49] and mast cells [50]. Various mechanisms are suggested for this attribute of IL-4 [48, 49, 51]. In cerebral ischemic-reperfusion injury, which is associated with hyperexcitablity of neurons and cell death, supplemental IL-4 has been able to increase viability of cortical neurons by reducing spontaneous neuronal firing and network burst activity [51]. This mechanism seems to be also involved in the preserving viability of neurons by IL-4 in our study. This suggestion is supported by the behavioral finding of our study as IL-4 could decrease brain hyperexcitablity and the accelerated rate of kindling epileptogenesis induced by TBI.
TBI is obviously associated with acute neuroinflammation and release of inflammatory cytokines. Neuroinflammation is the main causative factor in PTE [11, 13]. Therefore, modulation of neuroinflammation would have major role in preventing PTE. Hence, we measured impact of IL-4 on the TBI-induced brain expression of TNF-α, as the typical inflammatory cytokine, and IL-10 as well as TGF-β, as the classic anti-inflammatory cytokines. Our results showed that TNF-α protein level increased in the traumatic brain to a level more than twice of the sham level at 3 and 6 h after TBI. Then the TNF-α level returned to sham level 24 h after TBI. This finding is consistent with previous studies demonstrating the increase of TNF-α protein level in rat brain during a 3–12 h period after CCI [33] and fluid percussion injury [52, 53]. We selected the 6 h post-CCI interval, and measured TNF-α brain expression after treatment of the CCI-injured rats with IL-4. IL-4 by itself increased TNF-α level in the sham-operated rats. This is the first report indicating increase in TNF-α level in sham (control) group by IL-4. This finding is in contrast to the previous report indicating that IL4 does not change TNF-α level in control conditions such as unstimulated monocytes [54]. The difference in doses of IL-4 and conditions of the two studies (in vivo versus in vitro) might be the underlying reason for the different findings. IL-4 is predominantly regarded as an anti-inflammatory cytokine. However, there are in vitro and in vivo evidence suggesting that IL-4 by itself can also promote the inflammatory response including TNF-α and IL-1β expression in parallel [55,56,57]. The target cell, the time of application, and concentration during various phases of immune responses are determinant of pro- or anti-inflammatory effects of IL-4. The TNF-α increase by IL-4 in the sham-operated rats, is in line with the kindling data of the present study. IL-4 increased the rate of epileptogenesis in sham-operated rats, which was marginally significant (p = 0.05). Given the proconvulsant effect of TNF-α [58,59,60], the increased expression of TNF-α (and other inflammatory cytokines such as IL-1β) seems to be mainly involved in the acceleration of kindling development by IL-4 in rats with no TBI. On the other hand, IL-4 blocked the increase in TNF-α level after TBI. Anti-inflammatory activity of IL-4 is attributed to suppression of TNF-α and interleukin 1-β production and activity [54, 61]. Considering the positive impact of TNF-α on seizure threshold and epileptogenesis [58, 62, 63] the suppressing effect of IL-4 on the acceleration of kindling development by TBI is mediated (in part) via inhibiting TNF-α increase in the CCI-injured brain area.
In order to verify impact of IL-4 on expression of the classic anti-inflammatory cytokines L-10 and TGF-β after TBI, the time course of expression of these cytokines was first determined. IL-10 level increased 12 h after TBI, and remained higher than basal level till 48 h. TGF-β showed different pattern of expression. The level increased 6 and 24 h after TBI but then decreased to the level less than control at 48 h after TBI. IL-10 is shown modest elevation in mice 24 h after CCI with the peak 3 days after CCI [64]. However, in a weight drop model of TBI in rats, IL-10 acutely raised in brain from 2 h after trauma followed by a progressive increase over 24 h [65]. These findings demonstrate variability in the degree of cytokine response based on the mechanism and severity of injury. Then, we determined the effect of IL-4 on brain expression of IL-10 and TGF-β at 24 h (the time of rise in both cytokines) and 48 h (the time of decline in both cytokines) after TBI. At both 24 h and 48 h after sham operation, IL-4 had no effect on TGF-β brain level but increased IL-10 brain level. IL-4 has induced IL-10 expression from stimulated T helper 1 cells [17] as well as endotoxin-stimulated monocytes/microglia [66, 67] but not from unstimulated (control) cells. We report for the first time that IL-4 is able to induce IL-10 production in control condition in vivo. Administration of IL-4 to the CCI-injured rats significantly intensified IL-10 expression at both 24 h and 48 h post-CCI periods. IL-10 is a well-known anti-inflammatory cytokine which is expressed following TBI. IL-10 inhibits development of epileptiform activity evoked by transient episodes of hypoxia in rat hippocampal slices [68], and focal convulsions in electrical kindling [69]. IL-10 can also increase threshold of temperature-induced seizures in rats [70]. Thus, amplification of IL-10 expression by IL-4 plays a role in suppressing the TBI–induced acceleration of kindling development. With regard to TGF-β, IL-4 did not affect the increased TGF-β expression in the CCI-injured rats 24 h after TBI but could significantly raise TGF-β expression at 48 h (the time of fall in the cytokine level after TBI) post-TBI period. TGF-β possesses both pro- and anti-inflammatory functions depending on the context. TGF-β is involved in epilepsy and PTE. Over-expression of cerebral TGF-β in transgenic mice causes development of a set of neuropathological complications including seizures [71]. Moreover, TGF-β and TGF-β signaling pathway are demonstrated among the main elements in development of epileptiform activity in rats after the brain injury induced by blood brain barrier disruption [72]. TGF-β is overexpressed 3 days after CCI in mice, and i.p. injection of TGF-β blocker is associated with a decrease in seizure behavior and EEG power spectrum [73]. On the other hand, TGF-β has shown neuroprotective action against glutamate neurotoxicity and ischemic brain injury [74]. The genetic defects in the TGF-β pathway is accompanied by epilepsy in humans [75]. Moreover, inhibition of TGF-β signaling blocks the anti-seizure effects of the oligonucleotide miRNA inhibitors (antagomirs) in three different rodent models of temporal lobe epilepsy [76]. Thus, strengthening the TGF-β expression by IL-4 also seems to play a role in suppression of TBI–induced acceleration of kindling development. Given the central role of neuroinflammation in pathogenesis of PTE [11, 13], amplification of anti-inflammatory response greatly contributes in the suppressing impact of IL-4 on TBI–induced acceleration of kindling development.
We found that administration of Il-4 shortly after TBI causes overexpression of IL-10 and TGF-β, and impedes expression of TNF-α. TNF-α is the typical proinflammatory cytokine released from M1 macrophage/microglia, whereas IL-10/TGF-β are the representative cytokines of M2 macrophage/microglia [77,78,79]. Therefore, we assessed potential polarization of macrophage/microglia to M2 phenotype by measuring brain expression of the classic marker of M2 macrophage/microglia Arg1. In our study, TBI itself was associated with Arg1 overexpression in 6–24 h but not 48 h after TBI. Overexpression of Arg1 6–24 h after TBI is comparable with the period of overexpression of M2 cytokines IL-10 and TGF-β after TBI as they also enhanced in our study during first 24 h after TBI. It is reported that in addition to M2 phenotype, Arg1 is also upregulated in a fraction of murine M1 macrophage as well [80]. Therefore, in addition to M2 phenotype, M1 inflammatory macrophage/microglia might be another source of Arg1 expression in the brain of TBI-injured rats. In a study similar to ours, Turtzo et al. utilized CCI model of trauma and measured the cytokine expression at post-transcription level [4]. However, they found a 5–7 days post-TBI period for microglia mixed M1 and M2 response in rats [4]. In contrast to our study, they used female rats. Macrophage/microglia response in vivo is highly complex depending on many factors, of which gender differences is critical and seems to be the key reason for different macrophage/microglia time response between the two studies.
In our study, administration of IL-4 to sham-operated rats significantly increased expression of Arg1 after 24 h. This stimulatory effect on Arg1 was not seen at 48 h after administration, which is most likely due to short half-life of IL-4 [81]. Our finding is in line with previous study indicating temporal upregulation of Arg1 in frontal cortex and striatum microglia of naïve rats 8-16 h after direct injection of IL-4 into the third ventricle [82]. In addition, lentiviral delivery of IL-4 into the fourth ventricle of naïve mice has been associated with overexpression of Arg1 of spinal cord microglia at posttranscriptional level [83]. In contrast to the time effect of IL-4 in naïve rats, we observed IL-4 administration to the TBI-injured rats could increase Arg1 expression 48 h but not 24 h after TBI. It is recently reported that injection of IL-4 to the hippocampus of mice 5 days after head-closed injury could not significantly increase M2-like macrophages after 48 h [47]. In our study overexpression of Arg1 in the TBI-injured rats 48 h after IL-4 administration is associated with increased TGF-β expression at this time point. However, IL-10 was overexpressed both 24 h and 48 h after IL-4 administration to the TBI-injured rats. There is a range of activation states for microglial cells that span from the M1 to the M2 phenotypes, and the phenotype of the activated microglia will fall somewhere along this spectrum depending on the signal encountered. Moreover, the in vivo effects of IL-4 are also complex, and the final response depends on the local environment, pathological state, and the doses used. These factors might be responsible for different pattern of expression of Arg1, IL-10 and TGF-β in the TBI-injured rats after IL-4 administration.
In order to further confirm influence of IL-4 on time-dependent polarization of macrophages/microglia after TBI, we assessed macrophages/microglia activation and also polarization to the M2 anti-inflammatory phenotype directly in the brain sections by double immunostaining with the specific marker of the macrophages/microglia Iba1, and the specific marker of the M2 phenotype Arg1. Results of the double immunostaining supported the western blot findings so that macrophage/microglia showed activation and polarization to M2 phenotype after TBI. IL-4 notably increased the M2 phenotype 48 h after TBI. It is reported that astrocytes also exhibit inflammatory/anti-inflammatory phenotypes and express Arg1 [84]. In order to find other potential sources of Arg1 than macrophage/microglia in our study, we assessed Arg1 expression in the astrocytes of the TBI-injured area by double immunostaining of the brain sections with GFAP as the specific marker of astrocytes, and Arg1. We observed a considerable astrogliosis in the trauma-injured region but no Arg1-positive astrocytes was detected. Moreover, IL-4 did not induce Arg1 expression in the astrocytes. Therefore, under the conditions of our study, the main element being impacted by IL-4 seems to be macrophage/microglia, and not astrocytes. Interestingly, we observed a decrease in GFAP-positive cells in the trauma-injured region by IL-4. It is reported that IL-4 can inhibit astrocyte activation [85]. IL-4 can also regulate reactive astrocytes and play a protective role against neuroinflammation [86]. Our finding is in line with these reports. Therefore, in addition to macrophage/microglia polarization, inhibition of astrocyte proliferation and activation seems to also be involved in the protective effect of IL-4 on the TBI epileptogenesis observed in our study.
Macrophages (both resident and the invaded blood borne) as well as microglia have critical role in TBI [87, 88]. Meanwhile, IL-4 directly or indirectly attracts monocytes/macrophages from the hematopoietic system to infiltrate the CNS [83]. Although macrophage may differ from microglia for their capability to respond to IL-4 signaling, they share common markers and secreted cytokines [83]. Therefore, distinguishing macrophages from microglia is challenging. One of the established techniques that distinguishes macrophages from microglia to an acceptable extent is flow cytometry. Using advantage of this technique, it has shown that Arg1 is expressed exclusively by macrophages and not by microglia after CCI [89], and SCI [90]. We did not discriminate the extent of contribution of microglia and macrophages to the reduction of epileptogenesis susceptibility after CCI by IL-4. This issue needs to be elucidated by further works.
In conclusion, this translational study indicates that IL-4 is capable to reverse TBI-induced acceleration of epileptogenesis in an animal model of TBI. Our data highlight the potential of IL-4 as a promising biological drug for the treatment of TBI neurological complications including PTE. Future pre- and clinical studies are required to assess this possibility.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Dewan, M.C., A. Rattani, S. Gupta, R.E. Baticulon, Y.C. Hung, et al. 2018. Estimating the global incidence of traumatic brain injury. Journal of Neurosurgery 130: 1080–1097. https://doi.org/10.3171/2017.10.JNS17352.
Woodcock, T., and M.C. Morganti-Kossmann. 2013. The role of markers of inflammation in traumatic brain injury. Frontiers in Neurology 4: 18. https://doi.org/10.3389/fneur.2013.00018.
Hunt, R.F., J.A. Boychuk, and B.N. Smith. 2013. Neural circuit mechanisms of post-traumatic epilepsy. Frontiers in Cellular Neuroscience 7: 89. https://doi.org/10.3389/fncel.2013.00089.
Turtzo, L.C., J. Lescher, L. Janes, D.D. Dean, M.D. Budde, et al. 2014. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. Journal of Neuroinflammation 11: 82. https://doi.org/10.1186/1742-2094-11-82.
Xiong, Y., A. Mahmood, and M. Chopp. 2015. Current understanding of neuroinflammation after traumatic brain injury and cell-based therapeutic opportunities. Chinese Journal of Traumatology 21: 137–151. https://doi.org/10.1016/j.cjtee.2018.02.003.
Cruz-Haces, M., J. Tang, G. Acosta, J. Fernandez, and R. Shi. 2017. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Translational Neurodegeneration 6: 20. https://doi.org/10.1186/s40035-017-0088-2.
Webster, K.M., M. Sun, P. Crack, T.J. O’Brien, S.R. Shultz, et al. 2017. Inflammation in epileptogenesis after traumatic brain injury. Journal of Neuroinflammation 14: 10. https://doi.org/10.1186/s12974-016-0786-1.
Global Epilepsy Report 2019. https://www.ilae.org/about-ilae/policy-and-advocacy/international-public-policy-activities/global-epilepsy-report-2019. Accessed 1 Apr 2023.
Perucca, P., and I.E. Scheffer. 2021. Genetic contributions to acquired epilepsies. Epilepsy Currents 21: 5–13. https://doi.org/10.1177/1535759720954254.
Fordington, S., and M. Manford. 2020. A review of seizures and epilepsy following traumatic brain injury. Journal of Neurology 267: 3105–3111. https://doi.org/10.1007/s00415-020-09926-w.
Mukherjee, S., G.M. Arisi, K. Mims, G. Hollingsworth, K. O’Neil, et al. 2020. Neuroinflammatory mechanisms of post-traumatic epilepsy. Journal of Neuroinflammation 17: 193. https://doi.org/10.1186/s12974-020-01854-w.
Sharma, S., G. Tiarks, J. Haight, and A.G. Bassuk. 2021. Neuropathophysiological mechanisms and treatment strategies for post-traumatic epilepsy. Frontiers in Molecular Neuroscience 14: 612073. https://doi.org/10.3389/fnmol.2021.612073.
Sun, L., W. Shan, H. Yang, R. Liu, J. Wu, et al. 2021. The role of neuroinflammation in post-traumatic epilepsy. Frontiers in Neurology 12: 646152. https://doi.org/10.3389/fneur.2021.646152.
Gadani, S.P., J.C. Cronk, G.T. Norris, and J. Kipnis. 2012. IL-4 in the brain: a cytokine to remember. Journal of Immunology (Baltimore, Md.: 1950) 189: 4213–4219. https://doi.org/10.4049/jimmunol.1202246.
Woodward, E.A., C.M. Prêle, S.E. Nicholson, T.B. Kolesnik, and P.H. Hart. 2010. The anti-inflammatory effects of interleukin-4 are not mediated by suppressor of cytokine signalling-1 (SOCS1). Immunology 131: 118–127. https://doi.org/10.1111/j.1365-2567.2010.03281.x.
Liu, X., J. Liu, S. Zhao, H. Zhang, W. Cai, et al. 2016. Interleukin-4 is essential for microglia/macrophage M2 polarization and long-term recovery after cerebral ischemia. Stroke 47: 498–504. https://doi.org/10.1161/STROKEAHA.115.012079.
Mitchell, R.E., M. Hassan, B.R. Burton, G. Britton, E.V. Hill, et al. 2017. IL-4 enhances IL-10 production in Th1 cells: implications for Th1 and Th2 regulation. Scientific Reports 7: 11315. https://doi.org/10.1038/s41598-017-11803-y.
Xiong, X., G.E. Barreto, L. Xu, Y.B. Ouyang, X. Xie, et al. 2011. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke 42: 2026–2032. https://doi.org/10.1161/STROKEAHA.110.593772.
Lee, S.I., S.R. Jeong, Y.M. Kang, D.H. Han, B.K. Jin, et al. 2010. Endogenous expression of interleukin-4 regulates macrophage activation and confines cavity formation after traumatic spinal cord injury. Journal of Neuroscience Research 88: 2409–2419. https://doi.org/10.1002/jnr.22411.
Girard, S., D. Brough, G. Lopez-Castejon, J. Giles, N.J. Rothwell, et al. 2013. Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia 61: 813–824. https://doi.org/10.1002/glia.22478.
Li, H.L., N. Kostulas, Y.M. Huang, B.G. Xiao, P. van der Meide, et al. 2001. IL-17 and IFN-gamma mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. Journal of Neuroimmunology 116: 5–14. https://doi.org/10.1016/s0165-5728(01)00264-8.
Zhao, X., H. Wang, G. Sun, J. Zhang, N.J. Edwards, et al. 2015. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. Journal of Neuroscience 35: 11281–11291. https://doi.org/10.1523/JNEUROSCI.1685-15.2015.
Pu, H., C. Ma, Y. Zhao, Y. Wang, W. Zhang, et al. 2021. Intranasal delivery of interleukin-4 attenuates chronic cognitive deficits via beneficial microglial responses in experimental traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism 41: 2870–2886. https://doi.org/10.1177/0271678X211028680.
Francos-Quijorna, I., J. Amo-Aparicio, A. Martinez-Muriana, and R. López-Vales. 2016. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia 64: 2079–2092. https://doi.org/10.1002/glia.23041.
Lima, R., S. Monteiro, J.P. Lopes, P. Barradas, N.L. Vasconcelos, et al. 2017. Systemic interleukin-4 administration after spinal cord injury modulates inflammation and promotes neuroprotection. Pharmaceuticals (Basel) 10: 83. https://doi.org/10.3390/ph10040083.
Pu, H., X. Zheng, X. Jiang, H. Mu, F. Xu, et al. 2020. Interleukin-4 improves white matter integrity and functional recovery after murine traumatic brain injury via oligodendroglial PPARγ. Journal of Cerebral Blood Flow and Metabolism 41: 511–529. https://doi.org/10.1177/0271678X20941393.
Li, T., X. Zhai, J. Jiang, X. Song, W. Han, et al. 2017. Intraperitoneal injection of IL-4/IFN-γ modulates the proportions of microglial phenotypes and improves epilepsy outcomes in a pilocarpine model of acquired epilepsy. Brain Research 1657: 120–129. https://doi.org/10.1016/j.brainres.2016.12.006.
Eslami, M., E. Ghanbari, M. Sayyah, F. Etemadi, S. Choopani, et al. 2016. Traumatic brain injury accelerates kindling epileptogenesis in rats. Neurological Research 38: 269–274. https://doi.org/10.1179/1743132815Y.0000000086.
Hesam, S., B. Khoshkholgh-Sima, H.G. Pourbadie, V. Babapour, M. Zendedel, et al. 2018. Monophosphoryl lipid A and Pam3Cys prevent the increase in seizure susceptibility and epileptogenesis in rats undergoing traumatic brain injury. Neurochemical Research 43: 1978–1985. https://doi.org/10.1007/s11064-018-2619-3.
Paxinos, G., and C. Watson. 2007. The rat brain in stereotaxic coordinates. San Diego: Elsevier, Academic press.
Cherry, J.D., J.A. Olschowka, and M.K. O’Banion. 2014. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. Journal of Neuroinflammation 11: 98. https://doi.org/10.1186/1742-2094-11-98.
Racine, R.J. 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalography and Clinical Neurophysiology 32: 281–294. https://doi.org/10.1016/0013-4694(72)90177-0.
Eslami, M., M. Sayyah, M. Soleimani, L. Alizadeh, and M. Hadjighassem. 2015. Lipopolysaccharide preconditioning prevents acceleration of kindling epileptogenesis induced by traumatic brain injury. Journal of Neuroimmunology 289: 143–151. https://doi.org/10.1016/j.jneuroim.2015.11.003.
Bradford, M.M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72: 248–254. https://doi.org/10.1006/abio.1976.9999.
Lively, S., and L.C. Schlichter. 2018. Microglia responses to pro-inflammatory stimuli (LPS, IFNγ+TNFα) and reprogramming by resolving cytokines (IL-4, IL-10). Frontiers in Cellular Neuroscience 12: 215. https://doi.org/10.3389/fncel.2018.00215.
Mukherjee, S., L.Y. Chen, T.J. Papadimos, S. Huang, B.L. Zuraw, et al. 2009. Lipopolysaccharide-driven Th2 cytokine production in macrophages is regulated by both MyD88 and TRAM. Journal of Biological Chemistry 284: 29391–29398. https://doi.org/10.1074/jbc.M109.005272.
Zhang, W., Y. Zhang, Y. He, X. Wang, and Q. Fang. 2019. Lipopolysaccharide mediates time-dependent macrophage M1/M2 polarization through the Tim-3/Galectin-9 signalling pathway. Experimental Cell Research 376: 124–132. https://doi.org/10.1016/j.yexcr.2019.02.007.
Orecchioni, M., Y. Ghosheh, A.B. Pramod, and K. Ley. 2019. Macrophage polarization: different gene signatures in M1 (LPS+) vs. classically and M2 (LPS-) vs. alternatively activated macrophages. Frontiers in Immunology 10: 1084. https://doi.org/10.3389/fimmu.2019.01084.
Leach, M.W., E.A. Snyder, D.P. Sinha, and I.Y. Rosenblum. 1997. Safety evaluation of recombinant human interleukin-4. I. Preclinical studies. Clinical Immunology and Immunopathology 83: 8–11. https://doi.org/10.1006/clin.1997.4303.
Vokes, E.E., R. Figlin, H. Hochster, M. Lotze, and M.E. Rybak. 1998. A phase II study of recombinant human interleukin-4 for advanced or recurrent non-small cell lung cancer. Cancer Journal from Scientific American 4: 46–51.
Mantovani, A., S.K. Biswas, M.R. Galdiero, A. Sica, and M. Locati. 2013. Microglia plasticity and polarization in tissue repair and remodeling. Journal of Pathology 229: 176–185. https://doi.org/10.1002/path.4133.
Salmon-Ehr, V., L. Ramont, G. Godeau, P. Birembaut, M. Guenounou, et al. 2000. Implication of interleukin-4 in wound healing. Laboratory Investigation: A Journal of Technical Methods and Pathology 80: 1337–1343. https://doi.org/10.1038/labinvest.3780141.
Başkaya, M.K., A. Doğan, C. Temiz, and R.J. Dempsey. 2000. Application of 2,3,5-triphenyltetrazolium chloride staining to evaluate injury volume after controlled cortical impact brain injury: role of brain edema in evolution of injury volume. Journal of Neurotrauma 17: 93–99. https://doi.org/10.1089/neu.2000.17.93.
Benedek, A., K. Móricz, Z. Jurányi, G. Gigler, G. Lévay, et al. 2006. Use of TTC staining for the evaluation of tissue injury in the early phases of reperfusion after focal cerebral ischemia in rats. Brain Research 1116: 159–165. https://doi.org/10.1016/j.brainres.2006.07.123.
Liszczak, T.M., E.T. Hedley-Whyte, J.F. Adams, D.H. Han, V.S. Kolluri, et al. 1984. Limitations of tetrazolium salts in delineating infarcted brain. Acta Neuropathologica 65: 150–157. https://doi.org/10.1007/BF00690469.
Whalen, M.J., T. Dalkara, Z. You, J. Qiu, D. Bermpohl, et al. 2008. Acute plasmalemma permeability and protracted clearance of injured cells after controlled cortical impact in mice. Journal of Cerebral Blood Flow and Metaboism 28: 490–505. https://doi.org/10.1038/sj.jcbfm.9600544.
Enam, S.F., S.R. Kader, N. Bodkin, J.G. Lyon, M. Calhoun, et al. 2020. Evaluation of M2-like macrophage enrichment after diffuse traumatic brain injury through transient interleukin-4 expression from engineered mesenchymal stromal cells. Journal of Neuroinflammation 17: 197. https://doi.org/10.1186/s12974-020-01860-y.
Zamorano, J., H.Y. Wang, L.M. Wang, J.H. Pierce, and A.D. Keegan. 1996. IL-4 protects cells from apoptosis via the insulin receptor substrate pathway and a second independent signaling pathway. Journal of Immunology 157: 4926–4934.
Reinhart, R., and T. Kaufmann. 2018. IL-4 enhances survival of in vitro-differentiated mouse basophils through transcription-independent signaling downstream of PI3K. Cell Death and Disease 9: 713. https://doi.org/10.1038/s41419-018-0754-z.
Burton, O.T., A.R. Darling, J.S. Zhou, M. Noval-Rivas, T.G. Jones, et al. 2013. Direct effects of IL-4 on mast cells drive their intestinal expansion and increase susceptibility to anaphylaxis in a murine model of food allergy. Mucosal Immunology 6: 740–750. https://doi.org/10.1038/mi.2012.112.
Chen, X., J. Zhang, Y. Song, P. Yang, Y. Yang, et al. 2020. Deficiency of anti-inflammatory cytokine IL-4 leads to neural hyperexcitability and aggravates cerebral ischemia-reperfusion injury. Acta pharmaceutica SinicaB 10: 1634–1645. https://doi.org/10.1016/j.apsb.2020.05.002.
Clausen, F., N. Marklund, and L. Hillered. 2019. Acute inflammatory biomarker responses to diffuse traumatic brain injury in the rat monitored by a novel microdialysis technique. Journal of Neurotrauma 36: 201–211. https://doi.org/10.1089/neu.2018.5636.
Taupin, V., S. Toulmond, A. Serrano, J. Benavides, and F. Zavala. 1993. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion. Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. Journal of Neuroimmunology 42: 177–185. https://doi.org/10.1016/0165-5728(93)90008-m.
Hart, P.H., G.F. Vitti, D.R. Burgess, G.A. Whitty, D.S. Piccoli, et al. 1989. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proceedings of the National Academy of Sciences USA 86: 3803–3807. https://doi.org/10.1073/pnas.86.10.3803.
Latta, C.H., T.L. Sudduth, E.M. Weekman, H.M. Brothers, E.L. Abner, et al. 2015. Determining the role of IL-4 induced neuroinflammation in microglial activity and amyloid-β using BV2 microglial cells and APP/PS1 transgenic mice. Journal of Neuroinflammation 12: 41. https://doi.org/10.1186/s12974-015-0243-6.
Ratthé, C., J. Ennaciri, D.M. Garcês Gonçalves, S. Chiasson, and D. Girard. 2009. Interleukin (IL)-4 induces leukocyte infiltration in vivo by an indirect mechanism. Mediators of Inflammation 2009: 193970. https://doi.org/10.1155/2009/193970.
Van Kampen, C., J. Gauldie, and S.M. Collins. 2005. Proinflammatory properties of IL-4 in the intestinal microenvironment. American Journal of Physiology Gastrointestinal and Liver Physiology 288: 111–117. https://doi.org/10.1152/ajpgi.00014.2004.
Chong, S.A., S. Balosso, C. Vandenplas, G. Szczesny, E. Hanon, et al. 2018. Intrinsic inflammation is a potential anti-epileptogenic target in the organotypic hippocampal slice model. Neurotherapeutics 15: 470–488. https://doi.org/10.1007/s13311-018-0607-6.
Lagarde, S., N. Villeneuve, A. Trébuchon, E. Kaphan, A. Lepine, et al. 2016. Anti-tumor necrosis factor alpha therapy (adalimumab) in Rasmussen’s encephalitis: an open pilot study. Epilepsia 57: 956–966. https://doi.org/10.1111/epi.13387.
Riazi, K., M.A. Galic, J.B. Kuzmiski, W. Ho, K.A. Sharkey, et al. 2008. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proceedings of the National Academy of Sciences USA 105: 17151–17156. https://doi.org/10.1073/pnas.0806682105.
Levings, M.K., and J.W. Schrader. 1999. IL-4 inhibits the production of TNF-alpha and IL-12 by STAT6-dependent and -independent mechanisms. Journal of Immunology 162: 5224–5229.
Patel, D.C., G. Wallis, E.J. Dahle, P.B. McElroy, K.E. Thomson, et al. 2017. Hippocampal TNFα signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. eNeuro 4: ENEURO.0105-17.2017. https://doi.org/10.1523/ENEURO.0105-17.2017.
Rana, A., and A.E. Musto. 2018. The role of inflammation in the development of epilepsy. Journal of Neuroinflammation 15: 144. https://doi.org/10.1186/s12974-018-1192-7.
Lagraoui, M., J.R. Latoche, N.G. Cartwright, G. Sukumar, C.L. Dalgard, et al. 2012. Controlled cortical impact and craniotomy induce strikingly similar profiles of inflammatory gene expression, but with distinct kinetics. Frontiers in Neurology 3: 155. https://doi.org/10.3389/fneur.2012.00155.
Kamm, K., W. Vanderkolk, C. Lawrence, M. Jonker, and A.T. Davis. 2006. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. The Journal of Trauma 60: 152–157. https://doi.org/10.1097/01.ta.0000196345.81169.a1.
Cao, S., J. Liu, L. Song, and X. Ma. 2005. The protooncogene c-Maf is an essential transcription factor for IL-10 gene expression in microglias. Journal of Immunology 174: 3484–3492. https://doi.org/10.4049/jimmunol.174.6.3484.
Kambayashi, T., C.O. Jacob, and G. Strassmann. 1996. IL-4 and IL-13 modulate IL-10 release in endotoxin-stimulated murine peritoneal mononuclear phagocytes. Cellular Immunology 171: 153–158. https://doi.org/10.1006/cimm.1996.0186.
Levin, S.G., and O.V. Godukhin. 2007. Protective effects of interleukin-10 on the development of epileptiform activity evoked by transient episodes of hypoxia in rat hippocampal slices. Neuroscience and Behavioral Physiology 37: 467–470. https://doi.org/10.1007/s11055-007-0036-1.
Godukhin, O.V., S.G. Levin, and E.Y. Parnyshkova. 2009. The effects of interleukin-10 on the development of epileptiform activity in the hippocampus induced by transient hypoxia, bicuculline, and electrical kindling. Neuroscience and Behavioral Physiology 39: 625–631. https://doi.org/10.1007/s11055-009-9187-6.
Ishizaki, Y., R. Kira, M. Fukuda, H. Torisu, Y. Sakai, et al. 2009. Interleukin-10 is associated with resistance to febrile seizures: genetic association and experimental animal studies. Epilepsia 50: 761–767. https://doi.org/10.1111/j.1528-1167.2008.01861.x.
Wyss-Coray, T., L. Feng, E. Masliah, M.D. Ruppe, H.S. Lee, et al. 1995. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1. American Journal of Pathology 147: 53–67.
Cacheaux, L.P., S. Ivens, Y. David, A.J. Lakhter, G. Bar-Klein, et al. 2009. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. Journal of Neuroscience 29: 8927–8935. https://doi.org/10.1523/JNEUROSCI.0430-09.2009.
Wang, F., X. Wang, L.A. Shapiro, M.L. Cotrina, W. Liu, et al. 2017. NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Structure and Function 222: 1543–1556. https://doi.org/10.1007/s00429-016-1292-z.
Prehn, J.H., C. Backhauss, and J. Krieglstein. 1993. Transforming growth factor-beta 1 prevents glutamate neurotoxicity in rat neocortical cultures and protects mouse neocortex from ischemic injury in vivo. Journal of Cerebral Blood Flow and Metabolism 13: 521–525. https://doi.org/10.1038/jcbfm.1993.67.
Kotlarz, D., B. Marquardt, T. Barøy, W.S. Lee, L. Konnikova, et al. 2018. Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nature Genetics 50: 344–348. https://doi.org/10.1038/s41588-018-0063-6.
Venø, M.T., C.R. Reschke, G. Morris, N.M.C. Connolly, J. Su, et al. 2020. A systems approach delivers a functional microRNA catalog and expanded targets for seizure suppression in temporal lobe epilepsy. Proceedings of National Academy of Science USA 117: 15977–15988. https://doi.org/10.1073/pnas.1919313117.
Laffer, B., D. Bauer, S. Wasmuth, M. Busch, T.V. Jalilvand, et al. 2019. Loss of IL-10 promotes differentiation of microglia to a M1 phenotype. Frontiers in Cellular Neuroscience 13: 430. https://doi.org/10.3389/fncel.2019.00430.
Zhou, X., B. Spittau, and K. Krieglstein. 2012. TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. Journal of Neuroinflammation 9: 210. https://doi.org/10.1186/1742-2094-9-210.
Yao, Y., X.H. Xu, and L. Jin. 2019. Macrophage polarization in physiological and pathological pregnancy. Frontiers in Immunology 10: 792. https://doi.org/10.3389/fimmu.2019.00792.
Amici, S.A., J. Dong, and M. Guerau-de-Arellano. 2017. Molecular mechanisms modulating the phenotype of macrophages and microglia. Frontiers in Immunology 8: 1520. https://doi.org/10.3389/fimmu.2017.01520.
Conlon, P.J., S. Tyler, K.H. Grabstein, and P. Morrissey. 1989. Interleukin-4 (B-cell stimulatory factor-1) augments the in vivo generation of cytotoxic cells in immunosuppressed animals. Biotechnology Therapeutics 1: 31–41.
Pepe, G., G. Calderazzi, M. De Maglie, A.M. Villa, and E. Vegeto. 2014. Heterogeneous induction of microglia M2a phenotype by central administration of interleukin-4. Journal of Neuroinflammation 11: 211. https://doi.org/10.1186/s12974-014-0211-6.
Rossi, C., M. Cusimano, M. Zambito, A. Finardi, A. Capotondo, et al. 2018. Interleukin 4 modulates microglia homeostasis and attenuates the early slowly progressive phase of amyotrophic lateral sclerosis. Cell Death and Disease 9: 250. https://doi.org/10.1038/s41419-018-0288-4.
Jang, E., J.H. Kim, S. Lee, J.H. Kim, J.W. Seo, et al. 2013. Phenotypic polarization of activated astrocytes: the critical role of lipocalin-2 in the classical inflammatory activation of astrocytes. Journal of Immunology 191: 5204–5219. https://doi.org/10.4049/jimmunol.1301637.
Brodie, C., N. Goldreich, T. Haiman, and G. Kazimirsky. 1998. Functional IL-4 receptors on mouse astrocytes: IL-4 inhibits astrocyte activation and induces NGF secretion. Journal of Neuroimmunology 81: 20–30. https://doi.org/10.1016/s0165-5728(97)00154-9.
Chen, L., L. Zhu, D. Lu, Z. Wu, Y. Han, et al. 2020. Interleukin 4 affects epilepsy by regulating glial cells: potential and possible mechanism. Frontiers in Molecular Neuroscience 13: 554547. https://doi.org/10.3389/fnmol.2020.554547.
Kumar, A., and D.J. Loane. 2020. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behavior and Immunity 26: 1191–1201. https://doi.org/10.1016/j.bbi.2012.06.008.
Hirschberg, D.L., and M. Schwartz. 1995. Macrophage recruitment to acutely injured central nervous system is inhibited by a resident factor: a basis for an immune-brain barrier. Journal of Neuroimmunology 61: 89–96. https://doi.org/10.1016/0165-5728(95)00087-i.
Hsieh, C.L., C.C. Kim, B.E. Ryba, E.C. Niemi, J.K. Bando, et al. 2013. Traumatic brain injury induces macrophage subsets in the brain. European Journal of Immunology 43: 2010–2022. https://doi.org/10.1002/eji.201243084.
Greenhalgh, A.D., R. Passos Dos Santos, J.G. Zarruk, C.K. Salmon, A. Kroner, et al. 2016. Arginase-1 is expressed exclusively by infiltrating myeloid cells in CNS injury and disease. Brain Behavior and Immunity 56: 61–67. https://doi.org/10.1016/j.bbi.2016.04.013.
Funding
Research reported in this publication was supported by Elite Researcher Grant Committee under award number [943697] from the National Institute for Medical Research Development (NIMAD), Tehran, Iran.
Author information
Authors and Affiliations
Contributions
Acquisition of data, statistical analysis, data presentation: M Radpour, B Khoshkroodian, T Asgari; Experimental design, statistical analysis, data interpretation: HG Pourbadie; Study concept and design, obtained funding, administrative, technical, and material support, study supervision, analysis and interpretation of data, drafting the manuscript: M Sayyah.
Corresponding author
Ethics declarations
Ethics Approval
All animal experiments were carried out in accordance with the Review Board and Ethics Committee of Pasteur Institute of Iran, the National Institute for Medical Research Development (Authorization code 943697), and Council Directive 2010/63EU of the European Parliament, and the Council of 22 September 2010 on the protection of animals used for scientific purposes.
Conflict of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Radpour, M., Khoshkroodian, B., Asgari, T. et al. Interleukin 4 Reduces Brain Hyperexcitability after Traumatic Injury by Downregulating TNF-α, Upregulating IL-10/TGF-β, and Potential Directing Macrophage/Microglia to the M2 Anti-inflammatory Phenotype. Inflammation 46, 1810–1831 (2023). https://doi.org/10.1007/s10753-023-01843-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10753-023-01843-0