
Abstract
CNS (central nervous system) trauma, which is classified as SCI (spinal cord injury) and TBI (traumatic brain injury), is gradually becoming a major cause of accidental death and disability worldwide. Many previous studies have verified that the pathophysiological mechanism underlying cell death and the subsequent neuroinflammation caused by cell death are pivotal factors in the progression of CNS trauma. Simultaneously, EVs (extracellular vesicles), membrane-enclosed particles produced by almost all cell types, have been proven to mediate cell-to-cell communication, and cell death involves complex interactions among molecules. EVs have also been proven to be effective carriers of loaded bioactive components to areas of CNS trauma. Therefore, EVs are promising therapeutic targets to cure CNS trauma. However, the link between EVs and various types of cell death in the context of CNS trauma remains unknown. Therefore, in this review, we summarize the mechanism underlying EV effects, the relationship between EVs and cell death and the pathophysiology underlying EV effects on the CNS trauma based on information in published papers. In addition, we discuss the prospects of applying EVs to the CNS as feasible therapeutic strategies for CNS trauma in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
CNS trauma, which is a serious organ injury, has become very common throughout the world in recent years. The National SCI Statistical Center reported that nearly 0.935 million people are newly diagnosed with traumatic CNS injury each year, and the incidence of this disease is increasing every year. Additionally, the morbidity rate of CNS is extremely high. Therefore, lifelong follow-up care is needed, and this is accompanied by considerable emotional and financial costs [1, 2]. CNS trauma is divided into SCI and TBI according to the clinical needs of the patient. Generally, CNS trauma progresses two stages. Stage one is characterized by primary injuries, which occur in a short window of time by external violence and can directly damage the structure of neuronal tissue and the vasculature [3, 4]. Stage two is characterized by secondary injuries caused by the hypoxia, oedema or ischaemia caused by the primary injuries. Secondary injuries cause more serious damage due to cell death and the release of inflammatory factors [2]. Alleviating the inflammatory response and preventing cell death are of great importance to minimize injuries to the brain and spinal cord tissue. Therefore, it seems critical to focus on pathobiological change during the secondary injury phase.
EVs are cell-derived lipid bilayer-encapsulated particles that are produced by nearly all cell types and transport bioactive components to target cells [5]. Based on their biogenesis and size, EVs include MVs (50–1000 nm), apoptotic bodies (ApoBDs) (1000–5000 nm) and exosomes (30–100 nm) [2, 5, 6]. MVs are released by “donor” cells through outward membrane budding, while exosomes are formed in the endolysosomal pathway and in multivesicular bodies (MVBs) before fusing with the plasma membrane, which subsequently releases them [7,8,9]. Apoptotic bodies, which originate from apoptotic cells, are shed from cells in the same manner as MVs [5]. As carriers that mediate communication among cells, EVs can be loaded with various contents, such as proteins, lipids and nucleic acids [7]. EVs are abundant in all bodily fluids, including blood, urine and cerebrospinal fluid (CSF), and are of great significance for regulation signalling among cells [1]. Because of the ability of EVs to cross the blood–CNS barrier, they have become alternatives to therapies based on cells for improving the microenvironment after CNS trauma [10].
In recent years, cell death has become a hotspot in the field of biomedical research as an important mechanism in all kinds of organ injuries. Previously, it was believed that there were two types of cell death, namely, necrosis and apoptosis [11]. Previous studies have demonstrated that apoptosis is the main form of cell death induced by CNS trauma [11, 12]. However, because of the limited understanding of programmed cell death, few studies have focused on the role of cell death in the context of CNS trauma. Recently, new types of programmed cell death have been identified, including autophagy, necroptosis, ferroptosis and pyroptosis. Increasing evidence over the past 10 years has revealed that cell death is an important process in the secondary injury phase in CNS trauma [3, 13, 14]. These studies revealed that cell death may be a pivotal cause of exacerbated secondary injury in CNS trauma. Additionally, many small molecules have been implicated in cell death and function as upstream or downstream signalling factors that regulate the cell microenvironment. EVs carry active contents such as RNA and protein that control signalling pathways, but few studies have discussed the roles of EVs in mediating cell death in CNS trauma circumstances. Thus, this review mainly summarizes the relationships between cell death and EVs in the context of CNS trauma for the purpose of evaluating the therapeutic potential of EVs and providing a feasible method for TBI and SCI treatment.
Mechanisms
Biogenesis of EVs
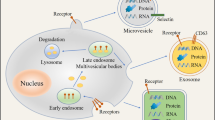
EVs are membrane-enclosed particles that are derived from the cell membrane. Based on their biogenesis and size, EVs are divided into MVs, exosomes and ApoBDs. EV biogenesis pathways also vary depending on the cell type that produces the EVs [6]. MVs are derived from all types of cells by direct blebbing of the cell plasma membrane [15] (Fig. 1). MVs are formed through various processes, such as phospholipid redistribution [4]. In MV biogenesis, plasma membrane molecules are rearranged; thus, the lipid and protein composition and Ca2+ level are changed [6]. Increased levels of Ca2+ catalysed by proteases promote the rearrangement of asymmetric membrane phospholipids; as a result, the membrane physically bends, and the underlying actin cytoskeleton is changed, leading to membrane budding and MV formation [6]. Cargoes located at the plasma membrane sites of MV budding are characterized by their respective plasma membrane anchors and higher order complexes [6]. MV release involves complex signalling pathways. By activating the ARF-PLD (phospholipase D)-ERK (extracellular signal-regulated kinase) pathway, myosin light chain kinase (MLCK) is phosphorylated and triggers the liberation of MVs carrying ARF6 (ADP-ribosylation factor 6), MHC-I, β1-integrin, VAMP3 (vesicle-associated membrane protein 3) and MT1MMP (membrane-type I matrix metalloproteinase) [4, 7]. Additionally, recent studies have revealed a new way in which MVs are released after the ESCRT-I subunit tumour susceptibility gene 101 (TSG101) is recruited to the plasma membrane [16, 17]. After ARRDC1 (Arrestin 1 domain-containing protein 1) is ubiquitinated by direct contact with the VPS4 ATPase with WWP2 (an E3 ubiquitin ligase), TSG101 binds to ARRDC1, and then, MVs that contain TSG101, ARRDC1 and other cellular proteins are released from cells [16, 17]. The release of MVs is likely fast, as the cargoes are intrinsic to the plasma membrane site where the MVs are formed, and subsequent MV release directly follows their generation and fission [6].

Mechanism of EV biogenesis. MVs are generated by the direct outward budding and fission of the plasma membrane. The first step of exosome formation is invagination of the plasma membrane to form early endosomes, and then, the Golgi complex can accumulate some exosomal proteins. Early endosomes fuse with vesicles derived from the budding of the Golgi complex to form late endosomes. Through ESCRT-dependent or ESCRT-independent pathways, cargoes are selectively sorted into ILVs, and then, late endosomes turn into MVBs. MVBs either fuse with lysosomes to degrade or fuse with the plasma membrane to release ILVs, which finally come into exosomes. ApoBDs are formed although membrane blebbing from apoptotic cells
Exosomes, small membrane vesicles derived from most cells, but largely associated with endothelial cells, dendritic cells (DCs), epithelial cells (ECs) and lymphocytes, are formed via the endolysosomal pathway [18]. Exosomes are formed through inward budding of the cell membrane and are released via fusion between multivesicular bodies (MVBs) and the plasma membrane, at which point they are called multiple intraluminal vesicles (ILVs) in MVBs [7, 18] (Fig. 1). Exosomes are ILVs in the extracellular space. In the first step, endosomes are formed through invagination of the plasma membrane. This type of endosome is named an early endosome [19]. Early endosomes are capable of fusing with endocytic vesicles from the Golgi complex to become recycling endosomes or late endosomes [7]. Recycled endosomes restore recycling contents and are involved in the process of recycling [20]. Via the collection of ILVs generated from endosomal membrane inwards budding, late endosomes can store proteins, nucleic acids and other active contents, followed by turning into MVBs [7]. Two endpoints determine the fate of MVBs: (1) they are sent to the lysosome for degradation with all components of the lysosome, and (2) they fuse with the plasma membrane of the cell to release their contents (exosomes) into the extracellular space [21]. However, it remains unknown which factors influence the outcome of MVBs. Regarding the regulation of exosomes, many studies have shown that exosome formation and release are closely related to the ESCRT (endosomal sorting complex required for transport) pathway [7, 21, 22]. ESCRT is an endosomal sorting complex, and four different ESCRT complexes have been identified to date. ESCRT-0 is capable of recognizing ubiquitinated proteins on the outer endosomal membrane [4]. ESCRT-I, ESCRT-II and ESCRT-III are capable of continuously recognizing monoubiquitinated transmembrane proteins and can induce these proteins to integrate into membrane domains, producing ILVs [23]. This cargo-sorting process of ILVs is mediated by endosomal sorting complexes. The completion of the process is followed by the dissociation of the ESCRT machinery from the MVB membrane, and the dissociated ESCRT machinery re-engages to conduct another round of protein sorting [23]. In addition, a study showed that MVB trafficking and MVB fusion with the cell membrane may be regulated by multiple Rab guanosine triphosphatase (GTPase) proteins and by cytoskeletal and molecular motor properties [24]. In addition to ESCRT-dependent pathway, distinct selective machinery in the ESCRT-independent pathway has been identified in multiple studies. The latter mechanism is mediated by the sphingolipid ceramide, which is generated during the hydrolysis of sphingomyelin by nSMase2 (neutral sphingomyelinase 2) [25]. However, the details of this regulatory process are poorly understood, and relevant studies need to be conducted.
ApoBDs are activated in cells undergoing apoptosis and facilitates the disintegration of cells into distinct membrane-enclosed vesicles with organelles [26] (Fig. 1). In contrast to MVs and exosomes, ApoBDs are assembled only during programmed cell death and are much larger than MVs and exosomes (1000–5000 nm) [26]. The morphological hallmarks of apoptotic cells are cytoplasmic cell shrinkage, plasma membrane budding, extracellular PS (phosphatidylserine) membrane exposure, chromatin condensation and DNA fragmentation, and throughout this process, the plasma membrane remains intact [27]. ApoBDs have been closely associated with membrane blebbing. However, it remains unclear whether ApoBDs are derived through membrane blebbing in apoptotic cells. Moreover, actin–myosin interactions may be involved in membrane blebbing [27], and some apoptosis-relevant proteins, such as caspase-9 and proapoptotic BH3-only proteins, may affect ApoBD formation because ApoBDs are associated with apoptosis. Finally, phagocytic cells engulf and eliminate ApoBDs in an orderly fashion to prevent apoptotic cell debris from damaging peripheral cells or tissues [28]. The discovery of ApoBDs revealed that apoptosis is not only a process of programmed cell death but also a mechanism underlying communication among cells. Recent studies have suggested that apoptotic vesicles derived from dying cells may be among the main regulators of the immune response [28,29,30]. This finding helps explain inflammation induced after cell death. Research on ApoBDs is greatly needed to expound on the inflammatory effects related to apoptosis.
Active Contents in EVs
The biogenesis of EVs suggests that they are not simply a simple lipid bilayer corpuscle but are loaded with a variety of molecules. Initially, it was believed that EV release partially constituted the mechanism by which unnecessary material was eliminated from cells. However, recent studies have shown that EVs promote communication among cells, which explains the action of the compounds loaded into EVs [6, 24, 31]. Generally, all EVs carry different proteins, lipids and nucleic acids. Because different cell types comprise specific contents, they induce a specific response when communicating with other cells [7].
EV proteins are capable of participating in EV biogenesis, sorting EV cargo, controlling EV release and promoting EV/recipient cell interactions. Common proteins loaded into EVs are related to biogenesis and release mechanisms [7]. As described in previous studies, MVs released through the ARF-PLD-ERK pathway specifically carry ARF6, β1-integrin, VAMP3 and MT1MMP, while recruitment of the ESCRT-I subunit TSG101 to the plasma membrane through its binding to a tetrapeptide protein within the Arrestin 1 domain-containing protein 1 (ARRDC1), resulting in the release of MVs containing TSG101 and ARRDC1. In addition, tetraspanins (CD63, CD53, CD37, CD81 and CD9), membrane proteins most abundant in EVs, clearly mediate ESCRT-independent pathway activation [32]. Rab GTPases are critical proteins that control the release of EVs, and a recent study showed the negative regulation of Rab 11 via transfection with plasmid DNA encoding genes that block EV secretion [32, 33].
Nuclide acids have been proven to be mediators in different cell types in recent studies. RNA in EVs has been extensively studied because it regulates the expression and modification of products after transcription. Cargo RNAs in EVs are characterized by different biotypes representing the RNA content in the source cell from which they were released. EVs preferentially carry small noncoding RNAs, although there are fragments and integrated mRNA, ribosomal RNA (rRNA) and long noncoding RNA (lncRNA) molecules in EVs [34]. Noncoding RNAs such as siRNAs, circular RNAs and microRNAs influence translation after transcription, thereby regulating biological behaviour at the genetic level. Among these RNAs, microRNAs have been the most extensively studied. A previous study showed that EVs containing microRNAs were involved in the regulation of cell physiological processes such as osteoblast differentiation [35]. By targeting the 3′ untranslated region (UTR) of specific mRNAs, microRNAs participate in the posttranscriptional modification of cells, thereby mediating a series of pathological or physiological processes. As a previous study described, M1-polarized macrophages accumulate in a lesion area and induce blood–spinal cord barrier breakdown, oxidative stress and neuroinflammation by secreting exosomal miR-155 after SCI [36]. Additionally, it has been reported that I/R injury in intestinal epithelial cells induces cortical neuron death by releasing paracrine mediators such as exosomal miRNAs associated with apoptosis, necroptosis and pyroptosis [37]. Thus, pathological or physiological processes in different cells may be mediated by secreted EVs containing microRNA. In addition, EVs carrying microRNA from different cells are neuroprotective, and EVs transport active RNA to nearby or distant cells, enabling communication among cells. Moreover, RNA contained in EVs is characteristic of the source cell type and physiological/pathological state, and some RNAs expressed in specific cells show curative or damaging effects on injured cells; therefore, EVs may serve as effective biomarkers for treating many diseases [34, 36, 38, 39].
Relationships Between EVs and Cell Death
EVs and Autophagy
Autophagy involves the formation of a phagocytic vesicle that encapsulates cytoplasmic proteins or organelles and then fuses with lysosomes to generate autophagolysosomes that degrade the contents in the phagosome, thereby meeting the needs of cells for metabolism and organelle renewal. There is a balance between EV secretion and macroautophagy because autophagosomes can fuse with lysosomes, which degrades their contents and inhibits the fusion of MVBs with autophagosomes necessary for the secretion of EVs [40]. Additionally, autophagy is regulated by a variety of signalling pathways and proteins, and EVs may play roles in transporting key signalling molecules to specific cells and induce or inhibit autophagy. Autophagy-related proteins (ATGs) are important for controlling autophagosome formation [41]. Two ubiquitin-like (UBL) systems, namely, ATG12-ATG5 and ATG8-LC3, regulate autophagosome initiation and elongation [42]. Shao et al. demonstrated that miR-454-3p in glioma cell exosomes may inhibit glioma cell proliferation, migration, invasion and autophagy, by targeting ATG12, while overexpression of ATG12 partially reversed the effects induced by miR-454-3p suppression [43]. Additionally, ATG5 has been proven to reduce the exosome biogenesis rate in breast cancer cells [44]. ATG4 is a protease that hydrolyses the C-terminus of ATG8 to form cytosolic LC3 I. Ni et al. demonstrated that exosomes from IL-β-treated human primary chondrocytes block autophagy in macrophages by inhibiting ATG4B activity, thereby further aggravating synovitis in osteoarthritis [45]. According to a recent study, ATG16L1 in combination with other ATG proteins confers protection against α-toxin by releasing ADAM10 from exosomes (EVs of endosomal cell origin) [46]. This recently discovered autophagy mechanism, called secretory autophagy, mediates the extracellular release of soluble and vesicle-bound substrates via the action of ATG [42]. This form of autophagy allows ATG machinery to prevent ADAM10 from accumulating on cells and promotes its inclusion on exosomes [46]. In the later stage of autophagy, autophagosomes fuse with lysosomes to degrade substrates. Blocking the combination of autophagosomes and lysosomes interrupts autophagic flux. Adipose-derived MSCs (ADMSCs) secrete EVs that contain miR-25-3p to enhance autophagic flux, thereby inducing neuroprotection [47]. In addition to EVs inducing intracellular autophagy, autophagy can regulate EV biogenesis and degradation. Notably, inhibition of ATG12–ATG3 formation changes MVB formation, disrupts late endosome trafficking and reduces exosome biogenesis [44]. In addition, a recent study demonstrated that EVs generated from damaged hepatocytes as well as sinusoidal endothelial cells (LSECs) were capable of inducing HSC activation and migration, which aggravated liver injury [48]. The activation of HSCs (hepatic stellate cells) mediated by PDGF (platelet-derived growth factor) and SHP2 resulted in a reduction in autophagic flux due to increased fibrogenic EV release [49]. Moreover, SHP2 (tyrosine phosphatase-2) in HSCs suppressed REDD1 (DNA damage response 1) and activated the mTOR (mammalian/mechanistic target of rapamycin) pathway to inhibit autophagy [49]. Thus, autophagy can eliminate MVs, and inhibiting SHP2-mTOR signalling might be a new strategy for treating liver fibrosis. All of this evidence suggests a link between autophagy and MVs, which may be helpful for understanding the progression of diseases and developing cures.
EVs and Necroptosis
Necroptosis refers to a form of regulated necrosis that proceed independent of caspase action and is mediated by receptor-interacting protein kinase 3 (RIPK3) together with mixed lineage kinase domain-like (MLKL) [50]. Similar to the apoptotic release of ApoBD, dying necroptotic cells also secrete EVs carrying pMLKL, ESCRT-III members and other proteins, and the DNA content in these EVs is lower than that in apoptotic bodies [51]. Studies demonstrated that active MLKL is capable of inducing EV formation at sites where it accumulates in the plasma membrane, and MLKL deficiency attenuates EV formation [51,52,53]. MLKL is phosphorylated by RIPK3 and then is translocated to the plasma membrane where it oligomerizes to form pores that cause plasma membrane rupture. Yoon et al. reported that RIPK3-dependent MLKL phosphorylation and conformational changes are indispensable in MLKL-mediated EV generation [54]. Thus, RIP kinases affect MLKL-mediated necroptotic EV biogenesis [51]. As we previously stated, EVs are able to mediate interactions among different cells; therefore, these findings prove that necroptosis may influence bioactivities in other cell by transporting EVs carrying active molecules. Shlomovitz et al. performed proteomic analysis on necroptotic EVs and found enriched ESCRT-III machinery and inflammatory signalling in EVs derived from necroptotic cells, and these proteins underwent phospholipid binding and vesicle-mediated transport [55]. These results revealed the vesicle transport and cell-targeting machinery of EVs derived from necroptotic cells. More importantly, PS-exposed necroptotic cells can be phagocytosed by macrophages and are capable of triggering increases in the levels of secreted TNF-α and CCL2 (C-C motif chemokine ligand) that exceed those secreted from apoptotic cells [51, 55]. Shlomovitz’s findings may explain why necroptosis aggravates injury through inflammation: their EVs are transported to impact recipient cells in the microenvironment.
Additionally, EVs from other cell types may trigger or inhibit the necroptosis of target cells. Notably, the oncobacterium Fusobacterium nucleatum (Fn) is capable of secreting EVs that carry many harmful molecules to the intestine and have the ability to change microbe–host interactions, particularly disrupting epithelial homeostasis in UC (ulcerative colitis) [56, 57]. Liu et al. discovered that in macrophage/Caco-2 cocultures, EVs from Fn profoundly upregulated the expression of receptor-interacting protein kinase 1 (RIPK1) and RIPK3, promoted the migration of RIPK1 and RIPK3 into necrosomes in Caco2 cells and thus facilitated epithelial barrier loss and oxidative stress damage [58]. This finding provides new insights into the toxic impact of bacteria on cells via the secretion of bacterial vesicles to mediate cell death and inflammation. In addition, hiPSC-MSCs (human-induced pluripotent stem cell-derived mesenchymal stromal cells)-derived EVs exerted a therapeutic effect in renal I/R injury through the delivery of SP1 to targeted renal cells, where it activated the SP1–SK1–S1P signalling pathway to inhibit necroptosis [59]. The effect of the necroptosis inhibitor Nec-1 as a pretreatment was the same as that of a single dose of hiPSC-MSCs-EVs, which indicates that hiPSC-MSCs confer renal protection by inhibiting necroptosis not inhibiting apoptosis [59]. These discoveries proved an interaction between necroptosis and EVs, but recent studies are rare. Some key mechanisms, such as how active compounds in EVs are precisely transported to target cells, need to be researched further.
EVs and Pyroptosis
Pyroptosis is a newly discovered mechanism of cell death that depends on caspase and is accompanied by the release of inflammatory factors [60]. For a long time, pyroptosis has been considered a form of monocyte death mediated by caspase-1 in response to specific bacterial insults but has been recharacterized as gasdermin-mediated programmed necrosis [60]. Pyroptosis is triggered in close association with inflammation, and EVs are considered pyroptotic inflammation mediators and biomarkers; therefore, we have assumed that EVs may be involved in pyroptosis [5]. Bacteria constitute the main cause of inflammation, and research has indicated that gram-negative bacteria-derived EVs induce pyroptosis by delivering LPS to the cytosol of recipient cells and inducing effector responses dependent on caspase-11 [61]. Previous studies indicated that LPS can be detected only at the cell surface by TLR4 (Toll-like receptor 4). Sivapriya’s research revealed a mechanism by which LPS from these gram-negative bacteria enter the cytosol to activate the inflammasome [61, 62]. In addition to those derived from bacteria, EVs derived from cells are also able to transport signalling molecules to target cells to trigger pyroptosis. Notably, mechanical ventilation-induced lung injury in preterm infants results from defective neurodevelopment, and Chavez et al. found that EV circulation after ventilation-induced lung injury resulted in brain injury and defective neurodevelopment in preterm infants by activating caspase-1 and GSDMD [63]. In addition, EVs from TBI patients can induce pyroptosis in lung endothelial cells [64]. Nadine found that in TBI patients, the serum-derived EV number and ASC (apoptosis-associated speck-like protein) level were profoundly increased, and these abundant proteins activated inflammasomes while inducing endothelial cell pyroptosis when cocultured with HMVEC-L (human lung microvascular endothelial cells) in vitro [64]. However, only head trauma from gunshots has been proven to be a neural trauma, and whether these EVs isolated from TBI patient serum were from injured brain cells is unknown. In conclusion, the characterization of EVs can explain how an injured organ influences another organ that is not closely related to it, such as systemic inflammatory response syndrome (SIRS), and the induction of inflammation. Future studies need to further explore the relationship between EVs and pyroptosis in different models of disease.
EVs and Apoptosis
Apoptosis is an autonomous and orderly cell death process controlled by genes, and it maintains a stable internal microenvironment. During apoptosis, cells undergo obvious morphological changes, and a dying cell is eventually disassembled into smaller fragments, that is, ApoBDs. ApoBDs are affiliated with EVs and were previously regarded as garbage bags containing genetic information and other substances from dying cells. Moreover, ApoBDs were considered to be biomarkers of apoptosis, but recent studies indicated that ApoBDs provide healthy recipient cells with necessary materials [65]. Billions of cells undergo apoptosis every day during normal development and homeostasis; as a result, many apoptotic bodies are generated [66]. This phenomenon suggests that apoptotic bodies may function in regulating tissue homeostasis. Apoptotic cells rarely accumulate under physiological conditions, which means that apoptotic cells undergo stable and rapid clearance [66]. Notably, macrophages, fibroblasts and specific phagocytes (Sertoli cells) can recognize and engulf apoptotic bodies to achieve apoptotic body clearance [67]. In one study, macrophages engulfed ApoBDs derived from MSCs, which triggered macrophage polarization towards the M2 phenotype to promote cutaneous wound healing [68]. Moreover, in Liu’s study, bone marrow MSCs were capable of engulfing apoptotic bodies to retain stem cell properties [69]. Systemic infusion of exogenous apoptotic bodies led to recycling of apoptotic body-derived RNF146 (the E3 ligase Ring figure protein 146) and miR-328-3p to activate the Wnt/β-catenin pathway, thereby rescuing damaged bone marrow MSCs [69]. These results showed that apoptotic bodies mediate the direct regulation of the Wnt/β-catenin pathway to maintain MSC homeostasis [69]. Similarly, in bone remodelling, osteoclasts undergo apoptosis in each bone turnover cycle, which results in the production of a large amount of ApoBDs. A study revealed the biological role of osteoclast-derived ApoBDs in bone remodelling via activation of receptor activator of NF-κB ligand (RANKL) reverse signalling in preosteoblasts [70]. All the evidence suggests that phagocytes not only eliminate superfluous ApoBDs but also acquire active ApoBD contents to maintain tissue homeostasis. Despite the small number of studies focusing on ApoBDs, ApoBDs obviously function as pivotal messengers that are released by dying cells to regulate cell clearance, tissue homeostasis and various other processes, implicating them in potential therapeutic applications.
EVs and Ferroptosis
Ferroptosis is a recently identified iron-dependent cell death pathway that is activated by iron overload and lipid peroxidation [71]. It has been genetically demonstrated that an imbalance in glutathione (GSH) synthesis via inhibition of system xc− (a cystine/glutamate antiporter containing subunits SLC7A11) and glutathione peroxidase 4 (GPX4) leads to lipid peroxidation [72]. Iron overload induced by environmental stress also triggers the excessive production of ROS (reactive oxygen species), causing further lipid peroxidation [71]. Therefore, the regulation of iron overload and lipid peroxidation controls ferroptosis activation. Recently, some studies have reported on the relationships between the extracellular system and ferroptosis. Zhang et al. found that plasma exosomes from lung adenocarcinoma patients specifically reduced lipid peroxidation rates and desensitized lung adenocarcinoma cells to ferroptosis [73]. Moreover, exosomes containing miR-522 that originated from cancer-associated fibroblasts (CAFs) blocked lipid ROS accumulation by targeting ALOX15 (arachidonate lipoxygenase 15) and thus inhibited ferroptosis [74, 75]. In obesity-induced cardiac injury, macrophages infiltrate adipose tissues, and exosomes derived from adipose tissue macrophages induce ferroptosis via glutathione synthesis inhibition mediated by targeting solute carrier family 7 member 11 (SLC7A11) [76]. In addition, prominin 2 alleviates iron overload in ferritin-containing MVBs and exosomes to stimulate iron transport out of cells and thereby inhibits ferroptosis [77]. In addition, cells undergoing ferroptosis are capable of generating EVs that influence other cells. One KRAS (Kirsten rat sarcoma viral oncogene homologue) mutant, KRASG12D, generated from tumour cells undergoing to autophagy-dependent ferroptosis was loaded into exosomes, and these exosomes caused macrophages to switch and acquire an M2-like pro-tumour phenotype via STAT3-dependent fatty acid oxidation [78]. All this evidence indicates that EVs are closely related to the process of ferroptosis. Thus, EVs may function as promising targets to regulate ferroptosis.
Cell Death Regulated by EVs in the CNS Trauma Context
Role of Cell Death in CNS Trauma
CNS trauma includes TBI and SCI, with increased incidence in recent years. CNS trauma is classified into two stages: the primary injury and secondary injury stages. Primary injuries occur at the time of direct external impact to the brain or spinal cord, causing mechanical damage to tissue, including blood vessels, axons and neural cell membranes [12, 79, 80]. Secondary injuries, which are more complex and severe consequences of CNS trauma, are described by the molecular, chemical and inflammatory cascades critical to the damage induced after the initial impact [12, 79, 80]. Thus, focusing on secondary injuries in the progression of CNS trauma is important to elucidate the causes of aggravated CNS trauma. In secondary injuries, free radicals, inflammatory mediators and other active factors mediate cell death [81]. Because of the irreversible damage to nerve cells, cell death may aggravate secondary CNS trauma injuries. Previous studies proved that programmed cell death, specifically apoptosis, may be involved in CNS injury [11, 12, 82]. Days or weeks after the initial trauma, oligodendrocytes may undergo cell suicide, another term for apoptosis, which impacts as many as 4 segments of a trauma site [12]. Although pathophysiological progression was previously attributed to ischaemia–reperfusion, excitotoxicity and calcium overload, these mechanisms are ultimately the causes of apoptosis [83]. However, preceding studies were rarely focused on the role of programmed cell death in CNS trauma due to limited understanding of programmed cell death. Recently, an increasing number of programmed cell death modalities have been discovered and have been proven to be critical factors in CNS trauma progression [11]. For example, the oxidative stress in damaged tissues induces microglial and astrocyte activation and the release of proinflammatory mediators, which promote blood–spinal cord barrier (BSCB) disruption and neutrophil influx and infiltration and contribute to inflammation and subsequent neural cell death after spinal cord ischaemia [84]. The infiltration of neutrophils causes a novel type of cell death involving the release of NETs (neutrophil extracellular traps) called NETosis, but this type of cell death has rarely been studied in the context of CNS trauma [85]. ROS produced after SCI cause pyroptosis, which further aggravates damage to the spinal cord [86]. Additionally, the production of downstream mature interleukin (IL)-1β and IL-18 triggers an inflammatory response [86]. Moreover, ROS profoundly affect ferroptosis. A study focused on how deferoxamine (DFO), which functions as a treatment to attenuate iron overload, affected SCI repair and revealed that DFO inhibited ferroptosis, which promotes functional recovery from SCI [11, 87]. This evidence demonstrated that ferroptosis does not favour SCI repair. As a self-degradative process, autophagy is involved in SCI and helps to eliminate damaged organelles. Most studies show that the activation of autophagy exerts neuroprotective effects on injured spinal cord tissue, revealing the important role of autophagy in SCI [88, 89]. Autophagy may function as a protective factor after SCI, and inhibition of autophagy may aggravate SCI. Necroptosis is mediated by RIPK1, RIPK3 and MLKL, which are regulated by intracellular signalling pathways, such as the caspase-independent pathway, and necroptotic cells exhibit the morphological characteristics similar to those exhibited by necrotic cells [90]. A previous study revealed that necroptosis contributed to neural cell death, and Nec-1 reduced functional and histopathological deficits in mice [11]. Necroptosis causes serious damage in injured spinal cords, and inhibiting necroptosis is a potential treatment of spinal cord injury. Similarly, previous studies proved that programmed cell death exerts the same effect as SCI in TBI [91,92,93]. These studies reveal that cell death may be an important cause of the tissue deterioration after CNS trauma.
According to previous studies, different types of cell death are mediated after CNS trauma in a time-dependent manner. For example, the levels of RIPK3 and MLKL obviously increase 6 h after TBI, while the levels of RIPK3 and MLKL protein increase markedly 1 day after SCI but decreased by 3 days after SCI [94,95,96]. These discoveries revealed that necroptosis follows a distinct temporal pattern in TBI and SCI. Similarly, the protein expression levels of pyroptosis markers, such as NLRP3 (NOD-like receptor protein 3), ASC and caspase-1, were elevated 1 day after brain tissue was injured, peaked at 3 days and gradually decreased over time after TBI [13, 97]. NLRP3, ASC and caspase-1 levels were found to be increased at 3 days after SCI, but the specific temporal pattern of pyroptosis-related molecule level changes is unknown [98]. Reportedly, autophagic indicators such as LC3II and Beclin1 in the lesion area were increasingly expressed within 1 h after injury, peaked 2 h after injury and returned to normal levels 72 h later in a model of SCI contusion [99]. In the context of TBI, the expression of Beclin-1 was increased 1 h post-TBI and peaked at 6 h, while the expression of LC3-II was upregulated rapidly and peaked at 48 h in injured cortex and hippocampus [100]. In apoptotic cells, caspase 3 levels increased 6 h after injury, peaked at 12 h and then slowly declined 2 weeks after SCI, while caspase 8 was expressed within 1 h and the caspase 3 level increased within 6 h in model rats with TBI [101, 102]. In cells undergoing ferroptosis, iron overload was detected 1 h after SCI [103]. Transferrin protein expression remained elevated 1 and 3 days after injury and returned to baseline on day 7, while lipid ROS levels and MDA (malondialdehyde) concentrations were significantly elevated 6 h after injury, peaked on day 3 and returned to baseline on day 7 after TBI [104]. All this evidence indicates that the programs in cells undergoing different types of death varied in terms of timing, and further experimental research is needed to investigate their similarities and differences at different time points. Although many cell death pathways, such as apoptosis, necroptosis and autophagy, have been identified in the context of CNS trauma, the major cell death pathway contributing to the imbalanced microenvironment in tissues after CNS trauma is still unknown [71]. Additionally, different types of cell death were found to be mediated in different cell types after CNS trauma. Therefore, future studies are needed.
Cell death is a complex process mediated by cell-to-cell communication. For example, it is known that the infiltration and recruitment of immune cells such as neutrophils and macrophages lead to the release of inflammatory factors, which induce neuronal death and demyelination [83]. These processes may involve interactions between EVs and target cells. Additionally, some studies have demonstrated that cells undergoing programmed cell death may release bioactive substances to induce the death of other cells via EV delivery; thus, necrotic neurons or brain cells may promote the death of other cells to aggravate the injury [37, 51, 105]. The aforementioned evidence proved relationships between EVs and cell death; therefore, we believe EVs may play important roles in the regulation of cell death in patients after CNS trauma. We examined recent studies related to cell death in the CNS trauma context in which EVs were released with the aim of elucidating the regulatory mechanisms underlying cell death and the roles of EVs in CNS trauma.
The Effects of EVs on Cell Death After TBI
TBI refers to disruptive brain function or other evidence of brain pathology resulting from the impact of an external physical force [106]. TBI has a yearly estimated incidence of 2.8 million cases in America; the mortality rates reach 30–40%, and nearly 60% of patients experience profound physical, social and psychosocial deficits [79, 106]. Secondary injuries, which constitute the most complex and severe stage of TBI, refer to the molecular, chemical and inflammatory cascades critical to further cerebral damage after the initial; secondary injuries include cerebral oedema, neuron degradation, hypoxia and increased intracranial pressure (ICP) [80, 107].
Organ injury is usually accompanied by cell death, and previous studies have explained concrete details of cell death after TBI [108]. Cell death is closely related to complex signalling pathways and molecules and is mediated by cell-to-cell communication. As mediators of cell communication, EVs are involved in the pathophysiological processes of cell death in the TBI context. Therefore, we examined a recent study that provided evidence of EV involvement in TBI. Reportedly, microcirculation in brains that are seriously damaged causes an increase in EV concentrations in the first 24 h following TBI [109]. By using nanoparticle tracking analysis to analyse 17 TBI patients and 18 healthy controls, the EV concentration was found to be the highest 1 day after TBI, and the EV size in CSF (cerebrospinal fluid) was obviously increase on days 4–7 [110]. Moreover, EVs crossed the BBB into peripheral circulation and were isolated from nearly all bodily fluids, including blood and the CSF, and by assessing EVs collected in blood or CSF, the severity of TBI was determined [111,112,113,114] (Fig. 2). EVs easily pass through the BBB due to their lipid solubility and ability to recognize specific proteins. Additionally, substantial evidence suggests that the tau protein released by injured neurons is carried by exosomes, which may be critical to chronic traumatic encephalopathy and neurodegenerative disorders [115,116,117]. These results reveal the possibility that EVs are broadly involved in cell-to-cell communication after TBI. The production of multiple EVs revealed active cell-to-cell communication — that is, EVs were implicated in driving both processes of pathology and recovery in TBI throughout the disease progression period. In terms of pathology, secondary injuries after TBI are related to inflammation and ischaemia. As described in a previous study, the initial inflammatory response is associated with protective and beneficial effects, such as tissue debris clearing and protection against pathogens. Moreover, successive bouts of neuroinflammation are harmful, as they may lead to the pathological progression after TBI by exacerbating the primary injury, inducing progressive neurodegeneration and delaying cell death [118]. EVs may function as bridges connecting inflammation signalling between injured cells and bystander cells. Recent experimental and human studies have shown that EVs exhibit immune-activating properties and inflammation-promoting activities by carrying and releasing numerous proinflammatory mediators, such as IL-6 and IL-1β, which may induce neuroinflammation under injury conditions [119] (Fig. 2). Notably, inflammasome activation is the reason for the release of EVs [120,121,122]. Inflammatory cascade-related inflammasomes, such as NLRP3, cause pyroptosis and the release of inflammatory cytokines critical for further injury in TBI (Fig. 2). Kumar and his colleagues found that microglia-derived MVs containing elevated concentrations of proinflammatory molecules such as IL-1β were released into the circulation following TBI via NLRP3 activation, inducing a systemic response and enhancing robust neuroinflammatory responses in the injured brain through microglial cell activation and increased expression of proinflammatory molecules [123] (Fig. 2). Proinflammatory cytokines induced inflammatory cell infiltration and exerted a detrimental impact on neurons (Fig. 2). Hazelton et al. demonstrated that peripheral inflammation was induced by manipulating the circulating EV population with ‘primed’ EVs to exacerbate CNS injury [124]. Inflammatory lesions in the brain activate a systemic acute phase response (APR) dependent on the release of EVs into the circulation, ultimately resulting in the regulation of leukocyte mobilization and subsequent recruitment to the brain [124,125,126]. The characteristics of the response depend on the nature of the EVs. In addition, the EV load is protected from degradation, which makes these molecules perfect biomarkers of upstream events. Additionally, EV release induced by inflammasomes exerts functional effects on bystander cells. For example, EVs released via NLRP3 contain IFN-β protein, which induced interferon-stimulated genes (ISGs) in recipient macrophages and limited inflammasome responses in unprimed EV recipient macrophages [120]. This was an interesting finding. Evidence from the work of Kumar et al. shows that microglia-derived MVs containing elevated concentrations of proinflammatory molecules (e.g. IL-1β and miR-155) were released into the circulation following TBI, thereby inducing a systemic response and enhancing robust neuroinflammatory responses in the injured brain through microglial cell activation and increased expression of proinflammatory molecules [123]. Similarly, related research reported that platelet-inflammasome activation led to the generation of IL-1β- and caspase-1-carrying platelet EVs that bound neutrophils and promoted platelet-neutrophil aggregation in lung arterioles in SCD mice in vivo and in human blood via microfluidic assays performed in vitro [127]. Therefore, EVs that were generated by inflammasome signalling may transport inflammatory factors to target cells to further aggravate TBI. In addition, an encapsulated inflammasome may be transferred from an original cell to other cells, causing target cell death. Using immunoblot analysis, NLRP1 inflammasome proteins have been identified in exosomes derived from the CSF of patients with severe TBI [128]. NLRP1 has been previously demonstrated to be an activator of pyroptosis, and transport of the inflammasome may be a reason for injury to nearby healthy neurons [129]. Moreover, cells undergoing cell death under the impact of stress and ontological components produced by dead neurons may function as antigens to activate specific reactions (Fig. 2). These antigens are formed by cell fragments called damage-associated molecular patterns (DAMPs). Animal studies have shown that DAMPs, such as HMGB1 (high mobility group box-1 protein), are carried by EVs after TBI and function as inflammasome activators that contribute to the development of pulmonary dysfunction [64] (Fig. 2). Additionally, serum-derived EVs from patients with severe TBI contribute to acute lung injury by activating pyroptosis via an EV-mediated neural-respiratory inflammasome axis [64]. Due to their small size and lipid bilayer, EVs can pass through the disrupted BBB into the peripheral circulation and contribute to acute lung injury after TBI. To some extent, this finding reveals the possibility that EVs from injured brain cells may induce cell death in bystander cells. Previous studies [130, 131] identified that EVs secreted by bacteria converged in a ubiquitous mechanism contributing to inflammasome activation, but whether DAMPs in patients with TBI exhibit the same function remains unknown. To date, studies related to the pathology of TBI and EVs are rare, and future research is needed to further explore current challenges.

The effect of EVs on cell death after TBI. (a) Neuron experiencing cell death and rupture to release EVs containing DAMPs, which fuse with nearby neurons and induce pyroptosis. Microglial experiencing pyroptosis may release EVs containing cytokines and inflammasome to induce further neuroinflammation. (b) EVs produced from brain cells can be transported to other organs by the bloodstream. (c) Serum-derived EVs from TBI patients cause ALI
The Effects of EVs on Cell Death After SCI
Patients suffering a SCI use a wheelchair and suffer lifelong medical issues for more than 10 years. The yearly estimated rate of SCI is in a range from 250,000 to 500,000 individuals [132]. Thus, SCI destroys neurons, which places an economic and physiological burden on patients. SCI refers to dysfunctions in motor, sensing and autonomic functions, as it exerts a comprehensive effect on various cell types (astrocytes, neurons, oligodendrocytes, microglia etc.) [132]. To some extent, patients with SCI experience primary injuries and secondary injuries in the same manner as patients with TBI, and a complete understanding of cerebral trauma and ischaemia ensures that we can better understand secondary events following the primary injury [12]. Although numerous important studies on SCI have been reported, the pathophysiology underlying SCI is still not well characterized.
Recent studies have suggested that EVs transport parent cell-specific EVs capable of changing recipient cell function within and beyond the CNS, thereby affecting secondary injury progression [1, 133]. Notably, damaged cells, blood vessels and axons are capable of releasing toxic chemicals to attack intact neighbouring cells, resulting in injury or death of nearby cells [12]. We assume that EVs transport these toxic chemicals. For instance, glutamate is a normal neurotransmitter that is released from the presynaptic membrane and binds to receptors on target neurons to stimulate impulses [12]. However, glutamate from damaged spinal neurons, axons and astrocytes overexcites neighbouring neurons [134] (Fig. 3). Overflowed glutamate may be encapsulated in EVs and transferred to cells to change the membrane potential (Fig. 3). Interestingly, Gosselin et al. found that astrocyte-derived EVs derived from astrocytes carry the functional glutamate transporters EAAT-1 and EAAT-2, which are important for maintaining low concentrations of extracellular glutamate via glutamate reuptake to maintain glutamate homeostasis [134] (Fig. 3). In a mouse model, exosomes from neurons were directly internalized by astrocytes and increased the astrocyte miR-124a level, which increased the level of GLT-1 (an EAAT-1 analogue in mice) protein levels [1] (Fig. 3). Neuronal dysfunction may decrease the release rate of miR-124a exosomes, causing dysfunctional glutamate transport. Dysfunctional glutamate transport after SCI may cause neurotoxicity. Overexcited cells allow waves of calcium ions to enter, leading to the production of many free radicals, such as ROS (reactive oxygen species) [12] (Fig. 3). As destructive bioactive components, ROS are critical to numerous modes of cell death, such as ferroptosis and pyroptosis [135, 136]. Reactive astrocyte-derived EVs enriched in small GTPases, proteins and miRNAs decrease neurite outgrowth and spike firing rates to inhibit neuronal function and may lead to neuronal apoptosis [1, 10, 137, 138]. A study demonstrated that small EVs encapsulating CCL2 derived from activated astrocytes promoted neuronal apoptosis and aggravated inflammation by interacting with CCR2 (C-C motif receptor 2) on both neurons and microglia [139] (Fig. 3). Activated microglia release IL-1β, which not only induces inflammatory reactions but also exerts an impact on neuronal cells, thereby further aggravating neural apoptosis (Fig. 3). Neuroinflammation is another factor that contributes to the aggravation of SCI in secondary injury. Similar to TBI described in the literature, EVs isolated from human CSF of patients with SCI also contain inflammasomes [128]. This evidence suggests that inflammation may be linked with EVs and that EVs may function as signal transmission vehicles to activate pyroptosis. The nuclear factor NF-κB pathway is a typical proinflammatory signalling pathway that mainly depends on the effect of NF-κB on proinflammatory gene expression (chemokines, cytokines, adhesion molecules etc.) [140]. A study reported that exosomal miR-155 derived from M1-polarized macrophages contributed to the EndoMT (endothelial-to-mesenchymal transition) and increased the generation of mitochondrial ROS in a model of SCI and then targeted downstream suppressor of cytokine signalling 6 (SOCS6) and suppressed the p65 ubiquitination and degradation mediated by SOCS6, thereby activating the NF-κB signalling pathway [36]. NF-κB is a signalling protein upstream of NLRP3, which is broadly involved in pyroptosis in numerous diseases [86, 141, 142]. Liu et al. demonstrated that kaempferol was capable of downregulating the ROS-MAPK-NF-κB and pyroptosis signalling pathways, hence reducing oxidative stress and weakening the inflammatory response, which clarified the relationship between NF-κB and pyroptosis in neuroinflammation [143]. Therefore, we presume that EVs activate the NF-κB signalling pathway to trigger pyroptosis. However, recent papers have focused mainly on the potential of EVs to deliver therapeutic components and thus contribute to recovery after SCI, and few researchers have focused on the cell-specific signalling cargo function of EVs participating in the progression of secondary injury. Future studies are needed to examine EV-mediated crosstalk.

The effect of EVs on cell death after SCI. EVs containing miR-124a secreted from normal neurons are able to upregulate astrocyte GLT-1 expression in mice, and EVs loaded with GLT-1 can maintain glutamate homeostasis. Glutamate that floods out of injured spinal neurons overexcites neighbouring neurons, causing superfluous production of ROS and finally inducing ferroptosis and pyroptosis. EVs containing miR-155 from M1-polarized macrophages induce ROS production in neurons, which can cause ferroptosis and pyroptosis. EVs encapsulating CCL2 from activated astrocytes promote neuronal apoptosis and aggravate inflammation by interacting with CCR2 on microglia and neurons
Therapeutic Insights into EVs in the Context of CNS Trauma
Potential Cell Sources of Therapeutic EVs
As described in previous studies, EVs can profoundly facilitate the regulation of factors involved in SCI, which helps in gaining a deeper understanding of the pathophysiology of SCI as well as in developing various corresponding treatment strategies. EVs are emerging as feasible candidates for the cell-to-cell transfer of bioactive contents to target cells. Despite the poor understanding of the different functional properties by EVs or the mechanism of action associated with parental cell type, diverse EV-related cargoes suggest a positive association between pleiotropic, complementary and/or synergistic effects and treatment results [1]. These findings reveal that the cell source of EVs may be a significant standard to consider during the development of EV-based treatment strategies specific to SCI and TBI.
MSCs are widely used to analyse therapies. MSCs are characterized by their differentiation potential, colony forming ability and self-renewal capacity [144]. MSCs are derived from adipose tissue, human umbilical cord blood, bone marrow etc. Initially, after injury, MSCs migrate to the damage site, undergo engraftment and differentiate into the cells needed for tissue regeneration, thereby exerting a therapeutic effect [145]. However, recent studies have revealed that MSCs may also repair damaged tissue by secreting bioactive contents, such as growth factors, cytokines and EVs [146, 147]. Notably, EVs are promising nanocarriers used for drug delivery and targeted therapy and may replace direct stem cell transplantation treatment. Researchers have shown that MSC-derived exosomes support tissue regeneration specific to many central nervous syndrome diseases, such as stroke, spinal cord injury, TBI, Parkinson’s disease and Alzheimer’s disease [31, 148,149,150,151]. Given that transplantation of MSCs has potential risks, such as the formation of tumours, EVs derived from MSCs may be regarded as feasible replacements of these cells [152]. It is easy to isolate EVs from MSCs of different origins, and these EVs carry biologically active molecules capable of being transferred to target cells where they can play roles in treatment, such as tissue injury regeneration, inflammatory response inhibition and immune system modulation. Additionally, MSC-derived EVs are involved in the regulation of cell death after CNS trauma, which makes them suitable therapy carriers for injury repair (Table 1).
Another widely used EV-derived cell type in research is the neural stem cell (NSC). As the cells of CNS origin, NSCs are capable of self-renewing and generating neurons and glia during mammalian CNS development [165]. According to some studies, NSC transplantation can ameliorate neuroinflammation and enhance neuronal plasticity and cell replacement [166,167,168]. However, stem cell transplantation exhibits the disadvantages of a low survival rate, tendency to dedifferentiate, immune rejection and formation of malignant tumours [152, 169]. Therefore, researchers have focused on EVs secreted from NSCs, and NSC exosomes are capable of protecting neuronal function and accelerating neurocognitive impairment and repair of SCI [153, 170]. NSCs have been extensively researched in the context of CNS injury, and EVs from NSCs show the potential to promote neurologic recovery by regulating cell death (Table 1). To some extent, EVs are dependent on the cell type of their origin, and NSCs share a close relationship with neurons; therefore, features of the cells of origin may contribute to EV localization to target cells. Additionally, the functions of NSC exosomes are similar to those of transplanted NSCs, which solves the limitations of direct stem cell transplantation, making NSCs perfect sources of EVs.
Macrophages are innate immune cells involved in CNS injury. Macrophages infiltrating damaged tissue exhibit functions that differ from those of resident microglia and can mediate beneficial and detrimental effects following injury. Under the influence of cytokines, infiltrated monocytes terminally differentiate into macrophages, which are classified into proinflammatory M1-like macrophages and anti-inflammatory M2-like macrophages [171]. As described in previous studies, M1-polarized macrophages induce the EndoMT while damaging mitochondrial function after spinal cord injury, which means that the persistence of M1 macrophages may aggravate the damage and impede cell regeneration [36]. In contrast, M2-like macrophages are capable of promoting cell proliferation and tissue growth, and EVs carrying a combination of nerve growth factor from M2 macrophages and curcumin promoted significant motor function recovery after SCI [172, 173]. EVs derived from M2 BMDMs (bone marrow-derived macrophages) or peripheral macrophages were able to increase autophagy and promote recovery after SCI [162, 163] (Table 1). These studies indicate that EVs from M2-like macrophages are potential therapeutic carriers in the context of CNS trauma. Some other cell types are also involved in CNS disease, but the scope of their therapeutic application is narrow. EVs derived from Schwann cells increased sciatic nerve axonal sprouting and remyelination and thus were successfully used to treat rodent peripheral neuropathy [174]. The Schwann cells derived from skin precursors activated the AKT/mTOR/p70S6K signalling pathway to enhance axonal outgrowth and motoneuron regeneration in an oxygen-glucose deprivation (OGD) model [175]. Astrocytes, which are broadly distributed in brain tissue, have also been proven to be effective source of carriers in cell death [155,156,157] (Table 1). Even neuron-derived exosomes protected traumatically injured spinal cord via the inhibition of M1 microglia and astrocyte activation in vivo and in vitro [176].
Altogether, different cell source-derived EVs are effective in regulating cell death, such as autophagy, apoptosis, pyroptosis and ferroptosis, and promote injury repair, but few studies have established a positive role for EVs in organ injury by inhibiting necroptosis (Table 1). In some pathological processes, EVs promote necroptosis and exacerbate tissue damage [58, 105]. Further research is needed to determine whether EVs derived from MSCs can alleviate damage by inhibiting necroptosis. In the future, studies are needed to compare the treatment effect and transport efficiency among EVs from these cell sources and then select the best cell type to use to generate EVs loaded with therapeutic cargo. It is also necessary to explore more efficient methods for EV separation and purification.
Therapeutic Use of EVs as Cargo Carriers
As perfect cargo carriers, EVs not only can carry therapeutic compounds but can also easily pass through the blood–brain barrier due to their lipid solubility. And receptors on EVs make it easier to cross blood–brain barrier or blood–spinal cord barrier by recognizing specific protein while direct drug delivery lacks such advantage. One study showed that exosomes derived from brain cells that expressed brain-specific surface proteins can cross the blood–brain barrier and deliver drugs to the other side [177]. This unique characteristic suggests new avenues for CNS trauma treatment. Our review includes recent studies on the application of EVs in TBI and SCI to elucidate the feasibility of using EVs in CNS trauma therapy by inhibiting cell death. As effective gene-regulating factors, microRNAs are the most widely explored noncoding RNAs in exosomes, and they were found to be capable of targeting the 3′ untranslated region (UTR) of specific mRNAs to inhibit their translation in TBI models [178]. MicroRNAs are capable of modifying the recipient cell phenotype or physiology by modulating cellular processes related to proliferation, differentiation and cell death [179]. We list the roles of some microRNAs enriched in EVs and applied to TBI treatment in Table 2. MiR-873a-5p, the major component of exosomes derived from astrocytes, suppressed the NF-κB signalling pathway to mediate microglial phenotype modulation, thereby attenuating the neuroinflammation mediated by microglia and enhancing neurological deficits following TBI [180]. Similarly, Exo-miR-124 treatment suppressed the TLR4 pathway, thereby facilitating microglial M2 polarization as well as enhancing hippocampal neurogenesis and functional recovery after brain injury [191]. After TBI, the inhibition of neuronal inflammation mediated by miR-124-3p in microglial exosomes promoted neuron outgrowth [181]. However, neuron-derived exosomes that contained high levels of miR-21-5p triggered M1 microglial polarization; increased the levels of neuroinflammatory factors such as TNF-α, IL-6 and IL-β; and induced neuronal apoptosis [182]. These results revealed that regulation of the polarization of M1/M2 microglia may be of great importance to inflammation control and functional recovery after TBI [192]. The treatment effect exhibited by miR-17-92 cluster-enriched exosomes derived from human bone MSC was much more robust, attenuating neuroinflammation, reducing the number of cells lost and enhancing angiogenesis and neurogenesis to greatly increase brain functional recovery [183]. Given that neuroinflammation is related to pyroptosis and necroptosis, miRNAs may be particularly useful to target genes involved in pyroptosis and necroptosis. Additionally, previous studies reported the downregulation of miR-212-5p in EVs after TBI, and the transfection of overexpressed miR-212-5p targeted Ptgs2 to reduce the rate of ferroptotic neuronal death, which demonstrated that miR-212-5p may be a suitable cargo to block ferroptosis after TBI [193, 194]. In addition, miR-124-3p- or miR-21-5p-enriched exosomes conferred neuroprotection by inhibiting autophagy [184, 195]. MicroRNAs exert the same effects on SCI, as described in Table 2. Interestingly, astrocyte-derived EVs carrying NF-κB-interacting lncRNA (NKILA) competitively bound to miR-195 and upregulated nucleotide-binding leucine-rich repeat that contained family member X1 (NLRX1) to reduce neuronal injury after TBI and further inhibit cell apoptosis [185]. Moreover, lncRNA MALAT1 derived from adipose-derived stem cells (hASCs) exerted a regulatory effect on mRNA and ncRNA expression during the inflammatory response and apoptosis and thus increased cell survival, MAPK pathway signalling and gene transcription [186]. LncRNAs (long noncoding RNAs) function through miRNA ‘sponges’ via miRNA response elements (MREs) to influence the expression of downstream target genes [196]. LncRNAs are thus excellent cargoes that impede the function of miRNAs that exacerbate injury during pathophysiological progression after CNS trauma. Exosomal miR-155 from M1-polarized macrophages impaired mitochondrial function after SCI; therefore, we can acquire a specific lncRNA sequence, load it onto EVs and transfer it to injured cells to silence related miRNAs and promote functional recovery. However, few studies have identified the miRNAs carried by EVs that aggravate disease progression after TBI, and future studies are needed to further explore this field and identify key targets of lncRNAs.
Many studies have also investigated the methods of EV cargo modification by specific drug loading (Table 2). These drugs may lead to severe adverse reactions during systemic administration or be impeded from reaching target cells. EV injection may be an alternative way to locally release EVs into a damaged area. The advantages of EVs as vehicles to deliver drugs include their weak or none cytotoxic effects, small size enabling penetration deep into tissues, ability to be cleared rapidly via the mononuclear or reticuloendothelial system and capability to penetrate the blood–brain barrier [197]. Chen et al. demonstrated that FTY720-NSC-Exos exerted a positive treatment in the context of SCI via PTEN/AKT pathway regulation [187]. FTY720 is a functional antagonist of sphingosine 1-phosphate receptor-1 (S1P1) and shows the ability to effectively inhibit the inflammatory response, neuronal apoptosis and spinal cord oedema [187]. Some growth factors have been proven to be effective in promoting functional recovery after SCI, and loading growth factors into EVs may lead to a better curative effect [198, 199]. Han et al. demonstrated that TGF-β in EVs shows the potential to promote the differentiation of NSCs into neurons and recovery after SCI. Although previous studies have confirmed that TGF-β functions as cytokine against inflammation, inhibiting TGF-β expression in an early stage of injury may lead to destructive inflammation, and the neuronal apoptosis rate in adjacent injury lesions be increased. TGF-β in MSC-derived EVs applied in the early stage of injury upregulated Smad 6 expression in NSCs, and Smad 6 regulates negative feedback by inhibiting BMP (bone morphogenetic protein)/Smad 1/5/8 signalling, which can decrease cell death after neuronal injury [188]. In addition, NGF engineered EVs accumulated precisely at the injury site in SCI model mice to increase the level of matrix metalloproteinase 9 (MMP9), which inhibits the inflammatory cascade response to reduce neuronal cell apoptosis and protect the spinal cord from secondary damage [172]. The results showed that EVs exerted a better curative effect than therapies delivered via direct administration. The key question is how to deliver EVs to an injured region after EVs fuse with injured cells. Intranasal delivery can effectively target EVs to access CNS tissues [150, 197]. As shown in an ischaemic stroke model, according to in vivo CT imaging of EVs labelled with gold nanoparticles, intranasal EV administration led to better EV accumulation than intravenous injection [200]. There may be an association between the migratory mechanisms underlying intranasal EV transport to CNS tissues and direct transport across the epithelial cell layer into the circulatory system and transport along the olfactory nerve [197]. We hope that future studies continue to explore the best method of EV delivery.
Acquisition of Therapeutic EVs
How to obtain targeted EVs carrying therapeutic genes or proteins has been a focus of EV therapy. Most of the previous experiments acquired high-throughput noncoding RNA-expressing EVs via target gene overexpression and then isolated purified EVs via ultracentrifugation [201, 202] (Fig. 4). However, a given molecule overexpressed in a cell may lead to unpredictable consequences, ultimately interfering with EV biogenesis. Thus, a great deal of research has been focused on finding a new strategy to acquire target EVs, and these strategies have mainly involved bioengineering approaches. Native EVs are characterized by low levels of accumulation in specific organs and tissues, difficulty in characterizing EV cargo and low expression levels of target-active contents, but engineering approaches may overcome these defects and be used for developing superior EVs for CNS trauma applications [203]. Membrane-permeabilizing strategies, such as electroporation (of both nucleic acids and drugs), heat shock or freeze-thaw procedures, detergent treatment and sonication, are applied for loading cells with exogenous material, and they have been broadly used in the EV field with certain success [203,204,205] (Fig. 4). Additionally, engineering approaches can be used to modify the surface molecules on EV membranes, which can lead to changes in the affinity of EVs, thereby remodelling EV biodistribution and targeting them to accumulate in injured tissues or organs (Fig. 4). Modifying EVs with polyethylene glycol (PEG) enhances EV stability in circulation and increases their uptake by specific cells [206]. Alternatively, modification of EV membranes with specific proteins or peptides via viral transfection to modify gene expression or using chemical approaches is a strategy for increasing selective and effective organ or tissue targeting [203]. However, genetic approaches may change the biological activity of the EVs because of genetic manipulation, and chemical modification is difficult to implement. The modification of EV surfaces requires further research.

Therapeutic methods of EVs as cargo carrier. EVs can obtain from MSCs, NSCs, macrophage etc. And then, there are two ways to acquire EVs containing effective contents: overexpressing the target gene or bioengineering approaches like electroporation, freeze-thaw procedures and sonication. Next, modifying the surface of EVs to better accumulated in injured tissues or organs through chemical or genetic approaches. Finally, administration of EVs to distribute in the target region
Conclusions and Perspectives
This review describes the biogenesis and contents of EVs, the relationships between EVs and cell death, the pathophysiologic mechanisms of EVs action in the context of CNS trauma and the potential therapeutic application of EVs. Herein, we analyse the possibilities of applying EVs to patients with CNS trauma by regulating cell death. The pathological process underlying CNS trauma is very complex and is a result of multiple factors, which may be related to EVs. By analysing recent literature, we found a close relationship between EVs and cell death. EVs are able to induce cell death, and EVs carry substances that are released after cell death, such as death via autophagy, pyroptosis, necroptosis, apoptosis and ferroptosis, and exert an impact on the cell microenvironment. In addition, the activity of EVs can be observed in CNS trauma, and EVs function as bridges connecting different cells with cell death modalities. Furthermore, EVs obtained from different cells, such as MSCs and NSCs, are capable of inhibiting cell death in the context of CNS trauma, and EVs carrying specific cell death inhibitors via bioengineering or chemical modification are effective in promoting recovery after CNS trauma. Neuronal death with poor cell regeneration is closely related to irrecoverable function after CNS trauma. Therefore, understanding the pathological mechanism underlying EVs and cell death may lead to the development of better treatments for patients with CNS trauma.
However, multiple obstacles are waiting to be overcome before EV therapy can be applied in the clinic. Selecting suitable cells from which to extract EVs is extremely important for EV-based therapy. MSCs and NSCs have been the most widely used stem cells in recent studies. Comparing the therapeutic effects of EVs among different cells and choosing appropriate cell sources for EV production are becoming the priorities of future work. Finding new standardized isolation and purification methods is another challenge for the clinical application of EVs. The perfect separation method of EVs that can be used for mass production will benefit clinical research and drug development. Moreover, how EVs are precisely localized to a lesion is unknown. Changing the receptors on the surface of EVs to make them more likely to accumulate in a lesion seems to be achievable via bioengineering approaches. Notably, genetic approaches carry a risk of affecting the biological activity, and chemical approaches are difficult to implement; therefore, bioengineering approaches to modify EV surfaces need to be improved. In addition to bioengineering technology, biomaterials can be applied to repair injury after CNS trauma. Biomaterials such as hydrogel scaffolds are used to load therapeutic drugs, and they are able to promote functional recovery after CNS trauma. Therefore, EVs or EV-produced cells can adhere to the same scaffolds and be targeted to the injured area. This manual positioning method seems simple and feasible; therefore, research into this approach may be an effective way in localizing EVs to the lesion area. Solving the problem of EV distribution will contribute to the best curative effect. Research on EVs proves that the selection of biologically relevant EVs as therapeutic cargo will aid in the development of next-generation therapeutic methods targeting CNS-specific cell death pathways and inducing functional regeneration following CNS trauma.
Data Availability
Not applicable
Abbreviations
- ATG:
-
Autophagy-related protein
- BBB:
-
Blood–brain barrier
- BMDM:
-
Bone morrow-derived macrophage
- CNS:
-
Central nervous system;
- CSF:
-
Cerebrospinal fluid
- DAMPs:
-
Damage-associated molecular patterns
- ERK:
-
Extracellular signal-regulated kinase
- ESCRT:
-
Endosomal sorting complexes required for transport
- EVs:
-
Extracellular vesicles
- GTPase:
-
Rab guanosine triphosphatase
- ILVs:
-
Intraluminal vesicles
- lncRNA:
-
Long noncoding RNA
- MLKL:
-
Mixed lineage kinase domain-like
- MREs:
-
MicroRNA response elements
- MSC:
-
Mesenchymal stem cell
- MVBs:
-
Multivesicular bodies
- MVs:
-
Microvesicles
- NGF:
-
Nerve growth factor
- NLRP:
-
NOD-like receptor protein
- NSC:
-
Neural stem cell
- OGD:
-
Oxygen-glucose deprivation
- PLD:
-
Phospholipase D
- PS:
-
Phosphatidylserine
- RIPK1:
-
Receptor-interacting protein kinase 1
- RIPK3:
-
Receptor-interacting protein kinase 3
- ROS:
-
Reactive oxygen species
- SCI:
-
Spinal cord injury
- SOCS6:
-
Cytokine signalling 6
- TBI:
-
Traumatic brain injury
- UTR:
-
3′ Untranslated region
References
Dutta D, Khan N, Wu J, Jay SM (2021) Extracellular vesicles as an emerging frontier in spinal cord injury pathobiology and therapy. Trends Neurosci 44(6):492–506. https://doi.org/10.1016/j.tins.2021.01.003
Yates AG, Anthony DC, Ruitenberg MJ, Couch Y (2019) Systemic Immune response to traumatic cns injuries-are extracellular vesicles the missing link? Front Immunol 10:2723. https://doi.org/10.3389/fimmu.2019.02723
Alizadeh A, Dyck SM, Karimi-Abdolrezaee S (2019) Traumatic spinal cord injury: an overview of pathophysiology, models and acute injury mechanisms. Front Neurol 10:282. https://doi.org/10.3389/fneur.2019.00282
Pearn ML, Niesman IR, Egawa J, Sawada A, Almenar-Queralt A, Shah SB, Duckworth JL, Head BP (2017) Pathophysiology associated with traumatic brain injury: current treatments and potential novel therapeutics. Cell Mol Neurobiol 37(4):571–585. https://doi.org/10.1007/s10571-016-0400-1
Sanwlani R, Gangoda L (2021) Role of extracellular vesicles in cell death and inflammation. Cells 10(10). https://doi.org/10.3390/cells10102663
van Niel G, D’Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19(4):213–228. https://doi.org/10.1038/nrm.2017.125
Abels ER, Breakefield XO (2016) Introduction to extracellular vesicles: biogenesis, RNA cargo selection, content, release, and uptake. Cell Mol Neurobiol 36(3):301–312. https://doi.org/10.1007/s10571-016-0366-z
Kang T, Atukorala I, Mathivanan S (2021) Biogenesis of extracellular vesicles. Subcell Biochem 97:19–43. https://doi.org/10.1007/978-3-030-67171-6_2
Théry C, Zitvogel L, Amigorena S (2002) Exosomes: composition, biogenesis and function. Nat Rev Immunol 2(8):569–579. https://doi.org/10.1038/nri855
Dickens AM, Tovar YRLB, Yoo SW, Trout AL, Bae M, Kanmogne M, Megra B, Williams DW et al (2017) Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci Signal 10(473). https://doi.org/10.1126/scisignal.aai7696
Shi Z, Yuan S, Shi L, Li J, Ning G, Kong X, Feng S (2021) Programmed cell death in spinal cord injury pathogenesis and therapy. Cell Prolif 54(3):e12992. https://doi.org/10.1111/cpr.12992
McDonald JW, Sadowsky C (2002) Spinal-cord injury. Lancet (London, England) 359(9304):417–425. https://doi.org/10.1016/s0140-6736(02)07603-1
Al Mamun A, Wu Y, Monalisa I, Jia C, Zhou K, Munir F, Xiao J (2021) Role of pyroptosis in spinal cord injury and its therapeutic implications. J Adv Res 28:97–109. https://doi.org/10.1016/j.jare.2020.08.004
Luo C, Tao L (2020) The function and mechanisms of autophagy in traumatic brain injury. Adv Exp Med Biol 1207:635–648. https://doi.org/10.1007/978-981-15-4272-5_46
Minciacchi VR, Freeman MR, Di Vizio D (2015) Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol 40:41–51. https://doi.org/10.1016/j.semcdb.2015.02.010
Nabhan JF, Hu R, Oh RS, Cohen SN, Lu Q (2012) Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc Natl Acad Sci U S A 109(11):4146–4151. https://doi.org/10.1073/pnas.1200448109
Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, Simpson RJ (2012) Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods (San Diego, Calif) 56(2):293–304. https://doi.org/10.1016/j.ymeth.2012.01.002
Zhang H, Wang L, Li C, Yu Y, Yi Y, Wang J, Chen D (2019) Exosome-induced regulation in inflammatory bowel disease. Front Immunol 10:1464. https://doi.org/10.3389/fimmu.2019.01464
Grant BD, Donaldson JG (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10(9):597–608. https://doi.org/10.1038/nrm2755
Stoorvogel W, Strous GJ, Geuze HJ, Oorschot V, Schwartz AL (1991) Late endosomes derive from early endosomes by maturation. Cell 65(3):417–427. https://doi.org/10.1016/0092-8674(91)90459-c
Doyle LM, Wang MZ (2019) Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 8(7). https://doi.org/10.3390/cells8070727
Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD (2002) Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3(2):271–282. https://doi.org/10.1016/s1534-5807(02)00220-4
de Gassart A, Géminard C, Hoekstra D, Vidal M (2004) Exosome secretion: the art of reutilizing nonrecycled proteins? Traffic (Copenhagen, Denmark) 5(11):896–903. https://doi.org/10.1111/j.1600-0854.2004.00223.x
He C, Zheng S, Luo Y, Wang B (2018) Exosome theranostics: biology and translational medicine. Theranostics 8(1):237–255. https://doi.org/10.7150/thno.21945
Maacha S, Bhat AA, Jimenez L, Raza A, Haris M, Uddin S, Grivel JC (2019) Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol Cancer 18(1):55. https://doi.org/10.1186/s12943-019-0965-7
Akers JC, Gonda D, Kim R, Carter BS, Chen CC (2013) Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neuro-Oncol 113(1):1–11. https://doi.org/10.1007/s11060-013-1084-8
Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G (2016) Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging 8(4):603–619. https://doi.org/10.18632/aging.100934
Kakarla R, Hur J, Kim YJ, Kim J, Chwae YJ (2020) Apoptotic cell-derived exosomes: messages from dying cells. Exp Mol Med 52(1):1–6. https://doi.org/10.1038/s12276-019-0362-8
Park SJ, Kim JM, Kim J, Hur J, Park S, Kim K, Shin HJ, Chwae YJ (2018) Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc Natl Acad Sci U S A 115(50):E11721–E11730. https://doi.org/10.1073/pnas.1811432115
Atkin-Smith GK, Tixeira R, Paone S, Mathivanan S, Collins C, Liem M, Goodall KJ, Ravichandran KS et al (2015) A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat Commun 6:7439. https://doi.org/10.1038/ncomms8439
Haney MJ, Klyachko NL, Zhao Y, Gupta R, Plotnikova EG, He Z, Patel T, Piroyan A et al (2015) Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Controlled Release : Official Journal of the Controlled Release Society 207:18–30. https://doi.org/10.1016/j.jconrel.2015.03.033
Qiu G, Zheng G, Ge M, Wang J, Huang R, Shu Q, Xu J (2019) Functional proteins of mesenchymal stem cell-derived extracellular vesicles. Stem Cell Res Ther 10(1):359. https://doi.org/10.1186/s13287-019-1484-6
Savina A, Vidal M, Colombo MI (2002) The exosome pathway in K562 cells is regulated by Rab11. J Cell Sci 115(Pt 12):2505–2515. https://doi.org/10.1242/jcs.115.12.2505
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO (2020) RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol 21(10):585–606. https://doi.org/10.1038/s41580-020-0251-y
Zhu S, Yao F, Qiu H, Zhang G, Xu H, Xu J (2018) Coupling factors and exosomal packaging microRNAs involved in the regulation of bone remodelling. Biological reviews of the Cambridge Philosophical Society 93(1):469–480. https://doi.org/10.1111/brv.12353
Ge X, Tang P, Rong Y, Jiang D, Lu X, Ji C, Wang J, Huang C et al (2021) Exosomal miR-155 from M1-polarized macrophages promotes EndoMT and impairs mitochondrial function via activating NF-κB signaling pathway in vascular endothelial cells after traumatic spinal cord injury. Redox Biol 41:101932. https://doi.org/10.1016/j.redox.2021.101932
Hsu CC, Huang CC, Chien LH, Lin MT, Chang CP, Lin HJ, Chio CC (2020) Ischemia/reperfusion injured intestinal epithelial cells cause cortical neuron death by releasing exosomal microRNAs associated with apoptosis, necroptosis, and pyroptosis. Sci Rep 10(1):14409. https://doi.org/10.1038/s41598-020-71310-5
Joyce DP, Kerin MJ, Dwyer RM (2016) Exosome-encapsulated microRNAs as circulating biomarkers for breast cancer. Int J Cancer 139(7):1443–1448. https://doi.org/10.1002/ijc.30179
Liu S, Chen J, Shi J, Zhou W, Wang L, Fang W, Zhong Y, Chen X et al (2020) M1-like macrophage-derived exosomes suppress angiogenesis and exacerbate cardiac dysfunction in a myocardial infarction microenvironment. Basic Res Cardiol 115(2):22. https://doi.org/10.1007/s00395-020-0781-7
Teng F, Fussenegger M (2020) Shedding light on extracellular vesicle biogenesis and bioengineering. Adv Sci (Weinheim, Baden-Wurttemberg, Germany) 8(1):2003505. https://doi.org/10.1002/advs.202003505
Levine B, Kroemer G (2019) Biological functions of autophagy genes: a disease perspective. Cell 176(1-2):11–42. https://doi.org/10.1016/j.cell.2018.09.048
Pleet ML, Branscome H, DeMarino C, Pinto DO, Zadeh MA, Rodriguez M, Sariyer IK, El-Hage N et al (2018) Autophagy, EVs, and infections: a perfect question for a perfect time. Front Cell Infect Microbiol 8:362. https://doi.org/10.3389/fcimb.2018.00362
Shao N, Xue L, Wang R, Luo K, Zhi F, Lan Q (2019) miR-454-3p is an exosomal biomarker and functions as a tumor suppressor in glioma. Mol Cancer Ther 18(2):459–469. https://doi.org/10.1158/1535-7163.mct-18-0725
Colletti M, Ceglie D, Di Giannatale A, Nazio F (2020) Autophagy and exosomes relationship in cancer: friends or foes? Front Cell Dev Biol 8:614178. https://doi.org/10.3389/fcell.2020.614178
Ni Z, Kuang L, Chen H, Xie Y, Zhang B, Ouyang J, Wu J, Zhou S et al (2019) The exosome-like vesicles from osteoarthritic chondrocyte enhanced mature IL-1β production of macrophages and aggravated synovitis in osteoarthritis. Cell Death Dis 10(7):522. https://doi.org/10.1038/s41419-019-1739-2
Keller MD, Ching KL, Liang FX, Dhabaria A, Tam K, Ueberheide BM, Unutmaz D, Torres VJ et al (2020) Decoy exosomes provide protection against bacterial toxins. Nature 579(7798):260–264. https://doi.org/10.1038/s41586-020-2066-6
Kuang Y, Zheng X, Zhang L, Ai X, Venkataramani V, Kilic E, Hermann DM, Majid A et al (2020) Adipose-derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR-25. J Extracell Vesicles 10(1):e12024. https://doi.org/10.1002/jev2.12024
Hirsova P, Ibrahim SH, Verma VK, Morton LA, Shah VH, LaRusso NF, Gores GJ, Malhi H (2016) Extracellular vesicles in liver pathobiology: small particles with big impact. Hepatology (Baltimore, Md) 64(6):2219–2233. https://doi.org/10.1002/hep.28814
Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, Cooper SA, Cao S et al (2020) Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol 73(5):1144–1154. https://doi.org/10.1016/j.jhep.2020.04.044
Khoury MK, Gupta K, Franco SR, Liu B (2020) Necroptosis in the pathophysiology of disease. Am J Pathol 190(2):272–285. https://doi.org/10.1016/j.ajpath.2019.10.012
Raden Y, Shlomovitz I, Gerlic M (2021) Necroptotic extracellular vesicles - present and future. Semin Cell Dev Biol 109:106–113. https://doi.org/10.1016/j.semcdb.2020.08.011
Gong YN, Guy C, Crawford JC, Green DR (2017) Biological events and molecular signaling following MLKL activation during necroptosis. Cell cycle (Georgetown, Tex) 16(19):1748–1760. https://doi.org/10.1080/15384101.2017.1371889
Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR (2017) ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 169(2):286–300.e216. https://doi.org/10.1016/j.cell.2017.03.020
Yoon S, Kovalenko A, Bogdanov K, Wallach D (2017) MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 47(1):51–65.e57. https://doi.org/10.1016/j.immuni.2017.06.001
Shlomovitz I, Erlich Z, Arad G, Edry-Botzer L, Zargarian S, Cohen H, Manko T, Ofir-Birin Y et al (2021) Proteomic analysis of necroptotic extracellular vesicles. Cell Death Dis 12(11):1059. https://doi.org/10.1038/s41419-021-04317-z
Abed J, Emgård JE, Zamir G, Faroja M, Almogy G, Grenov A, Sol A, Naor R et al (2016) Fap2 mediates fusobacterium nucleatum colorectal adenocarcinoma enrichment by binding to tumor-expressed Gal-GalNAc. Cell Host Microbe 20(2):215–225. https://doi.org/10.1016/j.chom.2016.07.006
Pocanschi CL, Apell HJ, Puntervoll P, Høgh B, Jensen HB, Welte W, Kleinschmidt JH (2006) The major outer membrane protein of Fusobacterium nucleatum (FomA) folds and inserts into lipid bilayers via parallel folding pathways. J Mol Biol 355(3):548–561. https://doi.org/10.1016/j.jmb.2005.10.060
Liu L, Liang L, Yang C, Zhou Y, Chen Y (2021) Extracellular vesicles of Fusobacterium nucleatum compromise intestinal barrier through targeting RIPK1-mediated cell death pathway. Gut Microbes 13(1):1–20. https://doi.org/10.1080/19490976.2021.1902718
Yuan X, Li D, Chen X, Han C, Xu L, Huang T, Dong Z, Zhang M (2017) Extracellular vesicles from human-induced pluripotent stem cell-derived mesenchymal stromal cells (hiPSC-MSCs) protect against renal ischemia/reperfusion injury via delivering specificity protein (SP1) and transcriptional activating of sphingosine kinase 1 and inhibiting necroptosis. Cell Death Dis 8(12):3200. https://doi.org/10.1038/s41419-017-0041-4
Shi J, Gao W, Shao F (2017) Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42(4):245–254. https://doi.org/10.1016/j.tibs.2016.10.004
Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, Rathinam VAK (2016) Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165(5):1106–1119. https://doi.org/10.1016/j.cell.2016.04.015
Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458(7242):1191–1195. https://doi.org/10.1038/nature07830
Chavez L, Meguro J, Chen S, de Paiva VN, Zambrano R, Eterno JM, Kumar R, Duncan MR et al (2021) Circulating extracellular vesicles activate the pyroptosis pathway in the brain following ventilation-induced lung injury. J Neuroinflammation 18(1):310. https://doi.org/10.1186/s12974-021-02364-z
Kerr NA, de Rivero Vaccari JP, Umland O, Bullock MR, Conner GE, Dietrich WD, Keane RW (2019) Human lung cell pyroptosis following traumatic brain injury. Cells 8(1). https://doi.org/10.3390/cells8010069
Xu X, Lai Y, Hua ZC (2019) Apoptosis and apoptotic body: disease message and therapeutic target potentials. Biosci Rep 39(1). https://doi.org/10.1042/bsr20180992
Phan TK, Ozkocak DC, Poon IKH (2020) Unleashing the therapeutic potential of apoptotic bodies. Biochem Soc Trans 48(5):2079–2088. https://doi.org/10.1042/bst20200225
Mesa KR, Rompolas P, Zito G, Myung P, Sun TY, Brown S, Gonzalez DG, Blagoev KB et al (2015) Niche-induced cell death and epithelial phagocytosis regulate hair follicle stem cell pool. Nature 522(7554):94–97. https://doi.org/10.1038/nature14306
Liu J, Qiu X, Lv Y, Zheng C, Dong Y, Dou G, Zhu B, Liu A et al (2020) Apoptotic bodies derived from mesenchymal stem cells promote cutaneous wound healing via regulating the functions of macrophages. Stem Cell Res Ther 11(1):507. https://doi.org/10.1186/s13287-020-02014-w
Liu D, Kou X, Chen C, Liu S, Liu Y, Yu W, Yu T, Yang R et al (2018) Circulating apoptotic bodies maintain mesenchymal stem cell homeostasis and ameliorate osteopenia via transferring multiple cellular factors. Cell Res 28(9):918–933. https://doi.org/10.1038/s41422-018-0070-2
Ma Q, Liang M, Wu Y, Ding N, Duan L, Yu T, Bai Y, Kang F et al (2019) Mature osteoclast-derived apoptotic bodies promote osteogenic differentiation via RANKL-mediated reverse signaling. J Biol Chem 294(29):11240–11247. https://doi.org/10.1074/jbc.RA119.007625
Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, Li B, Fan BY, Wang X, Li WX, Fu XH, Hu Y, Liu C, Kong XH, Feng SQ (2019) Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen Res 14(3):532–541. https://doi.org/10.4103/1673-5374.245480
Jiang X, Stockwell BR, Conrad M (2021) Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22(4):266–282. https://doi.org/10.1038/s41580-020-00324-8
Zhang X, Xu Y, Ma L, Yu K, Niu Y, Xu X, Shi Y, Guo S, Xue X, Wang Y, Qiu S, Cui J, Wang H, Tian X, Miao Y, Meng F, Qiao Y, Yu Y, Wang J (2022) Essential roles of exosome and circRNA_101093 on ferroptosis desensitization in lung adenocarcinoma. Cancer communications (London, England) 42(4):287–313. https://doi.org/10.1002/cac2.12275
Wu S, Li T, Liu W, Huang Y (2021) Ferroptosis and Cancer: Complex Relationship and Potential Application of Exosomes. Front Cell Dev Biol 9:733751. https://doi.org/10.3389/fcell.2021.733751
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, Zhang Q, Lin D, Ge S, Bai M, Wang X, Zhang L, Li H, Yang Y, Ji Z, Wang H, Ying G, Ba Y (2020) CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer 19(1):43. https://doi.org/10.1186/s12943-020-01168-8
Zhao X, Si L, Bian J, Pan C, Guo W, Qin P, Zhu W, Xia Y, Zhang Q, Wei K (2022) Adipose tissue macrophage-derived exosomes induce ferroptosis via glutathione synthesis inhibition by targeting SLC7A11 in obesity-induced cardiac injury. Free Radic Biol Med 182:232–245. https://doi.org/10.1016/j.freeradbiomed.2022.02.033
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, Baer CE, Dixon SJ, Mercurio AM (2019) Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev Cell 51(5):575–586.e574. https://doi.org/10.1016/j.devcel.2019.10.007
Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, Zeh HJ, Kang R, Wang J, Tang D (2020) Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 16(11):2069–2083. https://doi.org/10.1080/15548627.2020.1714209
Taylor CA, Bell JM, Breiding MJ, Xu L (2017) Traumatic brain injury-related emergency department visits, hospitalizations, and deaths - United States, 2007 and 2013. MMWR Surveill Summ (Washington, DC : 2002) 66(9):1–16. https://doi.org/10.15585/mmwr.ss6609a1
Zusman BE, Kochanek PM, Jha RM (2020) Cerebral edema in traumatic brain injury: a historical framework for current therapy. Curr Treat Options Neurol 22:(3). https://doi.org/10.1007/s11940-020-0614-x
Lu J, Ashwell KW, Waite P (2000) Advances in secondary spinal cord injury: role of apoptosis. Spine 25(14):1859–1866
Song B, Zhou T, Liu J, Shao L (2016) Involvement of programmed cell death in neurotoxicity of metallic nanoparticles: recent advances and future perspectives. Nanoscale Res Lett 11(1):484. https://doi.org/10.1186/s11671-016-1704-2
Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS (2001) Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 24(5):254–264. https://doi.org/10.1097/00002826-200109000-00002
Zhou K, Sansur CA, Xu H, Jia X (2017) The temporal pattern, flux, and function of autophagy in spinal cord injury. Int J Mol Sci 18(2). https://doi.org/10.3390/ijms18020466
Feng Z, Min L, Liang L, Chen B, Chen H, Zhou Y, Deng W, Liu H et al (2021) Neutrophil extracellular traps exacerbate secondary injury via promoting neuroinflammation and blood-spinal cord barrier disruption in spinal cord injury. Front Immunol 12:698249. https://doi.org/10.3389/fimmu.2021.698249
Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C, Zou L, Ding R et al (2020) Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation 17(1):90. https://doi.org/10.1186/s12974-020-01751-2
Hao J, Li B, Duan HQ, Zhao CX, Zhang Y, Sun C, Pan B, Liu C et al (2017) Mechanisms underlying the promotion of functional recovery by deferoxamine after spinal cord injury in rats. Neural Regen Res 12(6):959–968. https://doi.org/10.4103/1673-5374.208591
Liao HY, Wang ZQ, Ran R, Zhou KS, Ma CW, Zhang HH (2021) Biological functions and therapeutic potential of autophagy in spinal cord injury. Front Cell Dev Biol 9:761273. https://doi.org/10.3389/fcell.2021.761273
Zhou K, Zheng Z, Li Y, Han W, Zhang J, Mao Y, Chen H, Zhang W et al (2020) TFE3, a potential therapeutic target for spinal cord injury via augmenting autophagy flux and alleviating ER stress. Theranostics 10(20):9280–9302. https://doi.org/10.7150/thno.46566
Yuan J, Amin P, Ofengeim D (2019) Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20(1):19–33. https://doi.org/10.1038/s41583-018-0093-1
Akamatsu Y, Hanafy KA (2020) Cell death and recovery in traumatic brain injury. Neurotherapeutics : J Am Soc Experimental NeuroTherapeutics 17(2):446–456. https://doi.org/10.1007/s13311-020-00840-7
Yin Y, Sun G, Li E, Kiselyov K, Sun D (2017) ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res Rev 34:3–14. https://doi.org/10.1016/j.arr.2016.08.008
Gao C, Yan Y, Chen G, Wang T, Luo C, Zhang M, Chen X, Tao L (2020) Autophagy activation represses pyroptosis through the IL-13 and JAK1/STAT1 pathways in a mouse model of moderate traumatic brain injury. ACS Chem Neurosci 11(24):4231–4239. https://doi.org/10.1021/acschemneuro.0c00517
Liu ZM, Chen QX, Chen ZB, Tian DF, Li MC, Wang JM, Wang L, Liu BH, Zhang SQ, Li F, Ye H, Zhou L (2018) RIP3 deficiency protects against traumatic brain injury (TBI) through suppressing oxidative stress, inflammation and apoptosis: Dependent on AMPK pathway. Biochem Biophys Res Commun 499(2):112–119. https://doi.org/10.1016/j.bbrc.2018.02.150
Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, Lipinski MM (2018) Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis 9(5):476. https://doi.org/10.1038/s41419-018-0469-1
Hu X, Xu Y, Zhang H, Li Y, Wang X, Xu C, Ni W, Zhou K (2022) Role of necroptosis in traumatic brain and spinal cord injuries. J Adv Res 40:125–134. https://doi.org/10.1016/j.jare.2021.12.002
Xu X, Yin D, Ren H, Gao W, Li F, Sun D, Wu Y, Zhou S, Lyu L, Yang M, Xiong J, Han L, Jiang R, Zhang J (2018) Selective NLRP3 inflammasome inhibitor reduces neuroinflammation and improves long-term neurological outcomes in a murine model of traumatic brain injury. Neurobiol Dis 117:15–27. https://doi.org/10.1016/j.nbd.2018.05.016
Zhang H, Ni W, Yu G, Geng Y, Lou J, Qi J, Chen Y, Li F, Ye H, Ma H, Xu H, Zhao L, Cai Y, Wang X, Xu H, Xiao J, Zhou K (2023) 3,4-Dimethoxychalcone, a caloric restriction mimetic, enhances TFEB-mediated autophagy and alleviates pyroptosis and necroptosis after spinal cord injury. Theranostics 13 (2):810–832. https://doi.org/10.7150/thno.78370
Liao HY, Wang ZQ, Ran R, Zhou KS, Ma CW, Zhang HH (2021) Biological Functions and Therapeutic Potential of Autophagy in Spinal Cord Injury. Front Cell Dev Biol 9:761273. https://doi.org/10.3389/fcell.2021.761273
Zhang L, Wang H (2018) Autophagy in Traumatic Brain Injury: A New Target for Therapeutic Intervention. Front Mol Neurosci 11:190. https://doi.org/10.3389/fnmol.2018.00190
Wong J, Hoe NW, Zhiwei F, Ng I (2005) Apoptosis and traumatic brain injury. Neurocrit Care 3(2):177–182. https://doi.org/10.1385/ncc:3:2:177
Xu G, Zhang J, Wang L, Cui Z, Sun X, Liu Z, Zhu Z, Qiu Y (2017) Up-regulation of TRAF2 Suppresses Neuronal Apoptosis after Rat Spinal Cord Injury. Tissue & cell 49(5):589–596. https://doi.org/10.1016/j.tice.2017.08.002
Feng Z, Min L, Chen H, Deng W, Tan M, Liu H, Hou J (2021) Iron overload in the motor cortex induces neuronal ferroptosis following spinal cord injury. Redox Biol 43:101984. https://doi.org/10.1016/j.redox.2021.101984
Xie BS, Wang YQ, Lin Y, Mao Q, Feng JF, Gao GY, Jiang JY (2019) Inhibition of ferroptosis attenuates tissue damage and improves long-term outcomes after traumatic brain injury in mice. CNS Neurosci Ther 25(4):465–475. https://doi.org/10.1111/cns.13069
Gupta K, Brown KA, Hsieh ML, Hoover BM, Wang J, Khoury MK, Pilli VSS, Beyer RSH, Voruganti NR, Chaudhary S, Roberts DS, Murphy RM, Hong S, Ge Y, Liu B (2022) Necroptosis is associated with Rab27-independent expulsion of extracellular vesicles containing RIPK3 and MLKL. J Extracell Vesicles 11(9):e12261. https://doi.org/10.1002/jev2.12261
Khellaf A, Khan DZ, Helmy A (2019) Recent advances in traumatic brain injury. J Neurol 266(11):2878–2889. https://doi.org/10.1007/s00415-019-09541-4
Galgano M, Toshkezi G, Qiu X, Russell T, Chin L, Zhao LR (2017) Traumatic brain injury: current treatment strategies and future endeavors. Cell Transplant 26(7):1118–1130. https://doi.org/10.1177/0963689717714102
Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB (2014) Transcranial amelioration of inflammation and cell death after brain injury. Nature 505(7482):223–228. https://doi.org/10.1038/nature12808
Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H (2014) Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma 31(23):1927–1933. https://doi.org/10.1089/neu.2013.3168
Kuharić J, Grabušić K, Tokmadžić VS, Štifter S, Tulić K, Shevchuk O, Lučin P, Šustić A (2019) Severe traumatic brain injury induces early changes in the physical properties and protein composition of intracranial extracellular vesicles. J Neurotrauma 36(2):190–200. https://doi.org/10.1089/neu.2017.5515
Beard K, Meaney DF, Issadore D (2020) Clinical applications of extracellular vesicles in the diagnosis and treatment of traumatic brain injury. J Neurotrauma 37(19):2045–2056. https://doi.org/10.1089/neu.2020.6990
Chen CC, Liu L, Ma F, Wong CW, Guo XE, Chacko JV, Farhoodi HP, Zhang SX et al (2016) Elucidation of exosome migration across the blood-brain barrier model in vitro. Cell Mol Bioeng 9(4):509–529. https://doi.org/10.1007/s12195-016-0458-3
van Niel G, Raposo G, Candalh C, Boussac M, Hershberg R, Cerf-Bensussan N, Heyman M (2001) Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 121(2):337–349. https://doi.org/10.1053/gast.2001.26263
Beard K, Yang Z, Haber M, Flamholz M, Diaz-Arrastia R, Sandsmark D, Meaney DF, Issadore D (2021) Extracellular vesicles as distinct biomarker reservoirs for mild traumatic brain injury diagnosis. Brain Comm 3(3):fcab151. https://doi.org/10.1093/braincomms/fcab151
Saman S, Kim W, Raya M, Visnick Y, Miro S, Saman S, Jackson B, McKee AC et al (2012) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287(6):3842–3849. https://doi.org/10.1074/jbc.M111.277061
Kondo K, Maruishi M, Ueno H, Sawada K, Hashimoto Y, Ohshita T, Takahashi T, Ohtsuki T et al (2010) The pathophysiology of prospective memory failure after diffuse axonal injury--lesion-symptom analysis using diffusion tensor imaging. BMC Neurosci 11:147. https://doi.org/10.1186/1471-2202-11-147
Werner JK, Stevens RD (2015) Traumatic brain injury: recent advances in plasticity and regeneration. Curr Opin Neurol 28(6):565–573. https://doi.org/10.1097/wco.0000000000000265
Kabadi SV, Faden AI (2014) Neuroprotective strategies for traumatic brain injury: improving clinical translation. Int J Mol Sci 15(1):1216–1236. https://doi.org/10.3390/ijms15011216
Mondello S, Thelin EP, Shaw G, Salzet M, Visalli C, Cizkova D, Kobeissy F, Buki A (2018) Extracellular vesicles: pathogenetic, diagnostic and therapeutic value in traumatic brain injury. Expert Rev Proteomics 15(5):451–461. https://doi.org/10.1080/14789450.2018.1464914
Budden CF, Gearing LJ, Kaiser R, Standke L, Hertzog PJ, Latz E (2021) Inflammasome-induced extracellular vesicles harbour distinct RNA signatures and alter bystander macrophage responses. J Extracell Vesicles 10(10):e12127. https://doi.org/10.1002/jev2.12127
Sarkar S, Rokad D, Malovic E, Luo J, Harischandra DS, Jin H, Anantharam V, Huang X et al (2019) Manganese activates NLRP3 inflammasome signaling and propagates exosomal release of ASC in microglial cells. Sci Signal 12(563). https://doi.org/10.1126/scisignal.aat9900
Qu Y, Franchi L, Nunez G, Dubyak GR (2007) Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol 179(3):1913–1925. https://doi.org/10.4049/jimmunol.179.3.1913
Kumar A, Stoica BA, Loane DJ, Yang M, Abulwerdi G, Khan N, Kumar A, Thom SR et al (2017) Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J Neuroinflammation 14(1):47. https://doi.org/10.1186/s12974-017-0819-4
Hazelton I, Yates A, Dale A, Roodselaar J, Akbar N, Ruitenberg MJ, Anthony DC, Couch Y (2018) Exacerbation of acute traumatic brain injury by circulating extracellular vesicles. J Neurotrauma 35(4):639–651. https://doi.org/10.1089/neu.2017.5049
Campbell SJ, Zahid I, Losey P, Law S, Jiang Y, Bilgen M, van Rooijen N, Morsali D et al (2008) Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology 55(5):780–787. https://doi.org/10.1016/j.neuropharm.2008.06.074
Campbell SJ, Hughes PM, Iredale JP, Wilcockson DC, Waters S, Docagne F, Perry VH, Anthony DC (2003) CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J 17(9):1168–1170. https://doi.org/10.1096/fj.02-0757fje
Vats R, Brzoska T, Bennewitz MF, Jimenez MA, Pradhan-Sundd T, Tutuncuoglu E, Jonassaint J, Gutierrez E et al (2020) Platelet extracellular vesicles drive inflammasome-IL-1β-dependent lung injury in sickle cell disease. Am J Respir Crit Care Med 201(1):33–46. https://doi.org/10.1164/rccm.201807-1370OC
de Rivero Vaccari JP, Brand F 3rd, Adamczak S, Lee SW, Perez-Barcena J, Wang MY, Bullock MR, Dietrich WD et al (2016) Exosome-mediated inflammasome signaling after central nervous system injury. J Neurochem 136(Suppl 1):39–48. https://doi.org/10.1111/jnc.13036
Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM (2019) Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol 40(11):1035–1052. https://doi.org/10.1016/j.it.2019.09.005
Zhao P, Cao L, Wang X, Dong J, Zhang N, Li X, Li J, Zhang X et al (2021) Extracellular vesicles secreted by Giardia duodenalis regulate host cell innate immunity via TLR2 and NLRP3 inflammasome signaling pathways. PLoS Negl Trop Dis 15(4):e0009304. https://doi.org/10.1371/journal.pntd.0009304
Wang X, Eagen WJ, Lee JC (2020) Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc Natl Acad Sci U S A 117(6):3174–3184. https://doi.org/10.1073/pnas.1915829117
Anjum A, Yazid MD, Fauzi Daud M, Idris J, Ng AMH, Selvi Naicker A, Ismail OHR, Athi Kumar RK et al (2020) Spinal cord injury: pathophysiology, multimolecular interactions, and underlying recovery mechanisms. Int J Mol Sci 21(20). https://doi.org/10.3390/ijms21207533
Rufino-Ramos D, Albuquerque PR, Carmona V, Perfeito R, Nobre RJ, Pereira de Almeida L (2017) Extracellular vesicles: novel promising delivery systems for therapy of brain diseases. J Control Release 262:247–258. https://doi.org/10.1016/j.jconrel.2017.07.001
Upadhya R, Zingg W, Shetty S, Shetty AK (2020) Astrocyte-derived extracellular vesicles: neuroreparative properties and role in the pathogenesis of neurodegenerative disorders. J Control Release 323:225–239. https://doi.org/10.1016/j.jconrel.2020.04.017
Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F (2020) Ferroptosis and cancer: mitochondria meet the “Iron Maiden” cell death. Cells 9(6). https://doi.org/10.3390/cells9061505
Luo Z, Xu X, Sho T, Zhang J, Xu W, Yao J, Xu J (2019) ROS-induced autophagy regulates porcine trophectoderm cell apoptosis, proliferation, and differentiation. Am J Physiol Cell Physiol 316(2):C198–c209. https://doi.org/10.1152/ajpcell.00256.2018
You Y, Borgmann K, Edara VV, Stacy S, Ghorpade A, Ikezu T (2020) Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J Extracell Vesicles 9(1):1706801. https://doi.org/10.1080/20013078.2019.1706801
Datta Chaudhuri A, Dasgheyb RM, DeVine LR, Bi H, Cole RN, Haughey NJ (2020) Stimulus-dependent modifications in astrocyte-derived extracellular vesicle cargo regulate neuronal excitability. Glia 68(1):128–144. https://doi.org/10.1002/glia.23708
Rong Y, Ji C, Wang Z, Ge X, Wang J, Ye W, Tang P, Jiang D et al (2021) Small extracellular vesicles encapsulating CCL2 from activated astrocytes induce microglial activation and neuronal apoptosis after traumatic spinal cord injury. J Neuroinflammation 18(1):196. https://doi.org/10.1186/s12974-021-02268-y
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651. https://doi.org/10.1101/cshperspect.a001651
Zhao W, Ma L, Cai C, Gong X (2019) Caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-κB and A2aR signaling in LPS-induced THP-1 macrophages. Int J Biol Sci 15(8):1571–1581. https://doi.org/10.7150/ijbs.34211
An Y, Zhang H, Wang C, Jiao F, Xu H, Wang X, Luan W, Ma F et al (2019) Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J 33(11):12515–12527. https://doi.org/10.1096/fj.201802805RR
Liu Z, Yao X, Sun B, Jiang W, Liao C, Dai X, Chen Y, Chen J et al (2021) Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radic Biol Med 168:142–154. https://doi.org/10.1016/j.freeradbiomed.2021.03.037
Smirnov SV, Harbacheuski R, Lewis-Antes A, Zhu H, Rameshwar P, Kotenko SV (2007) Bone-marrow-derived mesenchymal stem cells as a target for cytomegalovirus infection: implications for hematopoiesis, self-renewal and differentiation potential. Virology 360(1):6–16. https://doi.org/10.1016/j.virol.2006.09.017
Keshtkar S, Azarpira N, Ghahremani MH (2018) Mesenchymal stem cell-derived extracellular vesicles: novel frontiers in regenerative medicine. Stem Cell Res Ther 9(1):63. https://doi.org/10.1186/s13287-018-0791-7
Madrigal M, Rao KS, Riordan NH (2014) A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Transl Med 12:260. https://doi.org/10.1186/s12967-014-0260-8
Phinney DG, Pittenger MF (2017) Concise review: MSC-derived exosomes for cell-free therapy. Stem Cells (Dayton, Ohio) 35(4):851–858. https://doi.org/10.1002/stem.2575
Xin H, Li Y, Cui Y, Yang JJ, Zhang ZG, Chopp M (2013) Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. J Cereb Blood Flow Metab 33(11):1711–1715. https://doi.org/10.1038/jcbfm.2013.152
Reza-Zaldivar EE, Hernández-Sapiéns MA, Minjarez B, Gutiérrez-Mercado YK, Márquez-Aguirre AL, Canales-Aguirre AA (2018) Potential effects of MSC-derived exosomes in neuroplasticity in Alzheimer’s disease. Front Cell Neurosci 12:317. https://doi.org/10.3389/fncel.2018.00317
Guo S, Perets N, Betzer O, Ben-Shaul S, Sheinin A, Michaelevski I, Popovtzer R, Offen D et al (2019) Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano 13(9):10015–10028. https://doi.org/10.1021/acsnano.9b01892
Williams AM, Dennahy IS, Bhatti UF, Halaweish I, Xiong Y, Chang P, Nikolian VC, Chtraklin K et al (2019) Mesenchymal stem cell-derived exosomes provide neuroprotection and improve long-term neurologic outcomes in a Swine model of traumatic brain injury and hemorrhagic shock. J Neurotrauma 36(1):54–60. https://doi.org/10.1089/neu.2018.5711
Jeong JO, Han JW, Kim JM, Cho HJ, Park C, Lee N, Kim DW, Yoon YS (2011) Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ Res 108(11):1340–1347. https://doi.org/10.1161/circresaha.110.239848
Rong Y, Liu W, Wang J, Fan J, Luo Y, Li L, Kong F, Chen J et al (2019) Neural stem cell-derived small extracellular vesicles attenuate apoptosis and neuroinflammation after traumatic spinal cord injury by activating autophagy. Cell Death Dis 10(5):340. https://doi.org/10.1038/s41419-019-1571-8
Lawson A, Snyder W, Peeples ES (2022) Intranasal administration of extracellular vesicles mitigates apoptosis in a mouse model of neonatal hypoxic-ischemic brain injury. Neonatology 119:1–9. https://doi.org/10.1159/000522644
Pei X, Li Y, Zhu L, Zhou Z (2019) Astrocyte-derived exosomes suppress autophagy and ameliorate neuronal damage in experimental ischemic stroke. Exp Cell Res 382(2):111474. https://doi.org/10.1016/j.yexcr.2019.06.019
Chen W, Wang H, Zhu Z, Feng J, Chen L (2020) Exosome-shuttled circSHOC2 from IPASs regulates neuronal autophagy and ameliorates ischemic brain injury via the miR-7670-3p/SIRT1 axis. Mol Ther Nucleic Acids 22:657–672. https://doi.org/10.1016/j.omtn.2020.09.027
Pei X, Li Y, Zhu L, Zhou Z (2020) Astrocyte-derived exosomes transfer miR-190b to inhibit oxygen and glucose deprivation-induced autophagy and neuronal apoptosis. Cell Cycle 19(8):906–917. https://doi.org/10.1080/15384101.2020.1731649
Chen Y, Li J, Ma B, Li N, Wang S, Sun Z, Xue C, Han Q et al (2020) MSC-derived exosomes promote recovery from traumatic brain injury via microglia/macrophages in rat. Aging 12(18):18274–18296. https://doi.org/10.18632/aging.103692
Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, Ge Z (2021) Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol 37(1):51–64. https://doi.org/10.1007/s10565-020-09530-8
Lin F, Chen W, Zhou J, Zhu J, Yao Q, Feng B, Feng X, Shi X et al (2022) Mesenchymal stem cells protect against ferroptosis via exosome-mediated stabilization of SLC7A11 in acute liver injury. Cell Death Dis 13(3):271. https://doi.org/10.1038/s41419-022-04708-w
Hu Z, Yuan Y, Zhang X, Lu Y, Dong N, Jiang X, Xu J, Zheng D (2021) Human umbilical cord mesenchymal stem cell-derived exosomes attenuate oxygen-glucose deprivation/reperfusion-induced microglial pyroptosis by promoting FOXO3a-dependent mitophagy. Oxidative Med Cell Longev 2021:6219715. https://doi.org/10.1155/2021/6219715
Zhang B, Lin F, Dong J, Liu J, Ding Z, Xu J (2021) Peripheral macrophage-derived exosomes promote repair after spinal cord injury by inducing local anti-inflammatory type microglial polarization via increasing autophagy. Int J Biol Sci 17(5):1339–1352. https://doi.org/10.7150/ijbs.54302
Wang J, Rong Y, Ji C, Lv C, Jiang D, Ge X, Gong F, Tang P et al (2020) MicroRNA-421-3p-abundant small extracellular vesicles derived from M2 bone marrow-derived macrophages attenuate apoptosis and promote motor function recovery via inhibition of mTOR in spinal cord injury. J Nanobiotechnol 18(1):72. https://doi.org/10.1186/s12951-020-00630-5
Huang J, Cao H, Cui B, Ma X, Gao L, Yu C, Shen F, Yang X, Liu N, Qiu A, Cai G, Zhuang S (2022) Mesenchymal Stem Cells-Derived Exosomes Ameliorate Ischemia/Reperfusion Induced Acute Kidney Injury in a Porcine Model. Front Cell Dev Biol 10:899869. https://doi.org/10.3389/fcell.2022.899869
Merkle FT, Alvarez-Buylla A (2006) Neural stem cells in mammalian development. Curr Opin Cell Biol 18(6):704–709. https://doi.org/10.1016/j.ceb.2006.09.008
Xiong LL, Hu Y, Zhang P, Zhang Z, Li LH, Gao GD, Zhou XF, Wang TH (2018) Neural stem cell transplantation promotes functional recovery from traumatic brain injury via brain derived neurotrophic factor-mediated neuroplasticity. Mol Neurobiol 55(3):2696–2711. https://doi.org/10.1007/s12035-017-0551-1
Fukuoka T, Kato A, Hirano M, Ohka F, Aoki K, Awaya T, Adilijiang A, Sachi M et al (2021) Neurod4 converts endogenous neural stem cells to neurons with synaptic formation after spinal cord injury. iScience 24(2):102074. https://doi.org/10.1016/j.isci.2021.102074
De Gioia R, Biella F, Citterio G, Rizzo F, Abati E, Nizzardo M, Bresolin N, Comi GP et al (2020) Neural stem cell transplantation for neurodegenerative diseases. Int J Mol Sci 21(9). https://doi.org/10.3390/ijms21093103
Koprivec S, Novak M, Bernik S, Voga M, Mohorič L, Majdič G (2021) Treatment of cranial cruciate ligament injuries in dogs using a combination of tibial tuberosity advancement procedure and autologous mesenchymal stem cells/multipotent mesenchymal stromal cells - a pilot study. Acta Vet Hung 68(4):405–412. https://doi.org/10.1556/004.2020.00063
Li WY, Zhu QB, Jin LY, Yang Y, Xu XY, Hu XY (2021) Exosomes derived from human induced pluripotent stem cell-derived neural progenitor cells protect neuronal function under ischemic conditions. Neural Regen Res 16(10):2064–2070. https://doi.org/10.4103/1673-5374.308665
Milich LM, Ryan CB, Lee JK (2019) The origin, fate, and contribution of macrophages to spinal cord injury pathology. Acta Neuropathol 137(5):785–797. https://doi.org/10.1007/s00401-019-01992-3
Zhang C, Li D, Hu H, Wang Z, An J, Gao Z, Zhang K, Mei X et al (2021) Engineered extracellular vesicles derived from primary M2 macrophages with anti-inflammatory and neuroprotective properties for the treatment of spinal cord injury. J Nanobiotechnol 19(1):373. https://doi.org/10.1186/s12951-021-01123-9
Mills CD, Shearer J, Evans R, Caldwell MD (1992) Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol 149(8):2709–2714
Wang L, Chopp M, Szalad A, Lu X, Zhang Y, Wang X, Cepparulo P, Lu M et al (2020) Exosomes derived from Schwann cells ameliorate peripheral neuropathy in type 2 diabetic mice. Diabetes 69(4):749–759. https://doi.org/10.2337/db19-0432
Wu X, Wang L, Cong M, Shen M, He Q, Ding F, Shi H (2020) Extracellular vesicles from skin precursor-derived Schwann cells promote axonal outgrowth and regeneration of motoneurons via Akt/mTOR/p70S6K pathway. Ann Transl Med 8(24):1640. https://doi.org/10.21037/atm-20-5965
Jiang D, Gong F, Ge X, Lv C, Huang C, Feng S, Zhou Z, Rong Y et al (2020) Neuron-derived exosomes-transmitted miR-124-3p protect traumatically injured spinal cord by suppressing the activation of neurotoxic microglia and astrocytes. J Nanobiotechnol 18(1):105. https://doi.org/10.1186/s12951-020-00665-8
Yang T, Martin P, Fogarty B, Brown A, Schurman K, Phipps R, Yin VP, Lockman P, Bai S (2015) Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res 32(6):2003–2014. https://doi.org/10.1007/s11095-014-1593-y
Yang Y, Ye Y, Su X, He J, Bai W, He X (2017) MSCs-derived exosomes and neuroinflammation, neurogenesis and therapy of traumatic brain injury. Front Cell Neurosci 11:55. https://doi.org/10.3389/fncel.2017.00055
Cocucci E, Meldolesi J (2015) Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol 25(6):364–372. https://doi.org/10.1016/j.tcb.2015.01.004
Long X, Yao X, Jiang Q, Yang Y, He X, Tian W, Zhao K, Zhang H (2020) Astrocyte-derived exosomes enriched with miR-873a-5p inhibit neuroinflammation via microglia phenotype modulation after traumatic brain injury. J Neuroinflammation 17(1):89. https://doi.org/10.1186/s12974-020-01761-0
Huang S, Ge X, Yu J, Han Z, Yin Z, Li Y, Chen F, Wang H et al (2018) Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J 32(1):512–528. https://doi.org/10.1096/fj.201700673R
Yin Z, Han Z, Hu T, Zhang S, Ge X, Huang S, Wang L, Yu J et al (2020) Neuron-derived exosomes with high miR-21-5p expression promoted polarization of M1 microglia in culture. Brain Behav Immun 83:270–282. https://doi.org/10.1016/j.bbi.2019.11.004
Zhang Y, Zhang Y, Chopp M, Pang H, Zhang ZG, Mahmood A, Xiong Y (2021) MiR-17-92 Cluster-enriched exosomes derived from human bone marrow mesenchymal stromal cells improve tissue and functional recovery in rats after traumatic brain injury. J Neurotrauma 38(11):1535–1550. https://doi.org/10.1089/neu.2020.7575
Li D, Huang S, Zhu J, Hu T, Han Z, Zhang S, Zhao J, Chen F et al (2019) Exosomes from MiR-21-5p-increased neurons play a role in neuroprotection by suppressing Rab11a-mediated neuronal autophagy in vitro after traumatic brain injury. Med Sci Monit 25:1871–1885. https://doi.org/10.12659/msm.915727
He B, Chen W, Zeng J, Tong W, Zheng P (2021) Long noncoding RNA NKILA transferred by astrocyte-derived extracellular vesicles protects against neuronal injury by upregulating NLRX1 through binding to mir-195 in traumatic brain injury. Aging 13(6):8127–8145. https://doi.org/10.18632/aging.202618
Patel NA, Moss LD, Lee JY, Tajiri N, Acosta S, Hudson C, Parag S, Cooper DR et al (2018) Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J Neuroinflammation 15(1):204. https://doi.org/10.1186/s12974-018-1240-3
Chen J, Zhang C, Li S, Li Z, Lai X, Xia Q (2021) Exosomes derived from nerve stem cells loaded with FTY720 promote the recovery after spinal cord injury in rats by PTEN/AKT signal pathway. J Immunol Res 2021:8100298. https://doi.org/10.1155/2021/8100298
Han T, Song P, Wu Z, Xiang X, Liu Y, Wang Y, Fang H, Niu Y et al (2022) MSC secreted extracellular vesicles carrying TGF-beta upregulate Smad 6 expression and promote the regrowth of neurons in spinal cord injured rats. Stem Cell Rev Rep 18(3):1078–1096. https://doi.org/10.1007/s12015-021-10219-6
Jia X, Huang G, Wang S, Long M, Tang X, Feng D, Zhou Q (2021) Extracellular vesicles derived from mesenchymal stem cells containing microRNA-381 protect against spinal cord injury in a rat model via the BRD4/WNT5A axis. Bone Joint Res 10(5):328–339. https://doi.org/10.1302/2046-3758.105.bjr-2020-0020.r1
Jiang Z, Zhang J (2021) Mesenchymal stem cell-derived exosomes containing miR-145-5p reduce inflammation in spinal cord injury by regulating the TLR4/NF-κB signaling pathway. Cell Cycle 20(10):993–1009. https://doi.org/10.1080/15384101.2021.1919825
Yang Y, Ye Y, Kong C, Su X, Zhang X, Bai W, He X (2019) MiR-124 enriched exosomes promoted the m2 polarization of microglia and enhanced hippocampus neurogenesis after traumatic brain injury by inhibiting TLR4 pathway. Neurochem Res 44(4):811–828. https://doi.org/10.1007/s11064-018-02714-z
Yao X, Liu S, Ding W, Yue P, Jiang Q, Zhao M, Hu F, Zhang H (2017) TLR4 signal ablation attenuated neurological deficits by regulating microglial M1/M2 phenotype after traumatic brain injury in mice. J Neuroimmunol 310:38–45. https://doi.org/10.1016/j.jneuroim.2017.06.006
Xiao X, Jiang Y, Liang W, Wang Y, Cao S, Yan H, Gao L, Zhang L (2019) miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol Brain 12(1):78. https://doi.org/10.1186/s13041-019-0501-0
Harrison EB, Hochfelder CG, Lamberty BG, Meays BM, Morsey BM, Kelso ML, Fox HS, Yelamanchili SV (2016) Traumatic brain injury increases levels of miR-21 in extracellular vesicles: implications for neuroinflammation. FEBS open bio 6(8):835–846. https://doi.org/10.1002/2211-5463.12092
Li D, Huang S, Yin Z, Zhu J, Ge X, Han Z, Tan J, Zhang S et al (2019) Increases in miR-124-3p in microglial exosomes confer neuroprotective effects by targeting FIP200-mediated neuronal autophagy following traumatic brain injury. Neurochem Res 44(8):1903–1923. https://doi.org/10.1007/s11064-019-02825-1
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495(7441):333–338. https://doi.org/10.1038/nature11928
Guo S, Redenski I, Levenberg S (2021) Spinal cord repair: from cells and tissue engineering to extracellular vesicles. Cells 10(8). https://doi.org/10.3390/cells10081872
Sas AR, Carbajal KS, Jerome AD, Menon R, Yoon C, Kalinski AL, Giger RJ, Segal BM (2020) A new neutrophil subset promotes CNS neuron survival and axon regeneration. Nat Immunol 21(12):1496–1505. https://doi.org/10.1038/s41590-020-00813-0
Ohori Y, Yamamoto S, Nagao M, Sugimori M, Yamamoto N, Nakamura K, Nakafuku M (2006) Growth factor treatment and genetic manipulation stimulate neurogenesis and oligodendrogenesis by endogenous neural progenitors in the injured adult spinal cord. J Neurosci 26(46):11948–11960. https://doi.org/10.1523/jneurosci.3127-06.2006
Betzer O, Perets N, Angel A, Motiei M, Sadan T, Yadid G, Offen D, Popovtzer R (2017) In vivo neuroimaging of exosomes using gold nanoparticles. ACS Nano 11(11):10883–10893. https://doi.org/10.1021/acsnano.7b04495
Zhang D, Ni N, Wang Y, Tang Z, Gao H, Ju Y, Sun N, He X et al (2021) CircRNA-vgll3 promotes osteogenic differentiation of adipose-derived mesenchymal stem cells via modulating miRNA-dependent integrin α5 expression. Cell Death Differ 28(1):283–302. https://doi.org/10.1038/s41418-020-0600-6
Song Y, Zhang C, Zhang J, Jiao Z, Dong N, Wang G, Wang Z, Wang L (2019) Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics 9(8):2346–2360. https://doi.org/10.7150/thno.29945
de Abreu RC, Fernandes H, da Costa Martins PA, Sahoo S, Emanueli C, Ferreira L (2020) Native and bioengineered extracellular vesicles for cardiovascular therapeutics. Nat Rev Cardiol 17(11):685–697. https://doi.org/10.1038/s41569-020-0389-5
Fuhrmann G, Serio A, Mazo M, Nair R, Stevens MM (2015) Active loading into extracellular vesicles significantly improves the cellular uptake and photodynamic effect of porphyrins. J Control Release 205:35–44. https://doi.org/10.1016/j.jconrel.2014.11.029
Kalani A, Chaturvedi P, Kamat PK, Maldonado C, Bauer P, Joshua IG, Tyagi SC, Tyagi N (2016) Curcumin-loaded embryonic stem cell exosomes restored neurovascular unit following ischemia-reperfusion injury. Int J Biochem Cell Biol 79:360–369. https://doi.org/10.1016/j.biocel.2016.09.002
Kooijmans SAA, Fliervoet LAL, van der Meel R, Fens M, Heijnen HFG, PMP v BEH, Vader P, Schiffelers RM (2016) PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. J Control Release 224:77–85. https://doi.org/10.1016/j.jconrel.2016.01.009
Acknowledgements
Figures 1–3 were illustrated by using Figdraw (www.figdraw.com).
Funding
This work was funded by grants from the National Natural Science Foundation of China (No. 82072192 to Kailiang Zhou); Wenzhou Science and Technology Bureau Foundation (No. Y20210438 to Kailiang Zhou); Public Welfare Technology Application Research Project of Zhejiang Province (LGF20H150003 to Kailiang Zhou); and Zhejiang Provincial Natural Science Foundation (No. LY17H060009 to Wenfei Ni).
Author information
Authors and Affiliations
Contributions
Yituo Chen, Haojie Zhang and Xinli Hu searched and reviewed literature and drafted manuscript and revision; Wanta Cai and Liting Jiang discussed and revised the manuscript; Yongli Wang, Yanqing Wu and Xiangyang Wang provided critical comments. Kailiang Zhou and Wenfei Ni designed and formulated the review theme and revised and finalized the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
All authors consent for publication.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Y., Zhang, H., Hu, X. et al. Extracellular Vesicles: Therapeutic Potential in Central Nervous System Trauma by Regulating Cell Death. Mol Neurobiol 60, 6789–6813 (2023). https://doi.org/10.1007/s12035-023-03501-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03501-w