
Abstract
Angiogenesis is the growth of new capillaries from the preexisting blood vessels. Glioblastoma (GBM) tumors are highly vascularized tumors, and glioma growth depends on the formation of new blood vessels. Angiogenesis is a complex process involving proliferation, migration, and differentiation of vascular endothelial cells (ECs) under the stimulation of specific signals. It is controlled by the balance between its promoting and inhibiting factors. Various angiogenic factors and genes have been identified that stimulate glioma angiogenesis. Therefore, attention has been directed to anti-angiogenesis therapy in which glioma proliferation is inhibited by inhibiting the formation of new tumor vessels using angiogenesis inhibitory factors and drugs. Here, in this review, we highlight and summarize the various molecular mediators that regulate GBM angiogenesis with focus on recent clinical research on the potential of exploiting angiogenic pathways as a strategy in the treatment of GBM patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gliomas arising from the glial cells in the central nervous system of adult brain are the most common primary intracranial tumors and account for 70–80% of all brain tumors [1,2,3]. Based on the recent classification of central nervous system tumors, diffuse gliomas are categorized into four grades (I–IV) according to World Health Organization (WHO): diffuse astrocytoma (IDH mutant, WHO grade II), oligodendroglioma (IDH mutant, WHO grade II), oligoastrocytoma (IDH mutant, WHO grade II), anaplastic astrocytoma, anaplastic oligodendroglioma (IDH mutant, WHO grade II), oligoastrocytoma (IDH mutant, WHO grade III), and glioblastoma multiforme (GBM or IDH mutant WHO grade IV) [4,5,6]. Furthermore, among all glioma cases diagnosed, astrocytoma grade III and GBM is considered to be the most aggressive and highly invasive as they spread into other parts of the brain quickly [7]. In spite of the aggressive treatments that include surgery combined with radiation, chemotherapy [8], and biological therapy [9], glioblastoma tumors remain as an enormous therapeutic challenge with survival rates following diagnosis of 12 to 15 months with less than 3 to 5% of people surviving longer than 5 years [10]. GBM tumors are also highly vascular brain tumors with very poor prognosis [11, 12]. Several angiogenic receptors and factors are upregulated in GBM and stimulate angiogenesis signaling pathways through activating oncogenes and/or downregulating tumor suppressor genes [13]. In this review, we will review the basic mechanisms of various molecular signaling events that regulate GBM angiogenesis and explore the potential of targeting angiogenic signaling as a therapeutic strategy for brain tumor pathogenesis.
Angiogenesis in Normal Physiology and in Tumor Progression
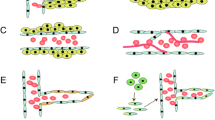
Physiological angiogenesis is a highly regulated process and is an essential one for the adequate supply of nutrients and oxygen to developing or healing tissues [14]. It is composed of many steps and is a combination of various components such as cells (endothelial cells and mural cells), soluble growth factors, proteolytic enzymes and adhesion proteins and matrix components (ECM) as shown in Fig. 1 [15, 16]. Hypoxia (low oxygen tension) is the main trigger which induce the activation of transcription factor, hypoxia-inducible factor-1 (HIF-1), which controls the expression of growth factors [17], matrix components [18, 19], adhesion molecules [20], and metabolic proteins [21]. The induction of angiogenesis relies on a balance between pro- and anti-angiogenic factors. Lack of oxygen in the cell simulates the release of pro-angiogenic growth factors like vascular endothelial growth factor (VEGF) [22], transforming growth factor-β (TGF-β) [23], fibroblast growth factors (FGFs) [24], angiopoietin-1 [25], and epidermal growth factor (EGF) [26]. These angiogenic factors bind to their receptors on the endothelial cell membrane resulting in the dissolution of the vessel wall and degradation of the endothelial cell basement membrane and extracellular matrix (ECM). Following the degradation of the basement membrane, specific proteases such as matrix metalloproteinases (MMPs) remodel the extracellular matrix components and a new matrix is synthesized by stromal cells which in turn foster the migration and proliferation of endothelial cells resulting in the formation of an endothelial tube-like structure [27]. Finally, a mature vascular basement membrane is formed around this newly formed the endothelial tube and the Mural cells (pericytes and smooth muscle cells) surrounding it resulting in a stable new vessel (Fig. 1) [28, 29].

Schematic representation of angiogenic events in GBM. (a) Angiogenesis processes are initiated by the angiogenic factors, which are being released from the GBM cells in the hypoxic tumor microenvironment. The major angiogenic factors are involved in GBM angiogenesis process which includes the VEGF, FGF, HIF1α, and Ang-1 and Ang-2. (b) These angiogenic factors bind to their receptors on endothelial cells and then start to initiate the endothelial cell proliferation and migration. During the endothelial cell proliferation and migration processes, the ECM start to degrade, and the endothelial cells are assembled into a tube/vessel-like structure. (c) The final step of GBM angiogenesis process is the maturation of the blood vessel wall, which is constructed by the recruitment of pericytes to cover the endothelial cells from its outside to form a new blood vessel formation
Angiogenesis is essential for tumor growth and progression. The tumor cells away from vessels experience hypoxia due to deficiency of blood and oxygen. Hypoxic environment induces cancer stem cells (CSCs) to differentiate toward endothelial progenitor cells and mature endothelium, which in turn generates new blood vessels inside the tumor. Tumors generate abnormal and functionally immature blood vessels due to deregulated factors such angiogenic growth factors, angiogenesis inhibitors, and other genetic factors by a process known as pathological angiogenesis [30]. Blood vessels developing in the primary tumor are larger than their normal counterparts and follow a criss-cross path, with irregular lumen diameters, dilated, highly permeable, and branch irregularly [31]. The tumor vasculature is also hyperpermeable to plasma and plasma proteins leading to local edema and extravascular clotting of plasma [32, 33]. This increase in the interstitial pressure in the tumor vasculature alters the blood flow and flux of leukocytes reaching the tumor site [34]. In addition, tumor cells can easily spread to the distant tissues due to the defective basement membrane and lack of normal perivascular connective tissue barrier [35]. Leakiness and compression of vessels leaves large volumes of tissue without blood flow in tumor and obstructs the delivery of blood-borne drugs, oxygen, and nutrients resulting in ischemia and necrotic regions within the tumor [36,37,38]. Ischemia leads to a hypoxic environment which in turn activates the HIF-1 resulting in new blood vessel formation [39]. Thus, the disorderly grown tumor vasculature observed in the tumors dramatically alters the tumor microenvironment and influences various aspects of tumor progression like tumor growth, allows easy penetration of the tumor cells and its ability to metastasize to distant sites, escape from the host immune system and response to anticancer therapies. Given the role of angiogenesis in tumor growth, targeting tumor vasculature and inhibition of growth factors/signaling pathways necessary for endothelial cell growth and proliferation is one of the practical approaches to inhibit tumor angiogenesis.
Factors Involved in Brain Tumor Angiogenesis
Brain tumor progression is closely associated with the formation of new vessels. Brain tumor angiogenesis is mediated through the action of many angiogenic factors, some of which are involved in normal angiogenesis (Fig. 2). The best-known angiogenesis regulators in GBM progression include VEGF, basic fibroblast growth factor (bFGF), hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), and TGF-β, MMPs, and angiopoietins (Angs). The expression levels of the angiogenic growth factors were shown to impact tumor progression. These angiogenic factors are upregulated by a variety of mechanisms like oncogene activation, loss of tumor suppressor gene function, and/or hypoxic microenvironments [40]. Moreover, fibroblast growth factor receptor (FGFR) modulates a series of angiogenic processes which includes FGF-mediated glioma endothelial cell migration and proliferation. In addition, FGFR plays an important role in the survival and angiogenesis of GBM cells through phosphatidylinositol 3-kinase (PI3K)/protein kinase B or AKT/mammalian target of rapamycin (mTOR) molecular signaling pathway [41,42,43,44,45]. FGF1, FGF2, and FGFR also activates the c-JUN/p38-MAPK pathway and STAT3/NF-κB signaling pathway; hence, all of these molecular signaling events are the most important events associated with GBM tumorigenesis, cell proliferation, migration, and angiogenesis [41,42,43,44,45] (Fig. 2). Previously, it has been reported that FGF2 is a prognostic biomarker of GBM patients [44]. All these different molecular effectors interact using various receptors equipped with tyrosine kinase activity on the endothelial cells membrane and transduce signals to activate multiple signaling pathways in GBM [46]. These signal transduction pathways regulate proliferation, migration, and differentiation of endothelial cells required for new vessel growth [47]. Moreover, the combination of VEGF-A with FGF-2 with/or without platelet-derived growth factor BB (PDGF-BB) [48], and that combination of FGF-2 and PDGF-BB [49] demonstrated synergistic effect in inducing neovascularization in vivo. The expression of these growth factors correlates with tumor progression with higher-grade tumors expressing higher levels of growth factors and their corresponding receptors when compared to low-grade tumors. Those factors that are well characterized in GBM neovascularization are summarized in Table 1 and are described below.

Angiogenic factors and receptors involved in GBM angiogenesis. VEGFR, PDGF, EGFR, and FGFR involved in the key molecular signaling events (RAS/RAF/MEK/MAPK signaling pathway and PI3K/AKT/mTOR signaling pathway) which plays an important role in glioma cells proliferation, migration, and survival
VEGF
Angiogenesis is fueled by several pro-angiogenic cytokines in malignant glioma, among which VEGF is the most important signaling molecule. This family of cytokines has six VEGF isoforms (VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor) [22]. VEGF-A is considered the main mediator in hypoxia-induced tumor growth. VEGF signaling is mediated through the receptor tyrosine kinases like VEGFR-1, VEGFR-2, and VEGFR-3 and mediates a variety of functions including pro-angiogenic activity, vascular permeability activity, and stimulate endothelial cell migration [50]. VEGF was shown to synergize with many growth factors and the effects of VEGF combinations with other factors exceeded those exerted by each factor alone in inducing angiogenesis [51, 52]. Binding of VEGF to its receptors on the endothelial cell membrane activates endothelial cells to secrete MMP into the surrounding tissue that are responsible for breakdown of ECM required for their proliferation and migration [53]. In addition, the combination of VEGF-A with FGF-2 or PDGF-BB was shown to have a potent synergistic effect in inducing angiogenesis in vitro and in vivo [48, 49].
VEGF plays an important role in the survival and proliferation of gliomas. VEGF mRNA expression was observed in low-grade gliomas with further upregulation in high-grade gliomas [54, 55]. Glioma formation occurs with the induction of VEGFR-1 mRNA in endothelial cells while progression toward malignancy is observed with the coordinated function of both the VEGFR-1/VEGFR-2 genes [56]. High levels of VEGF mRNA expression were observed in the necrotic regions in glioblastoma tumors [57, 58] which in turn promotes vascular proliferation and tumor progression of human glioblastoma [34, 59]. Overexpression of VEGF and VEGF-R1 in the low-grade astrocytomas was significantly associated with the same dismal prognosis as high-grade lesion, suggesting that VEGF and VEGFR expression can serve as a prognostic biomarker and provide useful information in determining the regime [60].
FGFs
FGF is another pro-angiogenic growth factor, which is both present the tumor cells and as well as stored in the vascular basement membrane for sustained release and is upregulated during angiogenesis. Two forms of FGFs, FGF-1 or acidic FGF (aFGF or FGF1) and FGF-2 or basic (bFGF or FGF2), bind most commonly to the receptor tyrosine kinases FGFR-1 or FGFR-2 [61]. FGF binding to its receptor activates signaling pathways mediated in part by protein kinase-C (PKC), phospholipase A2 [62], and increases endothelial cell migration and capillary formation promotes capillary morphogenesis [63]. FGF-2 also mediates proteolysis of matrix components and enhances the synthesis of collagen, fibronectin, and proteoglycans by endothelial cells demonstrating its effects on ECM remodeling during angiogenesis [63].
FGF-2 is implicated in brain tumor progression and localizes in the microvasculature as well as in the tumor cells in human gliomas [64,65,66]. bFGF levels correlate with the degree of glioma malignancy and vascularity as determined by immunohistochemical analysis [65]. It has previously shown that antibodies against bFGF were shown to inhibit glioma growth in vivo model and led to reduced blood vessel densities in glioma tumors of treated animals [67].
PDGF
PDGF family proteins are 45-kDa molecules and consist of four polypeptide chains (PDGF-A, PDGF-B (c-Sis form), PDGF-C, and PDGF-D) and were originally purified from platelets. All the PDGF family polypeptides have a highly conserved growth factor domain, called the PDGF/VEGF homology domain involved in forming bisulphite bridges to form the PDGF dimers PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD [68]. PDGF proteins regulate angiogenesis by binding to and activating two cell surface receptor tyrosine kinase (RTK) receptors, PDGFR-α and PDGFR-β, which leads to receptor dimerization, transphosphorylation, and subsequent activation of intracellular signaling pathways, such as PI3K/AKT and RAS/MAPK [69]. PDGF-B and PDGFR-β axis stimulates the proliferation of cultured smooth muscle cells and pericytes to the site of newly sprouting vessels and aids in establishing a new basement membrane [70]. In addition, PDGF-BB-induced erythropoietin (EPO), a hormone that stimulates erythropoiesis is elevated during tissue hypoxia through activation of the HIF-1α and promotes angiogenesis, vascular stability, and endothelial cell survival [71, 72]. Therefore, PDGF exert its pro-angiogenic effects by direct induction of endothelial cell proliferation and new vessel formation, and by endocrine stimulation of extramedullary hematopoiesis leading to increased oxygen perfusion and protection against tumor-induced hypoxia.
Several studies demonstrate that gliomas express all the PDGF ligands [73,74,75]. It was suggested that the growth factors produced by endothelial cells, such as PDGF-BB attract the glioma cells to the surrounding vasculature [76]. It was observed that the expression of PDGF ligand correlates with poor prognosis factors such as age at GBM diagnosis, phosphatase and tensin homolog deletion (PTEN), and isocitrate dehydrogenase 1 (IDH1) mutation in glioblastoma patients [77]. In situ hybridization studies indicated differential expression of the PDGF ligands and their receptors in glial cell of the tumor mass and the endothelial cells in the tumor areas suggesting the presence of autocrine and paracrine stimulatory loops affecting glioma angiogenesis. High expressions of both PDGF-B and PDGFR-β mRNA were found in the endothelial cells present in the tumor tissue; these were thought to stimulate the autocrine loop with the PDGFR-β receptor, while PDGF-A mRNA and PDGF-α were observed only in the glial tumor cells stimulating the autocrine/paracrine loop with the PDGFR-α receptor [73]. PDGFR-β is preferentially expressed in GBM stem cells, and genetic or pharmacological targeting of PDGFR-β (not PDGFR-α) attenuated glioma stem cell (GSC) self-renewal, survival, and GBM progression [78, 79].
HGF/SF
Hepatocyte growth factor/scatter factor (HGF/SF) is a heparin-binding mesenchyme-derived cytokine consisting of a 60-kDa α-chain and a 30-kDa β-chain. It transduces signals by binding to its receptor and is a transmembrane tyrosine kinase encoded by c-MET. SF and c-MET are strongly increased in several tumors and is often associated with poor prognosis [80]. HGF is a potent angiogenic molecule, and its angiogenic activity stimulates endothelial cell proliferation and migration in vitro and increases organization into capillary-like tubes in vivo. HGF/SF regulates angiogenesis by simultaneous upregulation a VEGF, a pro-angiogenic factor and suppressing thrombospondin 1 (TSP-1), an endogenous inhibitor of angiogenesis [81]. HGF/SF can also induce angiogenesis independently of VEGF through the direct activation of the AKT and ERKs to induce endothelial proliferation [82].
SF/HGF and its receptor tyrosine kinase c-MET are expressed in brain tumors and were shown to promote tumor proliferation, migration, invasion, and angiogenesis. This ligand-receptor pair expression levels correlate with tumor grade, tumor blood vessel density, and poor prognosis [83]. Inhibition of SF/HGF and c-met expression anti-SF and anti-c-MET U1/ribozymes promotes tumor cell apoptosis and inhibits tumor angiogenesis in an in vivo glioma model [84]. Suppression of both MET and VEGF exhibited a synergistic effect in the inhibition GBM growth compared to single treatment alone in an intracranial glioma mode [85].
Ang
The angiopoietins are glycosylated proteins that bind to Tie-2 (Tyr kinase with Ig and epidermal growth factor homology domains) receptors [86]. Four types of angiopoietins have been identified (Ang-1 to Ang-4) and were shown to play a role in angiogenesis. All the angiopoietins bind the same receptor, tunica interna endothelial cell kinase 2 (Tie-2), but appear to have differential and counteracting effects on the vasculature. Ang-1 induced new vessel formation with angiogenic actions that are distinct from VEGF and stabilizes them through reciprocal interactions between the endothelium and surrounding ECM [25]. Ang-2 is upregulated by the both hypoxia and VEGF and enhances the VEGF-mediated endothelial cell migration and proliferation. In the absence of VEGF, Ang-2 functions as an antagonist to Ang-1 which mediates blood vessel regression and contributes to leakiness and fragility of tumor vessels [87]. Therefore, Angs induce context dependent pro- or anti-angiogenic effects. Furthermore, it has been established that tetrameric or higher orders of aggregation of Angs is required for Tie-2-mediated signaling, suggesting the presence of monomeric or dimeric angiopoietins that may bind to their receptor and serve as inhibitors of Tie-2 [88]. Ang-3 is maintained as a monomeric form and exerts anti-angiogenic and anti-cancer activity [89]. A study reported by Cam et al. [90] showed that targeting the angiopoietin 1 (ANGPT1)/Tie-2 axis by using a highly potent, orally available small molecular inhibitor (rebastinib) in GBM extents survival. In addition, rebastinib (DCC-2036) is a selective inhibitor of the Tie-2 immunokinase and currently in clinical trials in combination with carboplatin (NCT03717415) or paclitaxel (NCT03601897) in patients including with GBM [90].
Ang-1 mRNA was localized in tumor cells while Ang-2 mRNA was detected in endothelial cells and causes blood vessel dissolution/destabilization, and it is identified as one the early marker of glioma-induced neovascularization [66, 91]. Ang-4 is upregulated in human GBM tissues and cells and was shown to have a more potent pro-angiogenic activity than Ang-1 and promotes intracranial growth in mouse model [92]. Tie-2 expression was observed in malignant human gliomas [93], and Ang-2 regulates VEGF expression at the transcriptional level in Tie-2-expressing glioma cells [94]. One preclinical study has demonstrated that combined anti-VEGF/anti-Ang-2 therapy can obliterate resistance to VEGF monotherapy by upregulation of Ang-2 in endothelial cells and had a synergistic effect in overall GBM survival [95].
TGF-β
The TGF-β family of structurally related polypeptides and control several pro-tumorigenic functions like proliferation, apoptosis, differentiation, epithelial-mesenchymal transition (EMT), and angiogenesis. They signal through heteromeric complexes of type I (activin receptor-like kinases, also known as TβRI) and type II (TβRII) transmembrane serine/threonine kinase receptors serine/threonine kinase receptor complexes which in turn triggers phosphorylation of the intracellular effectors, Smads (derived from proteins “Sma” and “Mad” from C. elegans and D. melanogaster) to regulate the expression of TGF-β target genes [96]. TGF-β1 is the most frequently overexpressed in carcinomas and elevated TGF-β activity has been associated with poor clinical outcome [97]. Smads interact with and modulate the functions of various transcription factors which mediate tumor-induced angiogenesis [98]. TGF-β regulates the expression of various ECM components that play a pivotal role in both the initiation and resolution phase of angiogenesis [99]. TGF-β modulates the levels of FGF-2 which is required in the formation of capillaries during angiogenesis by suppressing the induction of a serine protease, urokinase plasminogen activator [100]. TGF-β acts in concert with VEGF promote endothelial cell apoptosis as part of capillary acts in concert with TGF-β1 to induce endothelial cell apoptosis [101]. TGF-β pathway also activates αvβ3, which binds to various secreted ECM proteins, such as von Willebrand factor, TSP-1, fibrinogen, proteolyzed collagen, fibronectin, and vitronectin and facilitates their degradation during vascular remodeling during angiogenesis [102]. Several approaches have been used to neutralize TGF-β signaling at distinct levels to suppress tumor growth and angiogenesis [103].
A high level of TGF-β correlates with poor prognosis in GBM and enhances the expression of several pro-angiogenic factors such as VEGF, FGF, and PDGF-β [104]. TGF-β1 increased glioma-induced angiogenesis via JNK pathway in zebrafish embryo/xenograft glioma model [105]. A cross-talk between TGF-β and VEGF/PLGF signaling in glioblastoma was shown to have both pro- and anti-angiogenic activities in human brain-derived microvascular endothelial cells (hCMECs) and glioblastoma-derived endothelial cells (GMECs). TGF-β induces VEGF and placental growth factor (PlGF) mRNA and protein expression in glioma cells inducing pro-angiogenic effects. In contrast, exogenous TGF-β had inhibitory effects on endothelial properties and induces endothelial-mesenchymal transition (EndoMT) in hCMEC and GMEC [106]. High levels of TGF-β work in conjunction with the PDGF-β to increase GSC proliferation [104]. TGF-β induces generation of pericytes from the GSC residing in the perivascular niches to support vessel formation and tumor growth [107].
MMPs
MMP are a family of zinc-dependent endopeptidase endopeptidases that selectively degrade components of the ECM and are implicated in tumor cell invasion angiogenesis and suppression of anti-tumor immune surveillance. An integral part of the angiogenic process is degradation of the vessel basement membrane and surrounding ECM which facilitates the invasion of endothelial cells. MMPs were also shown to stimulate the proliferation and activation of pericytes through the release of growth factor bound to the ECM and aid in their migration to the new formed vessels leading to vessel stabilization [108].
Gelatinase-A (MMP-2) and gelatinase-B (MMP-9) are highly expressed in patients with WHO grade III brain tumors [109]. Both these proteases were shown to have a synergistic effect on endothelial basement membrane degradation in gliomas [110]. MMP-9-mediated liberation of matrix-sequestered VEGF induced the angiogenic switching in a pre-malignant tumor; this effect was observed in several transgenic mouse models including glioblastoma [111].
Angiogenic Regulators and Targets for Anti-angiogenesis Therapy in GBM
It has been previously mentioned that angiogenesis is one of the most obvious hallmarks of most tumors including adult brain tumor (GBM), which significantly contrasts GBM from normal brain tissues [112, 113]. Hence, anti-angiogenesis therapy has become the most effective strategy in the treatment of GBM patients. Previously, it has been shown that VEGF plays an essential role in the angiogenesis of GBM, and inhibiting the expression of VEGF always known to be the most effective therapeutic strategy to GBM growth in patients [58, 114]. Moreover, vasculogenic mimicry (VM) is a newly discovered tube-like vascular structure which was found to be among the potential therapies for GBM [115]. Additionally, anti-angiogenesis by the VEGF mono-antibody, bevacizumab, showed minimal efficacy and enhanced tumor invasiveness triggered by hypoxia induction, which may be partially due to VM. Several studies have reported that VM is endothelial cell-independent, consisting of tumor cells and extracellular matrix, and is found to be associated with poor prognosis in GBM patients [11, 116, 117]. In addition, these studies showed that the VM-associated mechanisms offered new insights compared to classical anti-angiogenesis therapies. These studies have also confirmed that there were a series of genes including molecular targets and molecular signaling pathways were involved in VM [115]. Hence, these molecular mechanisms of VM may provide potential targets for anti-angiogenesis therapy in GBM. For example, VEGFR-2 kinase inhibitors (SU1498 and AZD2171) have been shown to reduce VM formation in GBM cell lines in vitro and in vivo, accompanied by reduction in chemotaxis, cell proliferation, and tumorigenicity [118].
It has been reported that hypoxia-inducible gene 2 (HIG2) is a marker of hypoxia and it can serve as a diagnostic biomarker for several cancers including GBM, as a potential target for anti-angiogenesis therapy [119]. Furthermore, Mao et al. [120] showed a positive correlation of HIG2 with VEGFA and HIF1α expression, which ultimately contributes to bevacizumab resistance in GBM [120]. Several studies have shown that STAT3 is a receptor that is activated by ligand interaction and overexpression of STAT3 constitutively activated in several tumors including GBM [121, 122]. Additionally, it has been shown that STAT3 inhibitor (AZD1480) combined with cediranib significantly reduced the volume and microvessel density of GBM, suggesting that the STAT3 molecular signaling pathway may mediate resistance to anti-angiogenic therapy, and regulating the STAT3 pathway might be useful in treating the condition in GBM patients [123]. Previously, it has been shown that the downregulation of HIF1α and mTOR signaling pathway through rapamycin, including mTOR siRNA, may inhibit VM formation in GBM [124]. Moreover, this study provides the evidence that mTOR as a potential therapeutic target in GBM. A study reported by Nicholas et al. [125] has shown that the epidermal growth factor receptor (EGFR) is associated with tumor growth and angiogenesis, and it is also found activated in all types of tumors including GBM [125]. In addition, this study also reveals that RAS/MAPK and PI3K/AKT/mTOR molecular signaling pathway regulates glioma cell proliferation, differentiation, tumor angiogenesis, and survival in GBM [42, 45]. Furthermore, targeting of the RTK/PI3K/AKT pathway enhances the cytotoxic effect of radiation and TMZ in malignant GBM cells [126].
A study reported by Francescone et al. [127] showed that targeting VEGFR2 using Flk-1 shRNA in GBM-derived cell lines significantly reduced VM formation and subsequently inhibited the development of tumors [127]. In addition, the results of this study suggest that the VEGFR2 plays an important role in the formation of VM in GBM as a possible therapeutic target [127]. There were several studies demonstrate that vincristine promotes an anti-angiogenic effect via the inhibition of HIF1α in GBM, and result of this study may provide a new therapeutic target for anti-angiogenesis therapy in GBM [128, 129].
A study reported that the PTEN molecular signaling act as a tumor suppressor gene, and it is often inactivated in several cancers including GBM [130]. This study also reveals that the loss of the PTEN signaling leads to VEGFR2 expression in tumor cells in GBM patients, which may contribute to resistance against anti-angiogenic treatments. Moreover, it has been shown that overexpression of VEGFR2 in tumor cells could develop early resistance to chemotherapy with TMZ and anti-angiogenesis therapy with bevacizumab, in GBM [131].
More and more emerging studies have suggested that the targeted gene knockout techniques with well-designed experimental strategy could be effective in the treatment of GBM patients and other human diseases. Moreover, newly designed targeted drug delivery systems circumvent multidrug resistance and demonstrated an enhanced efficacy for GBM patient [132, 133]. Additionally, the use of strategies targeting multiple molecular signaling pathways in a combination with drug targets may lead to increased therapeutic efficiency, and studies on VM as a novel and distinct regulating target contribute significantly to the future of anti-angiogenesis treatment in GBM patients.
Clinical Trials of Angiogenesis Targets in GBM
Several preclinical studies suggested that anti-angiogenic therapeutic agents enhance the efficacy of conventional treatments. A number of anti-angiogenic therapies have been evaluated in clinical trials as an alternative or complementary to conventional cancer treatments. Table 2 summarizes clinical drug trials targeting angiogenesis in primary and secondary brain tumors, and the detail information about the clinical trial, drug target concentration, number of patient population, and the current clinical trial phase for the drug approval process. Most of the anti-angiogenic agents currently in phase I/II trials for brain tumors target the VEGF pathway as VEGF family and its receptors function as the central signaling pathway of glioma angiogenesis. On this basis, the majority of these clinical trials are targeting VEGF signaling (Table 3) with monoclonal antibodies against VEGF-A (bevacizumab), a small-molecule tyrosine kinase inhibitors (TKIs) that inhibit VEGFR-2 tyrosine kinase activity (cediranib, sunitinib, vandetanib) and soluble decoy receptors developed from VEGFR-1 that selectively inhibit VEGF activity (aflibercept).
The first anti-angiogenesis agent approved for clinical use for brain cancer is a drug called bevacizumab or avastin (Genentech, South San Francisco, CA). Bevacizumab is a monoclonal antibody, and it functions like the physiological antibodies that the human body naturally produces as part of the adaptive immune system. Bevacizumab binds to VEGF and blocks signaling of the molecule and suppresses the formation of new blood vessel growth. Several phase II clinical trials have studied the therapeutic efficacy of bevacizumab as a single agent or in combination with chemotherapy or radiation for recurrent GBM. Bevacizumab as a single agent had significant anti-glioma activity in patients with recurrent glioblastoma [134] (Table 2). A phase I study with a small number of patients suggested that bevacizumab in combination with irinotecan, an inhibitor of topoisomerase I, can be safely administered to patients with malignant gliomas and bevacizumab plus irinotecan achieved a significant improvement in radiographic response (changes in the density of the tumor area) as well as significant increases in progression-free survival among recurrent GBM patients. These studies observed anti-edema induced by bevacizumab treatment augmented the efficacy of the cytotoxic drug by improving the distribution of the drug in these tumors [135, 136]. Previously, the BRAIN study was completed in 2007 for bevacizumab drug trial in recurrent GBM patients, and the outcome of this study was reported the median overall survival (OS) rate of 9.3% and 8.7% with progression-free survival (PFS-6) rate of 43% and 50.3%, respectively, with compared to bevacizumab to bevacizumab plus irinotecan, an inhibitor of topoisomerase I [137]. Later, similar clinical trial was performed by the National Cancer Institute (NCI) for the use of bevacizumab in recurrent GBM patients and they found the median OS rate of 7.8% and PFS-6 rate of 29% [134]. There have been several studies investigated the use of bevacizumab drug target in combination with other drug products to treat recurrent GBM. For example, the BELOB clinical trial was initiated as a randomized phase-II clinical trial and this study used lomustine with bevacizumab or lomustine and bevacizumab alone for the treatment of recurrent GBM patients. The BELOB clinical trial reported that the combination of both drug products (bevacizumab and lomustine) resulted in a PFS-6 of 42% compared to 11% and 18% with OS at 9 months of 12% compared to 7.8% and 8% for lomustine and bevacizumab alone, respectively [138]. Furthermore, based on the BELOB study results, a phase III clinical trial (EORTC 26101) was performed to compare lomustine alone versus lomustine with bevacizumab. In conclusion of the EORTC study, they did find any significant difference in OS for combination treatment versus lomustine alone in recurrent GBM patients [139]. Previously, it has been mentioned that the AVAglio used the revised Response Assessment in Neuro-oncology (RANO) criteria to assess the GBM disease progression in the newly diagnosed GBM patients. The AVAglio clinical trial was performed based on this revised RANO criteria and the clinical trial concluded that bevacizumab prolong the maintenance of performance status in GBM patients, it also reported that decreased in steroid utilization, and prolonged time to deterioration in prespecified cognitive domains of the newly diagnosed GBM patients [140]. Moreover, the similar study has been performed in newly diagnosed GBM patients using the randomized phase II GLAIRUS study, and this study compared the standard care of chemoradiation with temozolomide (TMZ) versus with bevacizumab and irinotecan in GBM patients whose tumors expressed the DNA repair enzyme O6-methyl guanine DNA methyltransferase (MGMT). This phase II GLAIRUS study also concluded that the loss of MGMT increased the sensitivity to therapy with TMZ in newly diagnosed GBM patients [141] (Table 2).
Some of these clinical trials also suggested that the anti-angiogenic therapeutic agents (e.g., VEGF/VEGFR therapeutic targets) (Table 3) enhance the efficacy of conventional treatments by other mechanisms apart from normalization of blood vessels. It was also observed that anti-angiogenic therapy disrupted the tumor vasculature and that the CSC niche microenvironment associated with the tumor blood vessels reduced the CSC which in turn contributes to the efficacy of anti-angiogenic cancer therapy [142]. In another phase II multicenter trial with one hundred sixty-seven patients with recurrent glioblastoma, bevacizumab, alone or in combination with irinotecan, was well tolerated and active in recurrent glioblastoma [137]. It was also suggested that bevacizumab therapy restore a balance between pro- and anti-angiogenic cytokines and induces more stability within the tumor blood vessels with structural and functional phenotype more reflective of normal blood vessels, thus allowing for more effective penetration and distribution of cytotoxic chemotherapeutic drugs within the tumor [143, 144]. A randomized controlled phase II trial of a single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma suggested improved OS as compared with monotherapies [138]. Similarly, cediranib (AZD2171, an oral, pan-VEGF receptor inhibitor) as a monotherapy was shown to induce “normalization time window” in tumor vessels in patients with recurrent GBM with significant clinical and functional consequences [145].
Limitation of Anti-VEGF Therapy and Future Directions
More recent phase 2 trial suggested that the combination of bevacizumab and lomustine did not confer a survival advantage over treatment with lomustine alone in patients with progressive GBM [139]. A randomized phase III trial in newly diagnosed GBM and recurrent grade III gliomas has failed to show an overall survival in [146, 147]. These studies suggest that although a combination of anti-angiogenic therapy with chemotherapy compared with chemotherapy alone produces favorable results with improvements in objective response and PFS in patients with recurrent GBM, a large portion of the patients benefit because of a several factors including changes in the tumor microenvironment (TME) toxicity and resistance. It was proposed that hypoxia caused by vessel regression upregulates hypoxia regulated pro-angiogenic factors like SDF1α leading to recruitment of bone marrow-derived cells (BMDCs) that have the capacity to induce new blood vessel growth leading to tumor progression and relapse [148, 149]. A pro-invasive adaption of the tumors was observed in a subset of GBM patients who had developed multifocal recurrence of tumor’s during anti-VEGF therapy with bevacizumab along with either irinotecan or carboplatin [150, 151]. Toxicity associated with anti-angiogenesis includes thromboembolic and hemorrhagic complications. In addition, gastrointestinal (GI) perforations and one case of reversible posterior leukoencephalopathy were also noted [148, 149]. A compensatory switch to alternative angiogenic pathways could lead to the acquisition of resistance to angiogenic therapy. For example, the PDGF signaling was shown to contribute to angiogenesis in tumors refractory to anti-VEGF treatment by activating tumor stromal cells. However, the anti-tumor effect obtained with a combination of anti-VEGF and anti-PDGF therapy was minimal under conditions of maximal VEGF antagonism, suggesting that inhibition of these two pathways might not be fully additive or synergistic [152].
As discussed above, although anti-VEGF treatment indeed altered several abnormal characteristics of tumor vessels and was generally well tolerated leading to devascularization that limits tumor growth, a large fraction of patients develops toxicity and resistance to this treatment. Moreover, prolonged exposure to anti-angiogenic drugs blocks blood supply to the tumors leading to hypoxic environment which in turn is known to induce chemo-resistance and tumor progression. Therefore, the dose and time of initiation of anti-angiogenic treatment could play a significant role on the therapeutic benefit as the angiogenic inhibitors suppress the tumor growth by inhibiting the growth of blood vessels but does not necessarily kill cancer cells. Single-agent bevacizumab seems to have significant effects on vascular permeability and cerebral edema, suggesting that future trials should focus on the role of bevacizumab as the initial treatment of GBM before starting the chemotherapy treatment [134]. The association between the survival benefit and increased oxygenation leading to vascular normalization in the phase II trials with anti VEGF therapy suggest that identification and validation of early imaging biomarkers and new imaging parameters could help identify the subset of patients who most likely will benefit with anti-angiogenic agents [153]. A baseline of high and low plasma levels of MMP-2 and MMP-9, respectively, were associated with a high response rate and prolonged PFS and OS in recurrent high-grade gliomas treated with Bevacizumab but not with other cytotoxic agents suggesting that it could be predictive biomarker and potentially allow initial patient selection for bevacizumab treatment [154]. Another study showed that the sensitivity to bevacizumab may depend on the relative amount of the various isoforms of VEGF which differ in different molecular weights and biologic properties [155]. Bevacizumab-induced hypertension demonstrated significantly better progression-free survival and OS, suggesting that it could be a physiologic marker of outcome in patients with recurrent GBM [156]. Further, developing patient-specific personalized therapies, based on cellular response of the endothelial cells from the primary brain tumor by screening for sensitivity/resistance to anti-angiogenic agents, can optimize anti-angiogenic therapy in GBM patients [157]. There is also a need to determine novel points of convergence of various signaling pathways in the initiation and development of tumor-induced angiogenesis for predicting and identifying new targets for anti-angiogenic therapy. Several clinical trials are ongoing to validate and expand these efforts, including multiple studies to evaluate non-VEGF anti-angiogenic strategies for malignant glioma patients.
References
Ohgaki H (2009) Epidemiology of brain tumors. Methods Mol Biol 472:323–342. https://doi.org/10.1007/978-1-60327-492-0_14
Ohgaki H, Kleihues P (2005) Epidemiology and etiology of gliomas. Acta Neuropathol 109(1):93–108. https://doi.org/10.1007/s00401-005-0991-y
Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS (2017) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol 19(suppl_5):v1–v88. https://doi.org/10.1093/neuonc/nox158
Erridge SC, Hart MG, Kerr GR, Smith C, McNamara S, Grant R, Gregor A, Whittle IR (2011) Trends in classification, referral and treatment and the effect on outcome of patients with glioma: a 20 year cohort. J Neuro-Oncol 104(3):789–800. https://doi.org/10.1007/s11060-011-0546-0
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820. https://doi.org/10.1007/s00401-016-1545-1
Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19(4):764–772. https://doi.org/10.1158/1078-0432.CCR-12-3002
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109. https://doi.org/10.1007/s00401-007-0243-4
Palanichamy K, Erkkinen M, Chakravarti A (2006) Predictive and prognostic markers in human glioblastomas. Curr Treat Options in Oncol 7(6):490–504
Grauer OM, Wesseling P, Adema GJ (2009) Immunotherapy of diffuse gliomas: biological background, current status and future developments. Brain Pathol 19(4):674–693. https://doi.org/10.1111/j.1750-3639.2009.00315.x
Dolecek TA, Propp JM, Stroup NE, Kruchko C (2012) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncology 14(Suppl 5):v1–v49. https://doi.org/10.1093/neuonc/nos218
El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, Marie Y, Mokhtari K et al (2010) A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain 133(Pt 4):973–982. https://doi.org/10.1093/brain/awq044
Weller M (2010) Angiogenesis in glioblastoma: just another moving target? Brain 133(Pt 4):955–956. https://doi.org/10.1093/brain/awq063
Takano S, Yamashita T, Ohneda O (2010) Molecular therapeutic targets for glioma angiogenesis. J Oncol 2010:351908. https://doi.org/10.1155/2010/351908
Chung AS, Lee J, Ferrara N (2010) Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat Rev Cancer 10(7):505–514. https://doi.org/10.1038/nrc2868
Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9(6):653–660. https://doi.org/10.1038/nm0603-653
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438(7070):932–936. https://doi.org/10.1038/nature04478
Liao D, Johnson RS (2007) Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 26(2):281–290. https://doi.org/10.1007/s10555-007-9066-y
Lin JL, Wang MJ, Lee D, Liang CC, Lin S (2008) Hypoxia-inducible factor-1alpha regulates matrix metalloproteinase-1 activity in human bone marrow-derived mesenchymal stem cells. FEBS Lett 582(17):2615–2619. https://doi.org/10.1016/j.febslet.2008.06.033
Petrella BL, Lohi J, Brinckerhoff CE (2005) Identification of membrane type-1 matrix metalloproteinase as a target of hypoxia-inducible factor-2 alpha in von Hippel-Lindau renal cell carcinoma. Oncogene 24(6):1043–1052. https://doi.org/10.1038/sj.onc.1208305
Fitsialos G, Bourget I, Augier S, Ginouves A, Rezzonico R, Odorisio T, Cianfarani F, Virolle T et al (2008) HIF1 transcription factor regulates laminin-332 expression and keratinocyte migration. J Cell Sci 121(Pt 18):2992–3001. https://doi.org/10.1242/jcs.029256
Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG et al (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A 108(49):19611–19616. https://doi.org/10.1073/pnas.1117773108
Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S et al (2001) VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med 7(2):186–191. https://doi.org/10.1038/84635
Pepper MS, Vassalli JD, Orci L, Montesano R (1993) Biphasic effect of transforming growth factor-beta 1 on in vitro angiogenesis. Exp Cell Res 204(2):356–363. https://doi.org/10.1006/excr.1993.1043
Rifkin DB, Moscatelli D (1989) Recent developments in the cell biology of basic fibroblast growth factor. J Cell Biol 109(1):1–6. https://doi.org/10.1083/jcb.109.1.1
Suri C, McClain J, Thurston G, McDonald DM, Zhou H, Oldmixon EH, Sato TN, Yancopoulos GD (1998) Increased vascularization in mice overexpressing angiopoietin-1. Science 282(5388):468–471
Gleave ME, Hsieh JT, Wu HC, Hong SJ, Zhau HE, Guthrie PD, Chung LW (1993) Epidermal growth factor receptor-mediated autocrine and paracrine stimulation of human transitional cell carcinoma. Cancer Res 53(21):5300–5307
Lakka SS, Rao JS (2008) Antiangiogenic therapy in brain tumors. Expert Rev Neurother 8(10):1457–1473. https://doi.org/10.1586/14737175.8.10.1457
Hong H, Chen F, Zhang Y, Cai W (2014) New radiotracers for imaging of vascular targets in angiogenesis-related diseases. Adv Drug Deliv Rev 76:2–20. https://doi.org/10.1016/j.addr.2014.07.011
Senger DR, Davis GE (2011) Angiogenesis. Cold Spring Harb Perspect Biol 3(8):a005090. https://doi.org/10.1101/cshperspect.a005090
Carmeliet P, Jain RK (2011) Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov 10(6):417–427. https://doi.org/10.1038/nrd3455
De Bock K, Cauwenberghs S, Carmeliet P (2011) Vessel abnormalization: Another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr Opin Genet Dev 21(1):73–79. https://doi.org/10.1016/j.gde.2010.10.008
Mazzone M, Dettori D, de Oliveira RL, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA et al (2009) Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136(5):839–851. https://doi.org/10.1016/j.cell.2009.01.020
Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF (2010) Heterogeneity of the tumor vasculature. Semin Thromb Hemost 36(3):321–331. https://doi.org/10.1055/s-0030-1253454
Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, Hoffman RM, Kerbel RS (2009) Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res 69(18):7243–7251. https://doi.org/10.1158/0008-5472.CAN-09-0167
Papetti M, Herman IM (2002) Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol 282(5):C947–C970. https://doi.org/10.1152/ajpcell.00389.2001
Baish JW, Stylianopoulos T, Lanning RM, Kamoun WS, Fukumura D, Munn LL, Jain RK (2011) Scaling rules for diffusive drug delivery in tumor and normal tissues. Proc Natl Acad Sci U S A 108(5):1799–1803. https://doi.org/10.1073/pnas.1018154108
Stylianopoulos T, Jain RK (2013) Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci U S A 110(46):18632–18637. https://doi.org/10.1073/pnas.1318415110
Tredan O, Galmarini CM, Patel K, Tannock IF (2007) Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 99(19):1441–1454. https://doi.org/10.1093/jnci/djm135
Semenza GL (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7(8):345–350
Siemann DW, Chaplin DJ, Horsman MR (2017) Realizing the potential of vascular targeted therapy: the rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Investig 35(8):519–534. https://doi.org/10.1080/07357907.2017.1364745
Batchelor TT, Reardon DA, de Groot JF, Wick W, Weller M (2014) Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res 20(22):5612–5619. https://doi.org/10.1158/1078-0432.CCR-14-0834
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M et al (2012) Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget 3(10):1068–1111. https://doi.org/10.18632/oncotarget.659
Onishi M, Kurozumi K, Ichikawa T, Date I (2013) Mechanisms of tumor development and anti-angiogenic therapy in glioblastoma multiforme. Neurol Med Chir (Tokyo) 53(11):755–763
Sooman L, Freyhult E, Jaiswal A, Navani S, Edqvist PH, Ponten F, Tchougounova E, Smits A et al (2015) FGF2 as a potential prognostic biomarker for proneural glioma patients. Acta Oncol 54(3):385–394. https://doi.org/10.3109/0284186X.2014.951492
Vogt PK, Hart JR (2011) PI3K and STAT3: a new alliance. Cancer Discov 1(6):481–486. https://doi.org/10.1158/2159-8290.CD-11-0218
Bai RY, Staedtke V, Riggins GJ (2011) Molecular targeting of glioblastoma: drug discovery and therapies. Trends Mol Med 17(6):301–312. https://doi.org/10.1016/j.molmed.2011.01.011
Linkous AG, Yazlovitskaya EM (2011) Angiogenesis in glioblastoma multiforme: navigating the maze. Anti Cancer Agents Med Chem 11(8):712–718
Richardson TP, Peters MC, Ennett AB, Mooney DJ (2001) Polymeric system for dual growth factor delivery. Nat Biotechnol 19(11):1029–1034. https://doi.org/10.1038/nbt1101-1029
Cao R, Brakenhielm E, Pawliuk R, Wariaro D, Post MJ, Wahlberg E, Leboulch P, Cao Y (2003) Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med 9(5):604–613. https://doi.org/10.1038/nm848
Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13(1):9–22
Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, Wu Y, Bono F et al (2001) Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 7(5):575–583. https://doi.org/10.1038/87904
Sun XT, Ding YT, Yan XG, Wu LY, Li Q, Cheng N, Qiu YD, Zhang MY (2004) Angiogenic synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in an in vitro quantitative microcarrier-based three-dimensional fibrin angiogenesis system. World J Gastroenterol 10(17):2524–2528. https://doi.org/10.3748/wjg.v10.i17.2524
Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100(6):782–794. https://doi.org/10.1161/01.RES.0000259593.07661.1e
Hatva E, Kaipainen A, Mentula P, Jaaskelainen J, Paetau A, Haltia M, Alitalo K (1995) Expression of endothelial cell-specific receptor tyrosine kinases and growth factors in human brain tumors. Am J Pathol 146(2):368–378
Samoto K, Ikezaki K, Ono M, Shono T, Kohno K, Kuwano M, Fukui M (1995) Expression of vascular endothelial growth factor and its possible relation with neovascularization in human brain tumors. Cancer Res 55(5):1189–1193
Plate KH, Risau W (1995) Angiogenesis in malignant gliomas. Glia 15(3):339–347. https://doi.org/10.1002/glia.440150313
Phillips H, Armani M, Stavrou D, Ferrara N, Westphal M (1993) Intense focal expression of vascular endothelial growth-factor messenger-RNA in human intracranial neoplasms - association with regions of necrosis. Int J Oncol 2(6):913–919
Plate KH, Breier G, Weich HA, Risau W (1992) Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature 359(6398):845–848. https://doi.org/10.1038/359845a0
Chi AS, Sorensen AG, Jain RK, Batchelor TT (2009) Angiogenesis as a therapeutic target in malignant gliomas. Oncologist 14(6):621–636. https://doi.org/10.1634/theoncologist.2008-0272
Yao Y, Kubota T, Sato K, Kitai R, Takeuchi H, Arishima H (2001) Prognostic value of vascular endothelial growth factor and its receptors Flt-1 and Flk-1 in astrocytic tumours. Acta Neurochir 143(2):159–166
Gerwins P, Skoldenberg E, Claesson-Welsh L (2000) Function of fibroblast growth factors and vascular endothelial growth factors and their receptors in angiogenesis. Crit Rev Oncol Hematol 34(3):185–194
Sa G, Murugesan G, Jaye M, Ivashchenko Y, Fox PL (1995) Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J Biol Chem 270(5):2360–2366. https://doi.org/10.1074/jbc.270.5.2360
Kanda S, Landgren E, Ljungstrom M, Claesson-Welsh L (1996) Fibroblast growth factor receptor 1-induced differentiation of endothelial cell line established from tsA58 large T transgenic mice. Cell Growth Differ 7(3):383–395
Brem S, Tsanaclis AM, Gately S, Gross JL, Herblin WF (1992) Immunolocalization of basic fibroblast growth factor to the microvasculature of human brain tumors. Cancer 70(11):2673–2680
Takahashi JA, Fukumoto M, Igarashi K, Oda Y, Kikuchi H, Hatanaka M (1992) Correlation of basic fibroblast growth factor expression levels with the degree of malignancy and vascularity in human gliomas. J Neurosurg 76(5):792–798. https://doi.org/10.3171/jns.1992.76.5.0792
Zagzag D, Miller DC, Sato Y, Rifkin DB, Burstein DE (1990) Immunohistochemical localization of basic fibroblast growth factor in astrocytomas. Cancer Res 50(22):7393–7398
Stan AC, Nemati MN, Pietsch T, Walter GF, Dietz H (1995) In vivo inhibition of angiogenesis and growth of the human U-87 malignant glial tumor by treatment with an antibody against basic fibroblast growth factor. J Neurosurg 82(6):1044–1052. https://doi.org/10.3171/jns.1995.82.6.1044
Fredriksson L, Li H, Eriksson U (2004) The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev 15(4):197–204. https://doi.org/10.1016/j.cytogfr.2004.03.007
Renner O, Tsimpas A, Kostin S, Valable S, Petit E, Schaper W, Marti HH (2003) Time- and cell type-specific induction of platelet-derived growth factor receptor-beta during cerebral ischemia. Brain Res Mol Brain Res 113(1–2):44–51
Ferrara N, Kerbel RS (2005) Angiogenesis as a therapeutic target. Nature 438(7070):967–974. https://doi.org/10.1038/nature04483
Ribatti D, Vacca A, Roccaro AM, Crivellato E, Presta M (2003) Erythropoietin as an angiogenic factor. Eur J Clin Investig 33(10):891–896
Sasaki K, Murohara T, Ikeda H, Sugaya T, Shimada T, Shintani S, Imaizumi T (2002) Evidence for the importance of angiotensin II type 1 receptor in ischemia-induced angiogenesis. J Clin Invest 109(5):603–611. https://doi.org/10.1172/JCI13055
Hermansson M, Nister M, Betsholtz C, Heldin CH, Westermark B, Funa K (1988) Endothelial cell hyperplasia in human glioblastoma: coexpression of mRNA for platelet-derived growth factor (PDGF) B chain and PDGF receptor suggests autocrine growth stimulation. Proc Natl Acad Sci U S A 85(20):7748–7752. https://doi.org/10.1073/pnas.85.20.7748
Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA (2002) Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res 62(13):3729–3735
Martinho O, Longatto-Filho A, Lambros MB, Martins A, Pinheiro C, Silva A, Pardal F, Amorim J et al (2009) Expression, mutation and copy number analysis of platelet-derived growth factor receptor A (PDGFRA) and its ligand PDGFA in gliomas. Br J Cancer 101(6):973–982. https://doi.org/10.1038/sj.bjc.6605225
Farin A, Suzuki SO, Weiker M, Goldman JE, Bruce JN, Canoll P (2006) Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia 53(8):799–808. https://doi.org/10.1002/glia.20334
Cantanhede IG, de Oliveira JRM (2017) PDGF family expression in glioblastoma multiforme: data compilation from Ivy glioblastoma atlas project database. Sci Rep 7(1):15271. https://doi.org/10.1038/s41598-017-15045-w
Cenciarelli C, Marei HE, Felsani A, Casalbore P, Sica G, Puglisi MA, Cameron AJ, Olivi A et al (2016) PDGFRalpha depletion attenuates glioblastoma stem cells features by modulation of STAT3, RB1 and multiple oncogenic signals. Oncotarget 7(33):53047–53063. https://doi.org/10.18632/oncotarget.10132
Kim Y, Kim E, Wu Q, Guryanova O, Hitomi M, Lathia JD, Serwanski D, Sloan AE et al (2012) Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev 26(11):1247–1262. https://doi.org/10.1101/gad.193565.112
Trusolino L, Comoglio PM (2002) Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer 2(4):289–300. https://doi.org/10.1038/nrc779
Zhang YW, Su Y, Volpert OV, Vande Woude GF (2003) Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A 100(22):12718–12723. https://doi.org/10.1073/pnas.2135113100
Sengupta S, Gherardi E, Sellers LA, Wood JM, Sasisekharan R, Fan TP (2003) Hepatocyte growth factor/scatter factor can induce angiogenesis independently of vascular endothelial growth factor. Arterioscler Thromb Vasc Biol 23(1):69–75
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-Oncology 7(4):436–451. https://doi.org/10.1215/S1152851705000050
Abounader R, Lal B, Luddy C, Koe G, Davidson B, Rosen EM, Laterra J (2002) In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J 16(1):108–110. https://doi.org/10.1096/fj.01-0421fje
Okuda T, Tasaki T, Nakata S, Yamashita K, Yoshioka H, Izumoto S, Kato A, Fujita M (2017) Efficacy of combination therapy with MET and VEGF inhibitors for MET-overexpressing glioblastoma. Anticancer Res 37(7):3871–3876. https://doi.org/10.21873/anticanres.11767
Thurston G (2003) Role of angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res 314(1):61–68. https://doi.org/10.1007/s00441-003-0749-6
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277(5322):55–60
Kim KT, Choi HH, Steinmetz MO, Maco B, Kammerer RA, Ahn SY, Kim HZ, Lee GM et al (2005) Oligomerization and multimerization are critical for angiopoietin-1 to bind and phosphorylate Tie2. J Biol Chem 280(20):20126–20131. https://doi.org/10.1074/jbc.M500292200
Xu Y, Liu YJ, Yu Q (2004) Angiopoietin-3 inhibits pulmonary metastasis by inhibiting tumor angiogenesis. Cancer Res 64(17):6119–6126. https://doi.org/10.1158/0008-5472.CAN-04-1054
Cam M, Charan M, Welker AM, Dravid P, Studebaker AW, Leonard JR, Pierson CR, Nakano I et al (2019) DeltaNp73/ETS2 complex drives glioblastoma pathogenesis- targeting downstream mediators by rebastinib prolongs survival in preclinical models of glioblastoma. Neuro-Oncology. https://doi.org/10.1093/neuonc/noz190
Zagzag D, Amirnovin R, Greco MA, Yee H, Holash J, Wiegand SJ, Zabski S, Yancopoulos GD et al (2000) Vascular apoptosis and involution in gliomas precede neovascularization: a novel concept for glioma growth and angiogenesis. Lab Investig 80(6):837–849
Brunckhorst MK, Wang H, Lu R, Yu Q (2010) Angiopoietin-4 promotes glioblastoma progression by enhancing tumor cell viability and angiogenesis. Cancer Res 70(18):7283–7293. https://doi.org/10.1158/0008-5472.CAN-09-4125
Lee OH, Xu J, Fueyo J, Fuller GN, Aldape KD, Alonso MM, Piao Y, Liu TJ et al (2006) Expression of the receptor tyrosine kinase Tie2 in neoplastic glial cells is associated with integrin beta1-dependent adhesion to the extracellular matrix. Mol Cancer Res 4(12):915–926. https://doi.org/10.1158/1541-7786.MCR-06-0184
Lee OH, Xu J, Fueyo J, Alonso MM, Liu D, Martin V, Jiang H, Piao Y et al (2008) Angiopoietin-2 decreases vascular endothelial growth factor expression by modulating HIF-1 alpha levels in gliomas. Oncogene 27(9):1310–1314. https://doi.org/10.1038/sj.onc.1210731
Scholz A, Harter PN, Cremer S, Yalcin BH, Gurnik S, Yamaji M, Di Tacchio M, Sommer K et al (2016) Endothelial cell-derived angiopoietin-2 is a therapeutic target in treatment-naive and bevacizumab-resistant glioblastoma. EMBO Mol Med 8(1):39–57. https://doi.org/10.15252/emmm.201505505
ten Dijke P, Miyazono K, Heldin CH (2000) Signaling inputs converge on nuclear effectors in TGF-beta signaling. Trends Biochem Sci 25(2):64–70
Leivonen SK, Kahari VM (2007) Transforming growth factor-beta signaling in cancer invasion and metastasis. Int J Cancer 121(10):2119–2124. https://doi.org/10.1002/ijc.23113
Miyazono K, ten Dijke P, Heldin CH (2000) TGF-beta signaling by Smad proteins. Adv Immunol 75:115–157
Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J 21(7):1743–1753. https://doi.org/10.1093/emboj/21.7.1743
Saksela O, Moscatelli D, Rifkin DB (1987) The opposing effects of basic fibroblast growth factor and transforming growth factor beta on the regulation of plasminogen activator activity in capillary endothelial cells. J Cell Biol 105(2):957–963. https://doi.org/10.1083/jcb.105.2.957
Ferrari G, Pintucci G, Seghezzi G, Hyman K, Galloway AC, Mignatti P (2006) VEGF, a prosurvival factor, acts in concert with TGF-beta1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A 103(46):17260–17265. https://doi.org/10.1073/pnas.0605556103
Platten M, Wick W, Wild-Bode C, Aulwurm S, Dichgans J, Weller M (2000) Transforming growth factors beta(1) (TGF-beta(1)) and TGF-beta(2) promote glioma cell migration via up-regulation of alpha(V)beta(3) integrin expression. Biochem Biophys Res Commun 268(2):607–611. https://doi.org/10.1006/bbrc.2000.2176
van Meeteren LA, Goumans MJ, ten Dijke P (2011) TGF-beta receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr Pharm Biotechnol 12(12):2108–2120
Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, Paris R, Tortosa A et al (2007) High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11(2):147–160. https://doi.org/10.1016/j.ccr.2006.11.023
Yang XJ, Chen GL, Yu SC, Xu C, Xin YH, Li TT, Shi Y, Gu A et al (2013) TGF-beta1 enhances tumor-induced angiogenesis via JNK pathway and macrophage infiltration in an improved zebrafish embryo/xenograft glioma model. Int Immunopharmacol 15(2):191–198. https://doi.org/10.1016/j.intimp.2012.12.002
Krishnan S, Szabo E, Burghardt I, Frei K, Tabatabai G, Weller M (2015) Modulation of cerebral endothelial cell function by TGF-beta in glioblastoma: VEGF-dependent angiogenesis versus endothelial mesenchymal transition. Oncotarget 6(26):22480–22495. https://doi.org/10.18632/oncotarget.4310
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE et al (2013) Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153(1):139–152. https://doi.org/10.1016/j.cell.2013.02.021
Chantrain CF, Henriet P, Jodele S, Emonard H, Feron O, Courtoy PJ, DeClerck YA, Marbaix E (2006) Mechanisms of pericyte recruitment in tumour angiogenesis: a new role for metalloproteinases. Eur J Cancer 42(3):310–318. https://doi.org/10.1016/j.ejca.2005.11.010
Wang M, Wang T, Liu S, Yoshida D, Teramoto A (2003) The expression of matrix metalloproteinase-2 and -9 in human gliomas of different pathological grades. Brain Tumor Pathol 20(2):65–72
Lakka SS, Gondi CS, Rao JS (2005) Proteases and glioma angiogenesis. Brain Pathol 15(4):327–341
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P et al (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744. https://doi.org/10.1038/35036374
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Wang N, Jain RK, Batchelor TT (2017) New directions in anti-angiogenic therapy for glioblastoma. Neurotherapeutics 14(2):321–332. https://doi.org/10.1007/s13311-016-0510-y
Cheng SY, Huang HJ, Nagane M, Ji XD, Wang D, Shih CC, Arap W, Huang CM et al (1996) Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci U S A 93(16):8502–8507. https://doi.org/10.1073/pnas.93.16.8502
Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, Trent JM, Meltzer PS et al (1999) Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol 155(3):739–752. https://doi.org/10.1016/S0002-9440(10)65173-5
Chen Y, Jing Z, Luo C, Zhuang M, Xia J, Chen Z, Wang Y (2012) Vasculogenic mimicry-potential target for glioblastoma therapy: an in vitro and in vivo study. Med Oncol 29(1):324–331. https://doi.org/10.1007/s12032-010-9765-z
Mao JM, Liu J, Guo G, Mao XG, Li CX (2015) Glioblastoma vasculogenic mimicry: signaling pathways progression and potential anti-angiogenesis targets. Biomarker Res 3:8. https://doi.org/10.1186/s40364-015-0034-3
Yao X, Ping Y, Liu Y, Chen K, Yoshimura T, Liu M, Gong W, Chen C et al (2013) Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by glioma stem-like cells. PLoS One 8(3):e57188. https://doi.org/10.1371/journal.pone.0057188
Kim SH, Wang D, Park YY, Katoh H, Margalit O, Sheffer M, Wu H, Holla VR et al (2013) HIG2 promotes colorectal cancer progression via hypoxia-dependent and independent pathways. Cancer Lett 341(2):159–165. https://doi.org/10.1016/j.canlet.2013.07.028
Mao XG, Wang C, Liu DY, Zhang X, Wang L, Yan M, Zhang W, Zhu J et al (2016) Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 7(30):47808–47820. https://doi.org/10.18632/oncotarget.10029
Abou-Ghazal M, Yang DS, Qiao W, Reina-Ortiz C, Wei J, Kong LY, Fuller GN, Hiraoka N et al (2008) The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res 14(24):8228–8235. https://doi.org/10.1158/1078-0432.CCR-08-1329
Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T et al (2001) Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A 98(13):7319–7324. https://doi.org/10.1073/pnas.131568898
de Groot J, Liang J, Kong LY, Wei J, Piao Y, Fuller G, Qiao W, Heimberger AB (2012) Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget 3(9):1036–1048. https://doi.org/10.18632/oncotarget.663
Huang M, Ke Y, Sun X, Yu L, Yang Z, Zhang Y, Du M, Wang J et al (2014) Mammalian target of rapamycin signaling is involved in the vasculogenic mimicry of glioma via hypoxia-inducible factor-1alpha. Oncol Rep 32(5):1973–1980. https://doi.org/10.3892/or.2014.3454
Nicholas MK, Lukas RV, Jafri NF, Faoro L, Salgia R (2006) Epidermal growth factor receptor - mediated signal transduction in the development and therapy of gliomas. Clin Cancer Res 12(24):7261–7270. https://doi.org/10.1158/1078-0432.CCR-06-0874
Choi EJ, Cho BJ, Lee DJ, Hwang YH, Chun SH, Kim HH, Kim IA (2014) Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 14:17. https://doi.org/10.1186/1471-2407-14-17
Francescone R, Scully S, Bentley B, Yan W, Taylor SL, Oh D, Moral L, Shao R (2012) Glioblastoma-derived tumor cells induce vasculogenic mimicry through Flk-1 protein activation. J Biol Chem 287(29):24821–24831. https://doi.org/10.1074/jbc.M111.334540
Escuin D, Kline ER, Giannakakou P (2005) Both microtubule-stabilizing and microtubule-destabilizing drugs inhibit hypoxia-inducible factor-1alpha accumulation and activity by disrupting microtubule function. Cancer Res 65(19):9021–9028. https://doi.org/10.1158/0008-5472.CAN-04-4095
Park KJ, Yu MO, Park DH, Park JY, Chung YG, Kang SH (2016) Role of vincristine in the inhibition of angiogenesis in glioblastoma. Neurol Res 38(10):871–879. https://doi.org/10.1080/01616412.2016.1211231
Di Cristofano A, Pandolfi PP (2000) The multiple roles of PTEN in tumor suppression. Cell 100(4):387–390. https://doi.org/10.1016/s0092-8674(00)80674-1
Kessler T, Sahm F, Blaes J, Osswald M, Rubmann P, Milford D, Urban S, Jestaedt L et al (2015) Glioma cell VEGFR-2 confers resistance to chemotherapeutic and antiangiogenic treatments in PTEN-deficient glioblastoma. Oncotarget 6(31):31050–31068. https://doi.org/10.18632/oncotarget.2910
Ju RJ, Mu LM, Lu WL (2013) Targeting drug delivery systems for circumventing multidrug resistance of cancers. Ther Deliv 4(6):667–671. https://doi.org/10.4155/tde.13.39
Li XY, Zhao Y, Sun MG, Shi JF, Ju RJ, Zhang CX, Li XT, Zhao WY et al (2014) Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials 35(21):5591–5604. https://doi.org/10.1016/j.biomaterials.2014.03.049
Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M et al (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27(5):740–745. https://doi.org/10.1200/JCO.2008.16.3055
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S et al (2007) Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res 13(4):1253–1259. https://doi.org/10.1158/1078-0432.CCR-06-2309
Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S et al (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25(30):4722–4729. https://doi.org/10.1200/JCO.2007.12.2440
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27(28):4733–4740. https://doi.org/10.1200/JCO.2008.19.8721
Taal W, Oosterkamp HM, Walenkamp AM, Dubbink HJ, Beerepoot LV, Hanse MC, Buter J, Honkoop AH et al (2014) Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol 15(9):943–953. https://doi.org/10.1016/S1470-2045(14)70314-6
Wick W, Brandes A, Gorlia T, Bendszus M, Sahm F, Taal W, Taphoorn MJ, Domont J et al (2016) EORTC 26101 phase III trial exploring the combination of bevacizumab and lomustine in patients with first progression of a glioblastoma. J Clin Oncol 34:2001–2001. https://doi.org/10.1200/JCO.2016.34.15_suppl.2001
Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K et al (2014) Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med 370(8):709–722. https://doi.org/10.1056/NEJMoa1308345
Herrlinger U, Schafer N, Steinbach JP, Weyerbrock A, Hau P, Goldbrunner R, Friedrich F, Rohde V et al (2016) Bevacizumab plus irinotecan versus temozolomide in newly diagnosed O6-methylguanine-DNA methyltransferase nonmethylated glioblastoma: the randomized GLARIUS trial. J Clin Oncol 34(14):1611–1619. https://doi.org/10.1200/JCO.2015.63.4691
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11(1):69–82. https://doi.org/10.1016/j.ccr.2006.11.020
Samant RS, Shevde LA (2011) Recent advances in anti-angiogenic therapy of cancer. Oncotarget 2(3):122–134. https://doi.org/10.18632/oncotarget.234
Tanne JH (2011) FDA cancels approval for bevacizumab in advanced breast cancer. BMJ 343:d7684. https://doi.org/10.1136/bmj.d7684
Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, Eichler AF, Drappatz J et al (2010) Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol 28(17):2817–2823. https://doi.org/10.1200/JCO.2009.26.3988
Ameratunga M, Pavlakis N, Wheeler H, Grant R, Simes J, Khasraw M (2018) Anti-angiogenic therapy for high-grade glioma. Cochrane Database Syst Rev 11:CD008218. https://doi.org/10.1002/14651858.CD008218.pub4
Jo J, Wen PY (2018) Antiangiogenic therapy of high-grade gliomas. Prog Neurol Surg 31:180–199. https://doi.org/10.1159/000467379
Norden AD, Drappatz J, Wen PY (2008) Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol 7(12):1152–1160. https://doi.org/10.1016/S1474-4422(08)70260-6
Norden AD, Drappatz J, Wen PY (2008) Antiangiogenic therapy in malignant gliomas. Curr Opin Oncol 20(6):652–661. https://doi.org/10.1097/CCO.0b013e32831186ba
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-Oncology 12(3):233–242. https://doi.org/10.1093/neuonc/nop027
Narayana A, Kelly P, Golfinos J, Parker E, Johnson G, Knopp E, Zagzag D, Fischer I et al (2009) Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J Neurosurg 110(1):173–180. https://doi.org/10.3171/2008.4.17492
Kuhnert F, Tam BY, Sennino B, Gray JT, Yuan J, Jocson A, Nayak NR, Mulligan RC et al (2008) Soluble receptor-mediated selective inhibition of VEGFR and PDGFRbeta signaling during physiologic and tumor angiogenesis. Proc Natl Acad Sci U S A 105(29):10185–10190. https://doi.org/10.1073/pnas.0803194105
Lu-Emerson C, Duda DG, Emblem KE, Taylor JW, Gerstner ER, Loeffler JS, Batchelor TT, Jain RK (2015) Lessons from anti-vascular endothelial growth factor and anti-vascular endothelial growth factor receptor trials in patients with glioblastoma. J Clin Oncol 33(10):1197–1213. https://doi.org/10.1200/JCO.2014.55.9575
Tabouret E, Boudouresque F, Barrie M, Matta M, Boucard C, Loundou A, Carpentier A, Sanson M et al (2014) Association of matrix metalloproteinase 2 plasma level with response and survival in patients treated with bevacizumab for recurrent high-grade glioma. Neuro-Oncology 16(3):392–399. https://doi.org/10.1093/neuonc/not226
D'Alessandris QG, Martini M, Cenci T, Capo G, Ricci-Vitiani L, Larocca LM, Pallini R (2015) VEGF isoforms as outcome biomarker for anti-angiogenic therapy in recurrent glioblastoma. Neurology 84(18):1906–1908. https://doi.org/10.1212/WNL.0000000000001543
Zhong J, Ali AN, Voloschin AD, Liu Y, Curran WJ Jr, Crocker IR, Shu HK (2015) Bevacizumab-induced hypertension is a predictive marker for improved outcomes in patients with recurrent glioblastoma treated with bevacizumab. Cancer 121(9):1456–1462. https://doi.org/10.1002/cncr.29234
Guarnaccia L, Navone SE, Trombetta E, Cordiglieri C, Cherubini A, Crisa FM, Rampini P, Miozzo M et al (2018) Angiogenesis in human brain tumors: screening of drug response through a patient-specific cell platform for personalized therapy. Sci Rep 8(1):8748. https://doi.org/10.1038/s41598-018-27116-7
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8):592–603. https://doi.org/10.1038/nrc2442
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R et al (2002) VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci U S A 99(17):11393–11398. https://doi.org/10.1073/pnas.172398299
Norden AD, Drappatz J, Muzikansky A, David K, Gerard M, McNamara MB, Phan P, Ross A et al (2009) An exploratory survival analysis of anti-angiogenic therapy for recurrent malignant glioma. J Neuro-Oncol 92(2):149–155. https://doi.org/10.1007/s11060-008-9745-8
Du R, Petritsch C, Lu K, Liu P, Haller A, Ganss R, Song H, Vandenberg S et al (2008) Matrix metalloproteinase-2 regulates vascular patterning and growth affecting tumor cell survival and invasion in GBM. Neuro-Oncology 10(3):254–264. https://doi.org/10.1215/15228517-2008-001
Xue Q, Cao L, Chen XY, Zhao J, Gao L, Li SZ, Fei Z (2017) High expression of MMP9 in glioma affects cell proliferation and is associated with patient survival rates. Oncol Lett 13(3):1325–1330. https://doi.org/10.3892/ol.2017.5567
Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP et al (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11(1):83–95. https://doi.org/10.1016/j.ccr.2006.11.021
Minniti G, Salvati M, Arcella A, Buttarelli F, D'Elia A, Lanzetta G, Esposito V, Scarpino S et al (2011) Correlation between O6-methylguanine-DNA methyltransferase and survival in elderly patients with glioblastoma treated with radiotherapy plus concomitant and adjuvant temozolomide. J Neuro-Oncol 102(2):311–316. https://doi.org/10.1007/s11060-010-0324-4
Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, Grabenbauer GG, Ochsenbein AF et al (2010) Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol 28(16):2712–2718. https://doi.org/10.1200/JCO.2009.26.6650
Minder P, Zajac E, Quigley JP, Deryugina EI (2015) EGFR regulates the development and microarchitecture of intratumoral angiogenic vasculature capable of sustaining cancer cell intravasation. Neoplasia 17(8):634–649. https://doi.org/10.1016/j.neo.2015.08.002
Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P (2009) Transforming growth factor-beta 1 (TGF-beta1) induces angiogenesis through vascular endothelial growth factor (VEGF)-mediated apoptosis. J Cell Physiol 219(2):449–458. https://doi.org/10.1002/jcp.21706
Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T et al (2013) Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol 31(26):3212–3218. https://doi.org/10.1200/JCO.2012.47.2464
Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, Mason W, Weller M et al (2010) Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol 28(7):1168–1174. https://doi.org/10.1200/JCO.2009.23.2595
de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, Yao J, Jackson EF et al (2011) Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol 29(19):2689–2695. https://doi.org/10.1200/JCO.2010.34.1636
Muhic A, Poulsen HS, Sorensen M, Grunnet K, Lassen U (2013) Phase II open-label study of nintedanib in patients with recurrent glioblastoma multiforme. J Neuro-Oncol 111(2):205–212. https://doi.org/10.1007/s11060-012-1009-y
Iwamoto FM, Lamborn KR, Robins HI, Mehta MP, Chang SM, Butowski NA, Deangelis LM, Abrey LE et al (2010) Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02). Neuro-Oncology 12(8):855–861. https://doi.org/10.1093/neuonc/noq025
Reardon DA, Groves MD, Wen PY, Nabors L, Mikkelsen T, Rosenfeld S, Raizer J, Barriuso J et al (2013) A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clin Cancer Res 19(4):900–908. https://doi.org/10.1158/1078-0432.CCR-12-1707
Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, Marcello J, Herndon JE 2nd et al (2011) Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neuro-Oncol 101(1):57–66. https://doi.org/10.1007/s11060-010-0217-6
Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, Fine HA (2013) Continuous daily sunitinib for recurrent glioblastoma. J Neuro-Oncol 111(1):41–48. https://doi.org/10.1007/s11060-012-0988-z
Kreisl TN, McNeill KA, Sul J, Iwamoto FM, Shih J, Fine HA (2012) A phase I/II trial of vandetanib for patients with recurrent malignant glioma. Neuro-Oncology 14(12):1519–1526. https://doi.org/10.1093/neuonc/nos265
Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A et al (2014) A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med 370(8):699–708. https://doi.org/10.1056/NEJMoa1308573
Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, Mazurkiewicz M, Salacz M et al (2015) Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncology 17(5):708–717. https://doi.org/10.1093/neuonc/nou356
Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, Aldape KD, Lhermitte B et al (2014) Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol 15(10):1100–1108. https://doi.org/10.1016/S1470-2045(14)70379-1
Pope WB, Lai A, Nghiemphu P, Mischel P, Cloughesy TF (2006) MRI in patients with high-grade gliomas treated with bevacizumab and chemotherapy. Neurology 66(8):1258–1260. https://doi.org/10.1212/01.wnl.0000208958.29600.87
Reardon DA, Egorin MJ, Desjardins A, Vredenburgh JJ, Beumer JH, Lagattuta TF, Gururangan S, Herndon JE 2nd et al (2009) Phase I pharmacokinetic study of the vascular endothelial growth factor receptor tyrosine kinase inhibitor vatalanib (PTK787) plus imatinib and hydroxyurea for malignant glioma. Cancer 115(10):2188–2198. https://doi.org/10.1002/cncr.24213
Funding
This work was supported by the University of Illinois Cancer Center, Department of Medicine, Chicago, IL 60612, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahir, B.K., Engelhard, H.H. & Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol Neurobiol 57, 2461–2478 (2020). https://doi.org/10.1007/s12035-020-01892-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01892-8