
Abstract
Malignant brain tumors, including glioblastoma (GBM), display growth, survival, and invasive properties that are coupled to blood vessels and vascular-derived factors. For example, GBM stem cells (GSCs) home to perivascular niches and invasive tumor cells commonly disperse through the brain microenvironment via extracellular matrix (ECM)-rich vascular basement membranes. Anti-vascular agents that target angiogenesis, and particularly those involving vascular endothelial cell growth factor-A (VEGF-A) and its receptors, improve progression-free survival in GBM patients. However, these benefits are often transient due to compensation by alternative angiogenic pathways. The detailed molecular mechanisms that couple GBM cells to blood vessels during tumor growth and progression as well as following anti-angiogenesis therapies are just beginning to be elucidated, with various cytokines, growth factors, and ECM proteins playing important roles. In this review we will highlight molecular pathways that link cerebral blood vessels and GBM cells during tumor growth, progression, and invasion. We will also discuss mechanisms underlying GBM-induced angiogenesis, with a particular focus placed on roles for integrin adhesion receptors and their ECM protein ligands. Therapies that target angiogenesis in GBM and other brain cancers will also be summarized.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Brain cancer
- Glioblastoma
- Growth factors
- Integrins
- Extracellular matrix
- Invasion
- Neurovascular unit
- Vascular niches
1 Introduction
The formation of new blood vessels via endothelial cell (EC) proliferation and sprouting, or angiogenesis, is essential for proper development and physiology of all mammalian organs (Adams and Alitalo 2007; Potente et al. 2011). This is particularly relevant in the central nervous system (CNS)—comprised of the brain, spinal cord, and retina—where neurons and glia regulate EC behaviors via direct cell–cell contacts as well as secreted growth factors and extracellular matrix (ECM) proteins (McCarty 2009a; Zacchigna et al. 2008). Aberrant regulation of angiogenesis occurs in various diseases including brain cancers such as gliomas (Bao et al. 2006; Calabrese et al. 2007; Gilbertson and Gutmann 2007). Hallmark features of malignant gliomas include pathological neovascularization, disruption of the intratumoral blood–brain barrier (BBB), and perivascular tumor cell dispersal (Gilbertson and Rich 2007; Jain et al. 2007a; Louis 2006). Gliomas afflict approximately 20,000 people within the United States each year (Holland 2001; Maher et al. 2001; Ohgaki 2005). They represent the most common type of primary brain tumors, and in their advanced stages they are one of the deadliest forms of cancer. Most high-grade gliomas are refractory to standard surgical, radiation, and chemotherapeutic interventions (Ware et al. 2003). Survival rates have changed little in the last few decades, with nearly 100 % of patients succumbing to the disease within 3 years after diagnosis. Hence, understanding the basic cellular and molecular pathways that contribute to glioma growth and invasiveness may lead to new therapeutic strategies to treat or prevent the pathogenesis of this insidious disease.
Gliomas can be subdivided into three major categories based on their histology and prognosis: astrocytomas, oligodendrogliomas, and ependymomas. Astrocytomas, or gliomas of presumptive astrocytic origin, can be further divided into four main grades (Ware et al. 2003). Grade I pilocytic astrocytomas develop mostly in young adults and are managed primarily via surgical interventions. Grade II astrocytomas are gliomas consisting of differentiated and invasive tumor cells. Grade III anaplastic astrocytoma and grade IV GBMs are poorly differentiated and highly infiltrative tumors (Louis 2006).
Multiple chromosomal abnormalities and gene expression defects have been identified in gliomas, and these alterations often correlate with histological grade and clinical prognosis (Ohgaki 2005; Phillips et al. 2006). In general, glioma initiation and progression involve gene mutations that (1) deregulate growth factor receptor tyrosine kinase signaling and (2) alter the cell cycle checkpoint machinery. Low-grade tumors often express high levels of the growth factors FGF2 and PDGF, as well as their cognate receptor tyrosine kinases (Holland 2001; Shih et al. 2004). Elevated receptor activation in turn leads to amplification of downstream signaling events, often involving Ras (Ding et al. 2001; Holland et al. 2000), and commonly correlates with loss of p53 tumor suppressor functions (Reilly et al. 2000). Disruption of the cell cycle regulatory network is linked to the progression of high-grade gliomas (Holland et al. 1998b; Uhrbom et al. 2002). For example, anaplastic astrocytomas often contain deletions of the tumor suppressors Ink4a/Arf and retinoblastoma (Rb) (Bachoo et al. 2004; Xiao et al. 2002). GBMs also commonly display amplification of EGFR signaling, which can lead to Ras hyperactivation (Holland et al. 1998a). Collectively, these alterations disrupt multiple intracellular signaling pathways that contribute to the progression of glioma from low grade to high grade (Wechsler-Reya and Scott 2001).
The genetic mutations that contribute to gliomagenesis are commonly mutated in other cancers. Thus, glioma progression is likely influenced by a combination of tumor cell-extrinsic factors (Fukumura et al. 2001; Winkler et al. 2004), as well as alterations in a distinct tumor-initiating cell of origin (Sanai et al. 2005; Shih and Holland 2004; Wechsler-Reya and Scott 2001). The exact cell type that gives rise to glioma remains uncertain. However, most neural cells in the adult brain are terminally differentiated. Thus, the tumor-initiating cell of origin for glioma is limited to those compartments that retain proliferative potential, i.e., neural stem cells, glial progenitors, and differentiated glia. Genetically engineered mouse models reveal that astrocytomas arise from presumptive neural stem cells (Alcantara Llaguno et al. 2009; Zheng et al. 2008) and/or oligodendroglial cells (Liu et al. 2011). These cells reside in various regions of the adult brain (Gilbertson and Gutmann 2007), and abnormal regulation of their proliferative and differentiative capabilities likely triggers glioma onset and progression (Aboody et al. 2000; Fomchenko and Holland 2006; Maher et al. 2001).
Most brain tumors, and particularly GBM, harbor a subpopulation of proliferative and multipotent tumor-initiating cells, or GSCs (Dirks 2006). GSCs have several similarities with nonmalignant neural stem cells, including expression of common molecular markers, for example, Nestin and CD133/Prominin-1 (Read et al. 2006). Neural stem cells and brain tumor stem cells also intimately associate with vascular basement membranes in vascular niches. Importantly, contact and communication events between brain tumor stem cells and angiogenic blood vessels positively regulate tumor growth and progression (Bao et al. 2006; Calabrese et al. 2007; Salmaggi et al. 2006). Recently, subpopulations of GBM cells have been shown to transdifferentiate to ECs and pericytes and contribute to vascular pathologies. These events are also influenced by cues within the microenvironment (Ricci-Vitiani et al. 2011; Soda et al. 2011b; Wang et al. 2011) although their pathophysiological significance remains to be determined (Rodriguez et al. 2012). The focus of this review is to highlight how glioma-derived growth factors and adhesion proteins impact angiogenesis in the brain tumor microenvironment.
2 Blood Vessel Pathologies in Glioma
Malignant gliomas are defined, in part, by the development of hallmark angiogenesis pathologies including florid microvascular cell proliferation leading to the formation of capillaries with glomeruloid-like tufts (Fischer et al. 2005; Jain et al. 2007b). These abnormal blood vessel morphologies are accompanied by enhanced vascular permeability due to loss of the intratumoral BBB (Jansen et al. 2004; Lopes 2003; Rong et al. 2006). Although traditionally defined as the tight junctions between ECs, the BBB is now considered just one component of a larger multicellular complex, or neurovascular unit (NVU) (Abbott et al. 2006), consisting of neurons and astrocytes, vascular ECs and pericytes, as well as various growth factors and extracellular matrix (ECM) proteins in vascular basement membranes (McCarty 2009b). Comprised mainly of EC tight junctions and multidrug resistance transporters, the BBB regulates the exchange of ions, molecules, and cells between the circulation and brain and is an impediment for drug delivery (Liebner et al. 2011; Pardridge 2002). The molecular mechanisms that control BBB development and physiology remain largely unknown, although Wnts (Daneman et al. 2009; Liebner et al. 2008; Stenman et al. 2008), G protein coupled receptors such as Gpr124 (Anderson et al. 2011; Cullen et al. 2011; Kuhnert et al. 2010), and integrin-activated TGFβs (McCarty et al. 2005; Proctor et al. 2005) play important roles as detailed below.
3 Growth Factors and Cell Adhesion Molecules in Glioma Angiogenesis
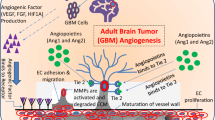
A balance between angiogenic activators and inhibitors regulates blood vessel growth, stability, and permeability (Hanahan and Folkman 1996). In glioma, this balance is disrupted by a number of pro-angiogenic and anti-angiogenic factors. Below we will discuss various growth factors and cell adhesion proteins that control angiogenesis during glioma initiation and progression (Fig. 7.1).

A summary of factors involved in glioma exploitation of angiogenesis. Glioma cells dynamically communicate with blood vessels via various secreted growth factors and ECM proteins. These factors control cerebral endothelial cell and pericyte survival, proliferation, and BBB permeability
3.1 VEGF
Vascular endothelial cell growth factor-A (VEGF-A) is a critical regulator of angiogenesis during organ development as well as tumor growth and progression. VEGF-A was first discovered by Dvorak and colleagues who initially named it vascular permeability factor for its ability to enhance permeability properties of blood vessels (Dvorak 2006). Efforts by Ferrara and colleagues revealed that VEGF-A also regulates vascular EC proliferation, migration, and survival (Leung et al. 1989). Subsequent studies by a number of independent groups identified a larger gene family consisting of at least six different VEGF family members (Jansen et al. 2004). VEGF genes express multiple protein isoforms and each can bind to distinct or shared transmembrane receptor tyrosine kinases including VEGFR1, VEGFR2, and VEGFR3 as well as Neuropilins (Bielenberg et al. 2006). These receptors are also expressed in glioma cells, suggesting autocrine VEGF signaling pathways (Ellis and Hicklin 2008).
VEGF-A expression is upregulated in glioma cells and this correlates with tumor growth and malignancy (Bulnes et al. 2012). Hypoxia-inducible factor 1a (HIF1α) is one of the major transcription factors that regulate VEGF-A gene expression (Kaelin and Ratcliffe 2008; Semenza 2003). In addition to HIF1α, other transcription factors also bind to the VEGF-A promoter to regulate gene expression. For example, p53 and VHL tumor suppressors form complexes with SP1 transcription factors and inhibit VEGF transcription (Kargiotis et al. 2006; Mukhopadhyay et al. 1997). p53 is commonly deleted in high-grade gliomas (Verhaak et al. 2010). EGFR signaling, which is often amplified in gliomas, also regulates VEGF-A expression via activation of the MAPK/ERK pathway (Woods et al. 2002). These effects were nullified by the inhibition using anti-EGFR antibodies (Goldman et al. 1993; Okamura et al. 1992; Valter et al. 1999). In addition, a truncated and constitutively active form of EGFR, EGFRvIII, has been shown to upregulate VEGF expression in glioma cells via Ras-dependent mechanisms (Feldkamp et al. 1999).
3.2 Notch/Delta
Cross talk between the VEGF-A and Notch pathways coordinately regulates blood vessel growth and stability (Chappell et al. 2009; Jakobsson et al. 2010). For example, VEGF-A stimulates Notch 1 expression which induces the formation of specialized endothelial “tip cells” found at the leading front of sprouting blood vessels (Hellstrom et al. 2007a). VEGFR2 signaling and tip cell formation are dampened by the anti-angiogenic Notch ligand Dll4 (Hellstrom et al. 2007b; Noguera-Troise et al. 2006). Deletion of one Dll4 allele or blockade of Notch activation with γ-secretase inhibitors induces similar phenotypes including hyperactive tip cell formation (Hellstrom et al. 2007b; Noguera-Troise et al. 2006; Siekmann and Lawson 2007). In contrast, Jag1 is a pro-angiogenic Notch ligand that counterbalances Dll4-Notch signaling and stimulates tip cell formation (Benedito et al. 2009). Exploitation of Jag1 by cancer cells has been reported; for example, epithelial carcinoma cells overexpress Jag1 and activate Notch in ECs (Zeng et al. 2005). Additionally, Jag1 in metastatic breast cancer cells mediates interactions with Notch in osteoblasts of the bone microenvironment (Sethi et al. 2011). Lastly, Notch signaling pathways are often hyperactivated in GBM (Fan et al. 2010; Hambardzumyan et al. 2008; Stockhausen et al. 2010). Inhibition of Notch activation diminishes mouse and human GSC self-renewal (Fan et al. 2010; Jeon et al. 2008) and can synergize with temozolomide to reduce glioma growth in xenograft models (Gilbert et al. 2010).
3.3 FGFs
Fibroblast growth factors (FGFs) are a family of structurally related proteins that regulate a wide range of developmental and pathophysiological processes (Friesel and Maciag 1995). Among the nine FGF family members, FGF-1 and FGF-2 are well characterized as angiogenic mediators and are often overexpressed in gliomas. FGF signaling has also been reported to promote VEGF expression in glioma cells (Friesel and Maciag 1995; Stefanik et al. 1991; Tsai et al. 1995). Degradation of ECM is an important step in blood vessel sprouting. FGF-2 facilitates EC migration through the ECM by upregulating urokinase-type plasminogen activator (uPA), which activates plasmin, a protease for many ECM protein components (Dunn et al. 2000).
3.4 PDGFs
Members of the platelet-derived growth factor (PDGF) family signal through different receptor tyrosine kinases (PDGFRs). Receptor binding activates multiple kinase cascades including PI3kinase, MAPK, JAK, SRC, and phospholipase C gamma (Fomchenko and Holland 2007). Gliomas express high levels of PDGFA and PDGFRα, but the tumor vasculature expresses low levels of PDGFRα. Instead, many glioma blood vessels express robust levels of PDGFRβ. These data suggest PDGFRα-dependent autocrine/paracrine signaling mechanisms in tumor cells and PDGFRβ-dependent paracrine signaling in ECs and pericytes (Hermanson et al. 1992). To study the effects of PDGFB in brain tumorigenesis, a mouse model was generated by overexpressing PDGFRβ in glial cells (Hermanson et al. 1992). These transgenic mice do not develop spontaneous tumors and showed normal brain development. However, when crossed to a p53−/− background mice developed tumors with pathologies similar to human GBMs including pseudopalisading necrosis, glomeruloid vessels, and BBB breakdown. Interestingly, overexpression of PDGFA in neurogenic regions of the adult mouse brain leads to premalignant gliomas via uncontrolled proliferation of neural stem and progenitor cells (Jackson et al. 2006). Interestingly, PDGFA is also a molecular marker for the classical GBM subtype (Verhaak et al. 2010). Using in vitro models, PDGFB was found to induce chemotaxis of rat brain microvascular ECs verifying the direct action of PDGFs during angiogenesis. PDGFs did not induce migratory effects on glioma cells, but were chemotactic for ECs (Brockmann et al. 2003).
3.5 TGFβs
The TGFβ superfamily of cytokines consists of bone morphogenetic proteins, Mullerian inhibiting substance, and activins. These proteins are involved in regulating a number of cellular processes ranging from proliferation to apoptosis (Massague et al. 2000). Members of the TGFβ family (TGFβ 1, 2, and 3) signal via canonical receptor serine/threonine kinases, TGFβR2 and TGFβR1. TGFβR2 is shared by all ligands and dimerizes with different TGFβR1s to form signaling-competent receptor complexes. Endoglin is a TGFβ co-receptor that facilitates ligand presentation to TGFβR1/TGFβR2 heterodimers (Massague and Gomis 2006). Immunohistochemical analysis of gliomas has shown upregulation of TGFβ as well as TGFβR1 and TGFβR2. TGFβ signaling via Smad transcription factors, or canonical TGFβ signaling, is also hyperactivated in many high-grade gliomas likely via uncontrolled growth and differentiation of GSCs (Bruna et al. 2007; Penuelas et al. 2009).
TGFβs often inhibit proliferation by cell cycle arrest in the G1 phase and this is mediated by regulation of INK4B expression. Interestingly, at higher TGFβ concentrations these growth inhibitory effects are negligible or in some cases potentiate glioma cell proliferation, in part owing to loss of p15 and p16 (Jen et al. 1994; Rich et al. 1999). The effects of TGFβs on angiogenesis remain controversial. In vitro studies using bovine aortic ECs treated with TGFβs showed an inhibitory effect while in vivo studies using angiogenesis system showed pro-angiogenic effects (Fajardo et al. 1996; Frater-Schroder et al. 1986). Ablation of TGFβ receptors in ECs leads to early lethality due to impaired yolk sac angiogenesis and cardiovascular development (Carvalho et al. 2007; Park et al. 2008). Additionally, TGFβ can stimulate VEGF production in glioma cells and pharmacological inhibition of TGFβR1 leads to decreased expression of VEGF and plasminogen activator inhibitor-1 (PAI-1) in gliomas (Hjelmeland et al. 2004; Koochekpour et al. 1996). PDGFA and PDGFB are downstream effectors of TGFβ in ECs while PDGFRβ expression is upregulated in vascular smooth muscle cells (Dunn et al. 2000; Helseth et al. 1988).
3.6 Angiopoietins
Angiopoietins (Ang1 and Ang2) play essential roles in regulating blood vessel development and stability. During embryogenesis Ang1 binds to its receptor tyrosine kinase, Tie2, and regulates stability of pericyte–EC interactions (Suri et al. 1996). Tumor cells also express Ang1, but Ang2 expression is generally limited to activated endothelium (Augustin et al. 2009). Ang2 competes with Ang1 for Tie2 binding and antagonizes Ang1 signaling (Maisonpierre et al. 1997). Hypoxia induces Ang2 expression in ECs, which disrupts Ang1–Tie2 signaling probably by acting as an antagonist to Ang1 (Holash et al. 1999). Antagonists that inhibit angiopoietin interactions with Tie receptors are currently being tested in clinical trials as anti-angiogenic agents (Peeters et al. 2013).
3.7 HGF
Scatter factor/hepatocyte growth factor (SF/HGF) signaling plays versatile roles in physiological and pathological processes including organogenesis and cancer (Abounader and Laterra 2005; Birchmeier and Gherardi 1998). SF/HGF and its receptor tyrosine kinase, c-Met, are expressed by glioma cells which correlate with malignancy and vascular pathologies (Koochekpour et al. 1997; Moriyama et al. 1998; Rosen et al. 1996). Overexpression of SF/HGF caused increased tumorigenesis and tumor angiogenesis while inhibition of c-Met signaling using blocking antibodies or siRNAs suppresses tumor growth (Abounader et al. 1999, 2002; Laterra et al. 1997). In addition to glioma cells, c-Met is also expressed in tumor-associated blood vessels suggesting paracrine signals from tumor cells lead to EC growth and sprouting (Ding et al. 2003; Nakamura et al. 1995).
HGF contributes to degradation of vascular basement membranes and promotes EC migration by upregulating expression of MMPs such as MT1-MMP, MMP2, and urokinase. Another possible way SF/HGF contributes to tumor angiogenesis is by promoting proliferation through MAPK/Stat3 pathway and inhibiting apoptosis of tumor ECs (Lamszus et al. 1998; Ma et al. 2002; Wang et al. 2004). In Matrigel assays using human umbilical vein ECs, SF/HGF induces EC tube formation in a dose-dependent manner. This effect was abolished by treating with anti-HGF antibodies. In another experiment when ECs and SF/HGF secreting keratinocytes were cocultured in an in vitro system it led to the formation of EC tubes (Jiang et al. 1999; Martin et al. 1999; Wojta et al. 1999).
3.8 IL-6 and IL-8
Interleukins are cytokines secreted by normal and tumor cells, and in gliomas they promote proliferation and directional migration (Brat et al. 2005). Many glioma cells are capable of secreting IL-6, which can activate Sp1 and Sp3 transcription factors to induce expression of VEGF-A mRNA. IL-8 is also expressed at high levels in many glioma cells (Van Meir et al. 1990, 1992). IL-8 is a potent mediator of tumor angiogenesis via its cell surface receptors CXCR1, CXCR2, and DARC (Holmes et al. 1991; Murphy and Tiffany 1991). Glioma cells express all three receptors while DARC, but not CXCR1 and CXCR2, is expressed in microvascular ECs. CXCR1 and CXCR2 are expressed in perivascular leukocytes; hence, the angiogenic properties of IL-8 involve inflammatory responses as well. Lastly, under hypoxic conditions, IL-8 expression is upregulated via Ap-1 binding to IL-8 promoter sequences (Brat et al. 2005; Desbaillets et al. 1997, 1999).
3.9 TNFα
Tumor necrosis factor alpha (TNFα) is a macrophage-derived cytokine that has pleiotropic effect on cells. At low concentrations TNFα is pro-angiogenic while at high concentrations it displays anti-angiogenic activities (Fajardo et al. 1992). In high-grade gliomas, TNFα is expressed in multiple cell types including tumor cells and ECs (Maruno et al. 1997), while its receptors are expressed by ECs (Slowik et al. 1993). Angiogenic effects of TNFα are mediated indirectly by inducing expression of a number of other pro-angiogenic molecules. For example, upon TNFα treatment VEGF-A expression is upregulated in glioma cells. TNFα also upregulates expression of VEGF, IL-8, and FGFs in human microvascular ECs in vitro and blocking antibodies directed against TNFα inhibit these effects (Kargiotis et al. 2006; Ryuto et al. 1996).
3.10 Other Pro-angiogenic Factors
Additional growth factors and cytokines play important yet less characterized roles in angiogenesis. For example, the inducible early response gene product Cyr61/CNN1 and connective tissue growth factor CTGF/CNN2 are growth factors belonging to CNN family that induce proliferative effects on glioma cells and are downstream targets of c-Met (Goodwin et al. 2010; Jedsadayanmata et al. 1999). Expression of these proteins correlates with glioma malignancy. Tumor-associated ECs also express CTGF, suggesting pro-angiogenic roles (Pan et al. 2002; Xie et al. 2004).
The cytokine stromal cell-derived factor 1 (SDF-1/CXCL12) and its chemokine receptor CXCR4 regulate glioma cell migration and tumor cell homing to blood vessels (Rao et al. 2012). Immunohistochemical analysis revealed expression and co-localization of SDF-1 and CXCR4 in glioma cells, with an increasing intensity correlating with tumor grade. Expression of these proteins was absent in normal brain (Rempel et al. 2000). This suggests that the SDF-1/CXCR4 signaling axis may be a novel target for inhibiting glioma growth and invasion.
Various signaling effectors that control neural development also play central roles in glioma growth and angiogenesis (Eichmann et al. 2005). For example, semaphorins have important functions in controlling axonal guidance and also regulate angiogenesis. Semaphorins bind to plexin as well as Nr cell surface receptors. Nrps are co-receptors for VEGF-A in ECs and tumor cells and promote cell proliferation. Whereas VEGFR2-dependent angiogenesis results in increased vascular permeability, plexin and Nrp elicit anti-angiogenic effects upon semaphorin binding. Additionally, application of anti-Nrp inhibitory antibodies in preclinical brain tumor models results in suppression of tumor growth (Snuderl et al. 2013). Lastly, Slit-Robo interactions are important regulatory pathways in angiogenesis. Depending on its interacting receptor, Slit has opposing roles in angiogenesis. For example, when Slit binding to Robo1 leads to pro-angiogenic effects, interactions with Robo4 have anti-angiogenic outcomes (Jain et al. 2007a; Tate and Aghi 2009).
3.11 Anti-angiogenic Growth Factors and Cytokines
A balance between pro-angiogenic and anti-angiogenic factors, termed the angiogenic switch, controls vessel growth and stability. Alterations in this switch, for example, overexpression of pro-angiogenic factors or diminished expression of anti-angiogenic factors, promote blood vessel growth and sprouting (Hanahan and Folkman 1996). Below we detail a partial list of anti-angiogenic molecules and their likely roles in regulating glioma angiogenesis.
Angiostatin is a 38 kDa fragment of plasminogen generated by cathepsin D and MMP activities. It was the first anti-angiogenic factor to be identified in mouse models of metastatic cancer (O’Reilly et al. 1994; Tate and Aghi 2009). Angiostatin is a ligand for αvβ3 integrin and downstream signaling leads to apoptosis of ECs and tumor cells (Kirsch et al. 1998; Nishida et al. 2006; Tarui et al. 2001). An angiostatin receptor is NG2, a chondroitin sulfate proteoglycan expressed by pericytes, oligodendrocytes, and tumor cells (Stallcup and Huang 2008). NG2 can bind and sequester angiostatin and impact angiogenesis by altering the angiogenic switch (Chekenya et al. 2002; Chekenya and Pilkington 2002). Another receptor for angiostatin is ATP synthase (Rege et al. 2005). Interactions with angiostatin inhibit the enzymatic activities of ATP synthase and reduce cellular ATP production (Moser et al. 2001). Angiomotin was also identified as an angiostatin binding partner in yeast two-hybrid assays. Angiostatin functions by antagonizing the normal pro-migratory and pro-invasive functions of angiomotin (Rege et al. 2005).
Endostatin is a C-terminal fragment of type XVIII collagen, a basement membrane protein, and is another protein with anti-angiogenic properties. Endostatin induces its effects by binding to fibronectin and α5β1 and αvβ3 integrins and potentially blocking the formation of endothelial focal adhesions (O’Reilly et al. 1997; Rehn et al. 2001; Wickstrom et al. 2002).
Thrombospondins are ECM proteins that induce pro- and anti-angiogenic outcomes. In the aortic ring assay, overexpression of thrombospondins inhibits vascular cell migration and blood vessel sprouting. These effects are mediated through the CLESH domain of the cells surface receptor CD36 and type I repeats of thrombospondins-1 and -2 (Klenotic et al. 2013). Thrombospondin knockout mice also display defective wound healing and tumor-induced angiogenesis (Lawler 2000).
Tissue inhibitors of matrix metalloproteases (TIMPs) negatively regulate MMP enzymatic activities; they control EC proliferation and downregulate expression of VEGF-A. TIMPs also have pro-angiogenic properties owing to their potential to block MMP activities. For example, reduced levels of MMP-dependent expression of angiostatin and endostatin result in anti-tumorigenic and anti-angiogenic properties (Jiang et al. 2002). Lastly, pigment epithelial-derived factor (PEDF) is a member of serpin family of serine proteases that regulate neuronal differentiation and survival and are also negative regulators of angiogenesis. A specific receptor pathway, through which PEDF contributes to anti-angiogenesis, has not revealed but a key pathway involves Fas signaling (Bouck 2002; Rege et al. 2005).
4 Integrins in Glioma Angiogenesis
Integrins are αβ heterodimeric receptors for many ECM protein ligands that play central roles in controlling cell growth, migration, and other responses (Hynes 2002). Integrin-ECM affinity is modulated by “inside-out” signaling mechanisms (Kim et al. 2011; Vinogradova et al. 2002) involving proteins such as talins (Calderwood et al. 1999; Tadokoro et al. 2003) and kindlins (Harburger et al. 2009; Ma et al. 2008) that bind to β integrin cytoplasmic domains and induce conformational changes in extracellular regions (Shattil et al. 2010; Takagi et al. 2002; Xiong et al. 2001). ECM adhesion subsequently triggers “outside-in” signaling via adhesion protein complexes and the cytoskeleton (Harburger and Calderwood 2009; Parsons et al. 2010). In vertebrates there are 26 different integrin genes: 18 genes encoding α subunits and 8 β subunit genes. The network of integrin–ligand interactions is vast: some integrins are ligand-specific while others bind many, sharing ligands. This overlap allows for one ECM ligand to have multiple effects on a cell via adhesion to different integrins.
5 Integrins in GBM Cells
The brain contains a rich milieu of extracellular matrix (ECM) proteins (Thiery 2003) and abnormal regulation of cell–ECM communication is associated with gliomagenesis (Bellail et al. 2004; Gladson 1999; Shi et al. 2007a); see also Chaps. 10 and 11. For example, glioma cells like nonmalignant neural stem cells migrate through the brain parenchyma along blood vessels and white matter tracts (Sanai et al. 2005). In fact, the infiltrative nature of these tumor cells is an important determinant in the poor prognosis associated with GBM. Most metazoan cells communicate with protein components of the ECM via a family of heterodimeric cell surface receptors known as integrins (Hynes 2002). In addition to their extracellular adhesion functions, integrins also regulate intracellular signal transduction pathways that control multiple cellular responses (Giancotti and Ruoslahti 1999). In vertebrates there are 26 distinct integrin genes: 18 genes encoding α subunits, and 8 genes that encode β subunits (Hynes 2002).
Various integrins and intracellular signaling partners have been linked to the onset and/or progression of glioma (Shi et al. 2007b; Tucker 2006; Uhm et al. 1999a). For example, the fibronectin receptor α5β1 integrin is expressed in human glioma cells and inhibition of α5β1 integrin with specific small molecule antagonists retards glioma cell proliferation (Maglott et al. 2006). Additionally, human glioma cell lines express the laminin receptors α3β1 and α6β1, and these integrins regulate migration on laminin substrates (Uhm et al. 1999b).
The five members of the αv integrin subfamily primarily recognize RGD tripeptide motifs present in many shared ECM ligands, most of which are abundantly expressed in the brain microenvironment. αv integrin ECM ligands include vitronectin and fibronectin (Kalluri 2003), collagen IV (Venstrom and Reichardt 1995), and the latent associated peptide of TGFβ1 (LAP-TGFβ1) (Moses and Serra 1996). Various data link abnormal regulation of αv integrin expression and function to glioma cell growth and invasiveness. For example, αvβ1 integrin expressed in U87 glioma cells binds to the extracellular matrix protein, Ang2, leading to enhanced glioma invasiveness (Hu et al. 2006). More recently, αvβ1 integrin was found to be upregulated in glioma cells treated with anti-vascular agents, with integrin expression promoting angiogenesis and tumor cell invasion (Carbonell et al. 2013; Jahangiri et al. 2013). Human malignant gliomas display elevated levels of αvβ3 and αvβ5 integrins, suggesting that these integrins contribute to glioma cell survival and invasion (Bello et al. 2001; Treasurywala and Berens 1998). Indeed, small molecule inhibitors of αvβ3 integrin reduce glioma growth and invasiveness in vitro and in vivo (Chatterjee et al. 2000). Pieper and colleagues have shown that transformed β3−/− astrocytes form abnormally large intracranial tumors, suggesting that αvβ3 integrin may act to suppress tumor cell growth (Kanamori et al. 2004). More recent studies reveal that αvβ3 integrin exerts opposing effects, depending on whether it is expressed in tumor cells or brain microenvironment (Kanamori et al. 2006).
6 αvβ8 in Glioma Angiogenesis and Tumor Cell Invasiveness
The normal brain depends on αvβ8 integrin and its interactions with the ECM. The blood vessels of mice null for αv or β8 dilate, the BBB is compromised, and the mice suffer from severe CNS hemorrhage (McCarty et al. 2002), (Zhu et al. 2002). αvβ8 integrin binds to ECM-associated latent-TGFβ ligands through RGD sites and mediates release of active TGFβs. The ligands then bind the TGFβRI/II and signal through Smads and other pathways resulting in a myriad of effects. In the context of glioma, αvβ8 integrin protein levels are critically important in angiogenesis and invasiveness (Fig. 7.2). Angiogenesis is more severe in tumors with low levels of endogenous β8 integrin and overexpression of the integrin diminishes these angiogenesis pathologies. Glioma cells expressing high endogenous levels of αvβ8 integrin generate less angiogenic tumors, yet the tumors are more invasive. Invasive pathologies can be attenuated by silencing integrin gene expression using lentiviral-delivered shRNAs (Tchaicha et al. 2011). More specifically, changes are detected in cell polarity and directional migration. In scratch-wound assays cells with low levels of β8 integrin had fewer ECM contacts and displayed delayed polarization into the wound region. In contrast, β8 integrin-expressing cells formed organized actin cytoskeletal networks and polarized in a uniform direction toward the wound. αvβ8 integrin control of glioma cell polarity and directional migration is mediated, in part, via binding to RhoGDI1 leading to regulation of the Rho GTPase signaling cascade (Reyes et al. 2013). It has also recently been seen that αvβ8 is negatively regulated by mir-93, leading to gliomas with increased size and neovascularization (Fang et al. 2011).

αvβ8 integrin regulation of blood vessel pathologies in glioma. (a) A human GBM section stained with hematoxylin and eosin (H&E) revealing distended, glomeruloid-like blood vessels. (b) Human GBM section immunostained with antibodies targeting β8 integrin. Note the enrichment of β8 integrin protein expression (brown stain) in perivascular tumor cells
7 αvβ8 Integrin in Neurovascular Development and Physiology
Neural cells and vascular cells within the brain microenvironment intimately interact and communicate to form multicellular structures, or neurovascular units (Ballabh et al. 2004; Iadecola 2004; McCarty 2005; Zlokovic 2005). Proper cell–cell communication at the neurovascular unit is essential for normal CNS development, and abnormal neurovascular functions are linked to various CNS pathologies (Abbott 2002; Ballabh et al. 2004; McCarty 2005). Cerebral blood vessels are entirely compartmentalized from the surrounding neural microenvironment via a vascular basement membrane that contains a rich assortment of ECM components (Marin-Padilla 1985). Astrocyte end feet associate with the ablumenal surfaces of nearly all cerebral blood vessels via direct contacts with the vascular basement membrane (Abbott 2002). Astrocyte–blood vessel communication plays important roles in regulating molecular transport across the BBB, and also modulates rates of cerebral blood flow in response to local metabolic demands (Begley and Brightman 2003; Engelhardt 2003; Neuwelt 2004; Simard et al. 2003; Zonta et al. 2003). Astrocytes express a variety of cell surface adhesion molecules, including several integrins. At least two integrins, α6β4 and αvβ8, mediate contact and communication between perivascular neural cells and ECM components of the vascular basement membrane (Milner and Campbell 2002).
The αv and β8 integrin subunits are absolutely essential for proper neurovascular development (McCarty et al. 2002, 2005). Mouse embryos completely null for the αv integrin gene, and thus lacking all five αv integrin family members, develop CNS-specific vascular defects that include abnormal angiogenesis and intracerebral hemorrhage (Bader et al. 1998; McCarty et al. 2002, 2005). Similar integrin-dependent phenotypes are detected in the neonatal retina, which is vascularized after birth (Hirota et al. 2011).
The β8 integrin subunit pairs exclusively with αv integrin. To study αvβ8 integrin functions in the postnatal CNS Nestin-Cre transgenic mice were used to ablate αv or β8 integrin gene expression specifically in CNS neural cells. Conditional αv integrin mutants develop embryonic intracerebral hemorrhage that is grossly apparent at birth (McCarty et al. 2005). However, unlike complete αv knockouts, Nestin-Cre conditional mutants live beyond the first day of birth and survive for several months. Using a GFAP-Cre transgene, we also induced hemorrhage in the embryonic and neonatal brain after αv gene ablation (McCarty et al. 2005). Similarly, the β8 integrin gene was selectively ablated in the CNS using an identical Nestin-Cre transgene (Proctor et al. 2005). These animals also develop embryonic and neonatal intracerebral hemorrhage that is phenotypically identical to that observed in the αv integrin mutants. Deletion of the other four αv integrin-associated β subunits does not yield similar CNS vascular phenotypes (Hynes 2002). Genetic ablation of αv or β8 integrin expression in vascular ECs using the Tie2-Cre transgene did not lead to intracerebral hemorrhage or other obvious neurovascular defects (McCarty et al. 2005). These αv and β8 integrin mutant mice actually develop intestinal autoimmunity due to activities of Tie2-Cre in hematopoietic stem cells. Subsequent studies have shown that αvβ8 integrin in dendritic cells regulates latent TGFβ activation and signaling to control intestinal homeostasis.
Collectively, these molecular genetic data prove that αvβ8 integrin in CNS neural cells, particularly astroglia, regulates proper neurovascular development. Loss of αvβ8 integrin expression on CNS glia leads to defective glial-vascular cell adhesion, resulting in abnormal brain angiogenesis and intracerebral hemorrhage. αv conditional mutants also display neurological phenotypes, including sporadic seizures and a rigid gait, and mice generally do not survive beyond 8 postnatal months (McCarty et al. 2005). Similar phenotypes have been reported for the β8 integrin mutants (Proctor et al. 2005), again suggesting that the neurological defects that develop in the αv mutants are due to the specific loss of αvβ8 integrin. Additional postnatal brain deficits in β8 integrin mutant mice include impaired neuronal migration in the rostral migratory stream and widespread perivascular reactive gliosis (Mobley and McCarty 2011; Mobley et al. 2009).
αvβ8 integrin is a receptor for LAP-TGFβ1, and adhesion to an RGD peptide sequence within LAP causes activation of TGFβ signaling pathways in ECs (Cambier et al. 2005; Mu et al. 2002). Genetic ablation of TGFβ receptors in ECs leads to neurovascular phenotypes that are identical to those that develop in Nestin-Cre αv or β8 integrin mutants (Allinson et al. 2012; Arnold et al. 2012; Nguyen et al. 2011). Interestingly, TGFβ1 stimulation of vascular ECs in vitro leads to the upregulation of various ECM proteins, such as thrombospondin-1 and plasminogen activator inhibitor-1, that play established roles in regulating developmental angiogenesis and postnatal neurovascular functions (Del Zoppo 2005; Lawler 2000). Lastly, human genetic data reveal that single nucleotide polymorphisms within the TGFβ1 gene are associated with elevated risk of age-related neurovascular diseases (Kim and Lee 2006). Hyperactivation of TGFβ1-mediated signaling pathways is detected in advanced stages of glioma (Bruna et al. 2007; Rich and Bigner 2004). Defective TGFβ activation and signaling are linked to various adult-onset CNS vascular pathologies, including Arteriovenous Malformations (Su et al. 2010), Hereditary Hemorrhagic Telangiectasia, and Pulmonary Arterial Hypertension (Orlova et al. 2011). A single nucleotide polymorphism in the TGFβ1 gene is linked to increased susceptibility to stroke (Kim and Lee 2006).
8 Anti-angiogenesis Therapies in Glioma
Significant progress has been made in understanding the molecular genetic events that lead to GBM initiation and progression (Furnari et al. 2007). However, only 5 % of the patients with GBM survive 5 years or more, and the medium overall survival time is about 15 months (Stupp et al. 2005; Taylor and Gerstner 2013). In addition to surgical resection, current standard-of-care treatments consist of radiation therapy and temozolomide (Stupp et al. 2005). Since gliomas are such highly vascularized neoplasms, targeting angiogenic pathways was thought to have powerful clinical benefits. Indeed, VEGF-A and VEGFRs, the main regulators of angiogenesis, as well as a number of other pro-angiogenic molecules (see above) are often overexpressed in malignant gliomas.
The US Food and Drug Administration approved the use of the anti-angiogenic antibody Bevacizumab/Avastin for the treatment of colon, lung, and breast cancers. Subsequently, in 2009 Bevacizumab was approved as a monotherapy for the treatment of gliomas (Mrugala et al. 2012). Bevacizumab is a humanized monoclonal antibody directed against VEGF-A but not other VEGF family members (Onishi et al. 2011). This antibody binds to all VEGF-A isoforms and proteolytic fragments with comparable affinities. In gliomas, Bevacizumab treatment gave promising results when combined with irinotecan. The treatments resulted in radiographic response rates of 28–40 % and a 6-month progression-free survival rate of 40–50 %. These efforts led to phase 2 clinical trials, which tested Bevacizumab as a monotherapy or in combination with irinotecan (Friedman et al. 2009; Vredenburgh et al. 2007). Combination therapies resulted in progression-free survival rates of 50.2 %, which was significantly higher than the 35 % response with Bevacizumab monotherapies. However, when compared to the medium overall survival, Bevacizumab showed promise with 9.7 months against 8.9 months in combination therapy, although progression-free survival rates were more pronounced with irinotecan. Overall survival was not significantly improved likely due to combined cytotoxic effects, leading to approval of Bevacizumab as a monotherapy for treating recurrent GBM in the United States (Kreisl et al. 2009; Taylor and Gerstner 2013). A recent publication described two patients displaying responses after receiving a combination treatment of radiation followed by temozolomide and bevacizumab, with ongoing progression-free survival of 37 and 47 months (Aguilera et al. 2013).
However, recent studies have revealed unexpected tumor cell behaviors resulting from Bevacizumab treatment. While Bevacizumab caused a reduction in tumor volumes, 30–50 % of patients developed highly infiltrative growth patterns. Inhibition of angiogenesis results in a shift in tumor growth properties toward more infiltrative (Norden et al. 2008; Shapiro et al. 2013). Preclinical mouse models showed similar results, with inhibition of VEGF signaling causing U87 satellite lesions to form distal to the primary tumor (de Groot et al. 2010; Lucio-Eterovic et al. 2009). A separate study also yielded similar results with a medium overall survival of 8.9 months (Sahebjam et al. 2013) and other studies showed comparable data (Demirci et al. 2013; Nagane et al. 2012).
8.1 Cilengitide
Activation of integrin signaling in concert with growth factor receptor tyrosine kinases regulates a number of cellular processes involved in angiogenesis as well as tumor cell growth and invasion (Hood and Cheresh 2002; Kurozumi et al. 2012; Schnell et al. 2008). Cilengitide is a cyclic peptide containing an RGD sequence that binds and inhibits integrin activation and signaling (Scaringi et al. 2012). This drug is capable of antagonizing αvβ3 integrin at sub-nanomolar concentrations, and in case of α3β1 and α5β1 integrins at low nanomolar concentrations. Cilengitide also induces detachment and apoptosis in αvβ3 and αvβ5 integrin-expressing cells in culture (Taga et al. 2002). Using human xenograft models of GBM, Cilengitide suppressed tumor growth and showed anti-angiogenic and anti-tumorigenic properties (Buerkle et al. 2002; MacDonald et al. 2001; Mitjans et al. 2000; Onishi et al. 2013). These cellular outcomes are achieved through cytotoxic, anti-angiogenic, as well as anti-invasive effects (Kurozumi et al. 2012). Phase III trials in glioma are ongoing. (Eskens et al. 2003; Hariharan et al. 2007; O’Donnell et al. 2012). Early data suggest that overall survival remains modest, even though Cilengitide effectively accesses integrin targets in glioma cells and intratumoral blood vessels (Gilbert et al. 2012).
8.2 Sorafenib
Sorafenib is a small molecule inhibitor of VEGFRs, PDGFRs, and other kinases (Siegelin et al. 2010). In phase I clinical trials Sorafenib tested as a monotherapy or in combination with bevacizumab (Scott et al. 2010) or with radiation and temozolomide (Den et al. 2013) resulted in only modest increases in overall survival, although to date the phase II trial results have not been reported. In vitro studies have shown that sorafenib treatment of glioma cells caused a marked reduction in cell proliferation and increased apoptosis that correlated with reduced phospho-MEK and phospho-MAPK levels (Du et al. 2012). The protein kinase C δ inhibitor rottlerin has also been reported to potentiate antigrowth effects of sorafenib (Jane et al. 2006).
8.3 Marimastat
During blood vessel sprouting and remodeling various ECM proteins within the vascular basement membrane must be degraded. These are made possible by a class of proteins known as matrix metalloproteinases (MMPs). MMP2 (gelatinase A) and MMP9 (gelatinase B) are particularly important in glioma angiogenesis. These proteinases are secreted as proactive molecules and membrane-bound MMPs cleave and activate these proteins (Markovic et al. 2009). In comparison to normal brain and low-grade astrocytomas, GBMs overexpress many MMPs, likely leading to increased invasiveness. For example, MMP9 expression was detected at very low levels in normal brain and low-grade astrocytomas, but strong protein expression was reported in GBM (Hagemann et al. 2012). In addition, MMPs actively contribute to tumor angiogenesis by facilitating pericyte release from vascular basement membranes, releasing ECM-bound growth factors, and releasing pro-migratory ECM components helping in directed migration and in disruption of EC–cell adhesion (Rundhaug 2005). Marimastat is an MMP inhibitor that is orally administered. Activation of MMPs has proven to be essential for the tumor cell migration and angiogenesis. Various clinical trials have been conducted in different types of cancer. A phase I study identified the toxicity level of this drug with mild to severe muscle and joint pain. A phase III trial performed in different cancers, including glioma, showed only minimal improvements in overall survival (Levin et al. 2006; Steward and Thomas 2000).
8.4 Other Anti-vascular Agents
Thalidomide is an angiogenesis inhibitor, although the exact mechanism of action is not completely understood. Reduced expression of αvβ3 and αvβ5 integrins, as well as VEGF-A, has been reported as possible modes of action. However, clinical trials conducted using thalidomide as a monotherapy failed to improve prognosis (D’Amato et al. 1994; Fine et al. 2000; Onishi et al. 2011). Imatinib is a tyrosine kinase inhibitor of PDGFR, c-Kit, Bcr-Abl, and other targets. This compound has been shown to induce apoptosis at high concentrations. Monotherapy using imatinib was unsuccessful and did not result in clinical benefits (Morris and Abrey 2010; Radford 2002). Tenascin-C is a pro-migratory protein overexpressed in tumor ECs in GBM. Inhibition of tenascin-C using neutralizing antibodies might block angiogenesis and thereby glioma progression. However, phase II clinical trial conducted using administration of neutralizing antibodies failed to achieve survival benefits to GBM patients (Reardon et al. 2002; Zagzag et al. 1996, 2002).
9 Future Directions
Targeting angiogenesis was one of the more promising strategies for inhibiting tumor growth and progression, particularly in highly vascularized gliomas. Early preclinical and clinical studies yielded promising results; however, the efficacies of anti-angiogenic therapies remain in question as many reports indicate recurrence of tumors with infiltrative and drug-resistant growth properties, especially in GBM. Activation of alternative signaling pathways or compensation by pro-angiogenic molecules likely accounts for tumor recurrence and drug resistance. For example, inhibition of VEGF resulted in upregulation of placental growth factor and FGF. VEGF was also upregulated after VEGFR or EGFR inhibition, and IL8 was upregulated after HIF1α gene deletion (Carmeliet 2005). Additional mechanisms of resistance include “angiogenic mimicry” where tumor cells can transdifferentiate to ECs and contribute to blood vessel functions. Hence, once the ECs are functionally inactive, the tumor cells adapt into the function of ECs integrating into the vessel wall (El Hallani et al. 2010; Ricci-Vitiani et al. 2010; Soda et al. 2011a). This transdifferentiation is not limited to ECs, but tumor cells also give rise to mural cells such as pericytes (Cheng et al. 2013; Scully et al. 2012). Dedifferentiation of neurons and astrocytes can also contribute to gliomagenesis (Friedmann-Morvinski et al. 2012).
Another mechanism by which glioma cells acquire resistance to anti-angiogenic therapies is via enhanced invasion to distal brain regions. Mechanisms of tumor cell invasion after Bevacizumab treatment are now under intense investigation. For example, Lu et al. have shown that this invasion is mediated through c-Met activation (Lu et al. 2012) while research from our laboratory has revealed the importance of integrin αvβ8 in tumor cell invasion (Reyes et al. 2013; Tchaicha et al. 2011). A better understanding is needed for how blood vessels develop and remodel under normal and neoplastic conditions and how their regulation is altered after anti-angiogenic therapies. Combination therapies that target angiogenic effectors or both angiogenic and invasive components may lead to more effective therapies for treating gliomas.
Abbreviations
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EGF:
-
Epidermal growth factor
- FGF:
-
Fibroblast growth factor
- HGF:
-
Hepatocyte growth factor
- GBM:
-
Glioblastoma
- GSC:
-
GBM stem cell
- IL:
-
Interleukin
- MAPK:
-
Mitogen-activated protein kinase
- MMP:
-
Matrix metalloproteinase
- MRP:
-
Multidrug resistance protein
- PDGF:
-
Platelet-derived growth factor
- RTK:
-
Receptor tyrosine kinase
- TGFβ:
-
Transforming growth factor β
- TIMP:
-
Tissue inhibitors of matrix metalloproteinase
- VEGF:
-
Vascular endothelial cell growth factor
References
Abbott NJ (2002) Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat 200:629–638
Abbott NJ, Ronnback L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53
Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W, Small JE, Herrlinger U, Ourednik V, Black PM et al (2000) Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci U S A 97:12846–12851
Abounader R, Lal B, Luddy C, Koe G, Davidson B, Rosen EM, Laterra J (2002) In vivo targeting of SF/HGF and c-met expression via U1snRNA/ribozymes inhibits glioma growth and angiogenesis and promotes apoptosis. FASEB J 16:108–110
Abounader R, Laterra J (2005) Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro Oncol 7:436–451
Abounader R, Ranganathan S, Lal B, Fielding K, Book A, Dietz H, Burger P, Laterra J (1999) Reversion of human glioblastoma malignancy by U1 small nuclear RNA/ribozyme targeting of scatter factor/hepatocyte growth factor and c-met expression. J Natl Cancer Inst 91:1548–1556
Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8:464–478
Aguilera DG, Mazewski C, Hayes L, Jordan C, Esiashivilli N, Janns A, Macdonald TJ (2013) Prolonged survival after treatment of diffuse intrinsic pontine glioma with radiation, temozolamide, and bevacizumab: report of 2 cases. J Pediatr Hematol Oncol 35:e42–e46
Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF (2009) Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell 15:45–56
Allinson K, Lee H, Fruttiger M, McCarty J, Arthur H (2012) Endothelial expression of TGFβ type II receptor is required to maintain vascular integrity during postnatal development of the central nervous system. PLos One 7(6):e39336
Anderson KD, Pan L, Yang XM, Hughes VC, Walls JR, Dominguez MG, Simmons MV, Burfeind P, Xue Y, Wei Y et al (2011) Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U S A 108:2807–2812
Arnold TD, Ferrero GM, Qiu H, Phan IT, Akhurst RJ, Huang EJ, Reichardt LF (2012) Defective retinal vascular endothelial cell development as a consequence of impaired integrin alphaVbeta8-mediated activation of transforming growth factor-beta. J Neurosci 32:1197–1206
Augustin HG, Koh GY, Thurston G, Alitalo K (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol 10:165–177
Bachoo RM, Kim RS, Ligon KL, Maher EA, Brennan C, Billings N, Chan S, Li C, Rowitch DH, Wong WH et al (2004) Molecular diversity of astrocytes with implications for neurological disorders. Proc Natl Acad Sci U S A 101:8384–8389
Bader BL, Rayburn H, Crowley D, Hynes RO (1998) Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95:507–519
Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis 16:1–13
Bao S, Wu Q, Sathornsumetee S, Hao Y, Li Z, Hjelmeland AB, Shi Q, McLendon RE, Bigner DD, Rich JN (2006) Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res 66:7843–7848
Begley DJ, Brightman MW (2003) Structural and functional aspects of the blood-brain barrier. Prog Drug Res 61:39–78
Bellail AC, Hunter SB, Brat DJ, Tan C, Van Meir EG (2004) Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int J Biochem Cell Biol 36:1046–1069
Bello L, Francolini M, Marthyn P, Zhang J, Carroll RS, Nikas DC, Strasser JF, Villani R, Cheresh DA, Black PM (2001) Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery 49:380–389, discussion 390
Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH (2009) The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137:1124–1135
Bielenberg DR, Pettaway CA, Takashima S, Klagsbrun M (2006) Neuropilins in neoplasms: expression, regulation, and function. Exp Cell Res 312:584–593
Birchmeier C, Gherardi E (1998) Developmental roles of HGF/SF and its receptor, the c-Met tyrosine kinase. Trends Cell Biol 8:404–410
Bouck N (2002) PEDF: anti-angiogenic guardian of ocular function. Trends Mol Med 8:330–334
Brat DJ, Bellail AC, Van Meir EG (2005) The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro Oncol 7:122–133
Brockmann MA, Ulbricht U, Gruner K, Fillbrandt R, Westphal M, Lamszus K (2003) Glioblastoma and cerebral microvascular endothelial cell migration in response to tumor-associated growth factors. Neurosurgery 52:1391–1399, discussion 1399
Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J et al (2007) High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11:147–160
Buerkle MA, Pahernik SA, Sutter A, Jonczyk A, Messmer K, Dellian M (2002) Inhibition of the alpha-nu integrins with a cyclic RGD peptide impairs angiogenesis, growth and metastasis of solid tumours in vivo. Br J Cancer 86:788–795
Bulnes S, Bengoetxea H, Ortuzar N, Argandona EG, Garcia-Blanco A, Rico-Barrio I, Lafuente JV (2012) Angiogenic signalling pathways altered in gliomas: selection mechanisms for more aggressive neoplastic subpopulations with invasive phenotype. J Signal Transduct 2012:597915
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M et al (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11:69–82
Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH (1999) The Talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem 274:28071–28074
Cambier S, Gline S, Mu D, Collins R, Araya J, Dolganov G, Einheber S, Boudreau N, Nishimura SL (2005) Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol 166:1883–1894
Carbonell WS, Delay M, Jahangiri A, Park CC, Aghi MK (2013) beta1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res 73:3145–3154
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Carvalho RL, Itoh F, Goumans MJ, Lebrin F, Kato M, Takahashi S, Ema M, Itoh S, van Rooijen M, Bertolino P et al (2007) Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice. J Cell Sci 120:4269–4277
Chappell JC, Taylor SM, Ferrara N, Bautch VL (2009) Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell 17:377–386
Chatterjee S, Matsumura A, Schradermeier J, Gillespie GY (2000) Human malignant glioma therapy using anti-alpha(v)beta3 integrin agents. J Neurooncol 46:135–144
Chekenya M, Hjelstuen M, Enger PO, Thorsen F, Jacob AL, Probst B, Haraldseth O, Pilkington G, Butt A, Levine JM et al (2002) NG2 proteoglycan promotes angiogenesis-dependent tumor growth in CNS by sequestering angiostatin. FASEB J 16:586–588
Chekenya M, Pilkington GJ (2002) NG2 precursor cells in neoplasia: functional, histogenesis and therapeutic implications for malignant brain tumours. J Neurocytol 31:507–521
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD et al (2013) Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153:139–152
Cullen M, Elzarrad MK, Seaman S, Zudaire E, Stevens J, Yang MY, Li X, Chaudhary A, Xu L, Hilton MB et al (2011) GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc Natl Acad Sci U S A 108:5759–5764
D’Amato RJ, Loughnan MS, Flynn E, Folkman J (1994) Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A 91:4082–4085
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA (2009) Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci U S A 106:641–646
de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12:233–242
Del Zoppo GJ (2005) Focal cerebral ischemia and hemostasis: a PAI-1 conundrum. J Thromb Haemost 3:1376–1378
Demirci U, Tufan G, Aktas B, Balakan O, Alacacioglu A, Dane F, Engin H, Kaplan MA, Gunaydin Y, Ozdemir NY et al (2013) Bevacizumab plus irinotecan in recurrent or progressive malign glioma: a multicenter study of the Anatolian Society of Medical Oncology (ASMO). J Cancer Res Clin Oncol 139(5):829–835
Den RB, Kamrava M, Sheng Z, Werner-Wasik M, Dougherty E, Marinucchi M, Lawrence YR, Hegarty S, Hyslop T, Andrews DW et al (2013) A phase I study of the combination of sorafenib with temozolomide and radiation therapy for the treatment of primary and recurrent high-grade gliomas. Int J Radiat Oncol Biol Phys 85:321–328
Desbaillets I, Diserens AC, de Tribolet N, Hamou MF, Van Meir EG (1999) Regulation of interleukin-8 expression by reduced oxygen pressure in human glioblastoma. Oncogene 18:1447–1456
Desbaillets I, Diserens AC, Tribolet N, Hamou MF, Van Meir EG (1997) Upregulation of interleukin 8 by oxygen-deprived cells in glioblastoma suggests a role in leukocyte activation, chemotaxis, and angiogenesis. J Exp Med 186:1201–1212
Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, Gutmann DH, Squire JA, Nagy A, Guha A (2001) Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res 61:3826–3836
Ding S, Merkulova-Rainon T, Han ZC, Tobelem G (2003) HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood 101:4816–4822
Dirks PB (2006) Cancer: stem cells and brain tumours. Nature 444:687–688
Du W, Zhou JR, Wang DL, Gong K, Zhang QJ (2012) Vitamin K1 enhances sorafenib-induced growth inhibition and apoptosis of human malignant glioma cells by blocking the Raf/MEK/ERK pathway. World J Surg Oncol 10:60
Dunn IF, Heese O, Black PM (2000) Growth factors in glioma angiogenesis: FGFs, PDGF, EGF, and TGFs. J Neurooncol 50:121–137
Dvorak HF (2006) Discovery of vascular permeability factor (VPF). Exp Cell Res 312:522–526
Eichmann A, Makinen T, Alitalo K (2005) Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev 19:1013–1021
El Hallani S, Boisselier B, Peglion F, Rousseau A, Colin C, Idbaih A, Marie Y, Mokhtari K, Thomas JL, Eichmann A et al (2010) A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain 133:973–982
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8:579–591
Engelhardt B (2003) Development of the blood-brain barrier. Cell Tissue Res 314:119–129
Eskens FA, Dumez H, Hoekstra R, Perschl A, Brindley C, Bottcher S, Wynendaele W, Drevs J, Verweij J, van Oosterom AT (2003) Phase I and pharmacokinetic study of continuous twice weekly intravenous administration of Cilengitide (EMD 121974), a novel inhibitor of the integrins alphavbeta3 and alphavbeta5 in patients with advanced solid tumours. Eur J Cancer 39:917–926
Fajardo LF, Kwan HH, Kowalski J, Prionas SD, Allison AC (1992) Dual role of tumor necrosis factor-alpha in angiogenesis. Am J Pathol 140:539–544
Fajardo LF, Prionas SD, Kwan HH, Kowalski J, Allison AC (1996) Transforming growth factor beta1 induces angiogenesis in vivo with a threshold pattern. Lab Invest 74:600–608
Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J et al (2010) NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28:5–16
Fang L, Deng Z, Shatseva T, Yang J, Peng C, Du WW, Yee AJ, Ang LC, He C, Shan SW et al (2011) MicroRNA miR-93 promotes tumor growth and angiogenesis by targeting integrin-beta8. Oncogene 30:806–821
Feldkamp MM, Lau N, Rak J, Kerbel RS, Guha A (1999) Normoxic and hypoxic regulation of vascular endothelial growth factor (VEGF) by astrocytoma cells is mediated by Ras. Int J Cancer 81:118–124
Fine HA, Figg WD, Jaeckle K, Wen PY, Kyritsis AP, Loeffler JS, Levin VA, Black PM, Kaplan R, Pluda JM et al (2000) Phase II trial of the antiangiogenic agent thalidomide in patients with recurrent high-grade gliomas. J Clin Oncol 18:708–715
Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D (2005) Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol 15:297–310
Fomchenko EI, Holland EC (2006) Origins of brain tumors–a disease of stem cells? Nat Clin Pract Neurol 2:288–289
Fomchenko EI, Holland EC (2007) Platelet-derived growth factor-mediated gliomagenesis and brain tumor recruitment. Neurosurg Clin N Am 18:39–58, viii
Frater-Schroder M, Muller G, Birchmeier W, Bohlen P (1986) Transforming growth factor-beta inhibits endothelial cell proliferation. Biochem Biophys Res Commun 137:295–302
Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733–4740
Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, Singer O, Ellisman MH, Verma IM (2012) Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 338(6110):1080–1084
Friesel RE, Maciag T (1995) Molecular mechanisms of angiogenesis: fibroblast growth factor signal transduction. FASEB J 9:919–925
Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK (2001) Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Res 61:6020–6024
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21:2683–2710
Giancotti FG, Ruoslahti E (1999) Integrin signaling. Science 285:1028–1032
Gilbert CA, Daou MC, Moser RP, Ross AH (2010) Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res 70:6870–6879
Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S et al (2012) Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol 106:147–153
Gilbertson RJ, Gutmann DH (2007) Tumorigenesis in the brain: location, location, location. Cancer Res 67:5579–5582
Gilbertson RJ, Rich JN (2007) Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7:733–736
Gladson CL (1999) The extracellular matrix of gliomas: modulation of cell function. J Neuropathol Exp Neurol 58:1029–1040
Goldman CK, Kim J, Wong WL, King V, Brock T, Gillespie GY (1993) Epidermal growth factor stimulates vascular endothelial growth factor production by human malignant glioma cells: a model of glioblastoma multiforme pathophysiology. Mol Biol Cell 4:121–133
Goodwin CR, Lal B, Zhou X, Ho S, Xia S, Taeger A, Murray J, Laterra J (2010) Cyr61 mediates hepatocyte growth factor-dependent tumor cell growth, migration, and Akt activation. Cancer Res 70:2932–2941
Hagemann C, Anacker J, Ernestus RI, Vince GH (2012) A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J Clin Oncol 3:67–79
Hambardzumyan D, Becher OJ, Holland EC (2008) Cancer stem cells and survival pathways. Cell Cycle 7:1371–1378
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86:353–364
Harburger DS, Bouaouina M, Calderwood DA (2009) Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem 284:11485–11497
Harburger DS, Calderwood DA (2009) Integrin signalling at a glance. J Cell Sci 122:159–163
Hariharan S, Gustafson D, Holden S, McConkey D, Davis D, Morrow M, Basche M, Gore L, Zang C, O’Bryant CL et al (2007) Assessment of the biological and pharmacological effects of the alpha nu beta3 and alpha nu beta5 integrin receptor antagonist, cilengitide (EMD 121974), in patients with advanced solid tumors. Ann Oncol 18:1400–1407
Hellstrom M, Phng LK, Gerhardt H (2007a) VEGF and Notch signaling: the yin and yang of angiogenic sprouting. Cell Adh Migr 1:133–136
Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N et al (2007b) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445:776–780
Helseth E, Unsgaard G, Dalen A, Vik R (1988) The effects of type beta transforming growth factor on proliferation and epidermal growth factor receptor expression in a human glioblastoma cell line. J Neurooncol 6:269–276
Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, Nister M (1992) Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res 52:3213–3219
Hirota S, Liu Q, Lee HS, Hossain MG, Lacy-Hulbert A, McCarty JH (2011) The astrocyte-expressed integrin alphavbeta8 governs blood vessel sprouting in the developing retina. Development 138:5157–5166
Hjelmeland MD, Hjelmeland AB, Sathornsumetee S, Reese ED, Herbstreith MH, Laping NJ, Friedman HS, Bigner DD, Wang XF, Rich JN (2004) SB-431542, a small molecule transforming growth factor-beta-receptor antagonist, inhibits human glioma cell line proliferation and motility. Mol Cancer Ther 3:737–745
Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ (1999) Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 284:1994–1998
Holland EC (2001) Gliomagenesis: genetic alterations and mouse models. Nat Rev Genet 2:120–129
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25:55–57
Holland EC, Hively WP, DePinho RA, Varmus HE (1998a) A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev 12:3675–3685
Holland EC, Hively WP, Gallo V, Varmus HE (1998b) Modeling mutations in the G1 arrest pathway in human gliomas: overexpression of CDK4 but not loss of INK4a-ARF induces hyperploidy in cultured mouse astrocytes. Genes Dev 12:3644–3649
Holmes WE, Lee J, Kuang WJ, Rice GC, Wood WI (1991) Structure and functional expression of a human interleukin-8 receptor. Science 253:1278–1280
Hood JD, Cheresh DA (2002) Role of integrins in cell invasion and migration. Nat Rev Cancer 2:91–100
Hu B, Jarzynka MJ, Guo P, Imanishi Y, Schlaepfer DD, Cheng SY (2006) Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the alphavbeta1 integrin and focal adhesion kinase signaling pathway. Cancer Res 66:775–783
Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110:673–687
Iadecola C (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5:347–360
Jackson EL, Garcia-Verdugo JM, Gil-Perotin S, Roy M, Quinones-Hinojosa A, VandenBerg S, Alvarez-Buylla A (2006) PDGFR alpha-positive B cells are neural stem cells in the adult SVZ that form glioma-like growths in response to increased PDGF signaling. Neuron 51:187–199
Jahangiri A, De Lay M, Miller LM, Carbonell WS, Hu YL, Lu K, Tom MW, Paquette J, Tokuyasu TA, Tsao S et al (2013) Gene expression profile identifies tyrosine kinase c-Met as a targetable mediator of anti-angiogenic therapy resistance. Clin Cancer Res 19(7):1773–1783
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT (2007) Angiogenesis in brain tumours. Nat Rev Neurosci 8:610–622
Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A et al (2010) Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12:943–953
Jane EP, Premkumar DR, Pollack IF (2006) Coadministration of sorafenib with rottlerin potently inhibits cell proliferation and migration in human malignant glioma cells. J Pharmacol Exp Ther 319:1070–1080
Jansen M, de Witt Hamer PC, Witmer AN, Troost D, van Noorden CJ (2004) Current perspectives on antiangiogenesis strategies in the treatment of malignant gliomas. Brain Res Brain Res Rev 45:143–163
Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, Lam SC (1999) Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin alpha(IIb)beta(3). J Biol Chem 274:24321–24327
Jen J, Harper JW, Bigner SH, Bigner DD, Papadopoulos N, Markowitz S, Willson JK, Kinzler KW, Vogelstein B (1994) Deletion of p16 and p15 genes in brain tumors. Cancer Res 54:6353–6358
Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, Park HJ, Joo KM, Park WY, Nam DH, DePinho RA et al (2008) Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev 22:2028–2033
Jiang WG, Hiscox SE, Parr C, Martin TA, Matsumoto K, Nakamura T, Mansel RE (1999) Antagonistic effect of NK4, a novel hepatocyte growth factor variant, on in vitro angiogenesis of human vascular endothelial cells. Clin Cancer Res 5:3695–3703
Jiang Y, Goldberg ID, Shi YE (2002) Complex roles of tissue inhibitors of metalloproteinases in cancer. Oncogene 21:2245–2252
Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30:393–402
Kalluri R (2003) Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer 3:422–433
Kanamori M, Kawaguchi T, Berger MS, Pieper RO (2006) Intracranial microenvironment reveals independent opposing functions of host alphaVbeta3 expression on glioma growth and angiogenesis. J Biol Chem 281:37256–37264
Kanamori M, Vanden Berg SR, Bergers G, Berger MS, Pieper RO (2004) Integrin beta3 overexpression suppresses tumor growth in a human model of gliomagenesis: implications for the role of beta3 overexpression in glioblastoma multiforme. Cancer Res 64:2751–2758
Kargiotis O, Rao JS, Kyritsis AP (2006) Mechanisms of angiogenesis in gliomas. J Neurooncol 78:281–293
Kim C, Ye F, Ginsberg MH (2011) Regulation of integrin activation. Annu Rev Cell Dev Biol 27:321–345
Kim Y, Lee C (2006) The gene encoding transforming growth factor {beta}1 confers risk of ischemic stroke and vascular dementia. Stroke 37(11):2843–2845
Kirsch M, Strasser J, Allende R, Bello L, Zhang J, Black PM (1998) Angiostatin suppresses malignant glioma growth in vivo. Cancer Res 58:4654–4659
Klenotic PA, Page RC, Li W, Amick J, Misra S, Silverstein RL (2013) Molecular basis of antiangiogenic thrombospondin-1 type 1 repeat domain interactions with CD36. Arterioscler Thromb Vasc Biol 33(7):1655–1662
Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, Resau JH, Vande Woude GF (1997) Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 57:5391–5398
Koochekpour S, Merzak A, Pilkington GJ (1996) Vascular endothelial growth factor production is stimulated by gangliosides and TGF-beta isoforms in human glioma cells in vitro. Cancer Lett 102:209–215
Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, Garren N, Mackey M, Butman JA, Camphausen K et al (2009) Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol 27:740–745
Kuhnert F, Mancuso MR, Shamloo A, Wang HT, Choksi V, Florek M, Su H, Fruttiger M, Young WL, Heilshorn SC et al (2010) Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330:985–989
Kurozumi K, Ichikawa T, Onishi M, Fujii K, Date I (2012) Cilengitide treatment for malignant glioma: current status and future direction. Neurol Med Chir (Tokyo) 52:539–547
Lamszus K, Schmidt NO, Jin L, Laterra J, Zagzag D, Way D, Witte M, Weinand M, Goldberg ID, Westphal M et al (1998) Scatter factor promotes motility of human glioma and neuromicrovascular endothelial cells. Int J Cancer 75:19–28
Laterra J, Nam M, Rosen E, Rao JS, Lamszus K, Goldberg ID, Johnston P (1997) Scatter factor/hepatocyte growth factor gene transfer enhances glioma growth and angiogenesis in vivo. Lab Invest 76:565–577
Lawler J (2000) The functions of thrombospondin-1 and-2. Curr Opin Cell Biol 12:634–640
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306–1309
Levin VA, Phuphanich S, Yung WK, Forsyth PA, Maestro RD, Perry JR, Fuller GN, Baillet M (2006) Randomized, double-blind, placebo-controlled trial of marimastat in glioblastoma multiforme patients following surgery and irradiation. J Neurooncol 78:295–302
Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M et al (2008) Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183:409–417
Liebner S, Czupalla CJ, Wolburg H (2011) Current concepts of blood-brain barrier development. Int J Dev Biol 55:467–476
Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L et al (2011) Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 146:209–221
Lopes MB (2003) Angiogenesis in brain tumors. Microsc Res Tech 60:225–230
Louis DN (2006) Molecular pathology of malignant gliomas. Annu Rev Pathol 1:97–117
Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ, Cheresh DA et al (2012) VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 22:21–35
Lucio-Eterovic AK, Piao Y, de Groot JF (2009) Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res 15:4589–4599
Ma H, Calderon TM, Fallon JT, Berman JW (2002) Hepatocyte growth factor is a survival factor for endothelial cells and is expressed in human atherosclerotic plaques. Atherosclerosis 164:79–87
Ma YQ, Qin J, Wu C, Plow EF (2008) Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol 181:439–446
MacDonald TJ, Taga T, Shimada H, Tabrizi P, Zlokovic BV, Cheresh DA, Laug WE (2001) Preferential susceptibility of brain tumors to the antiangiogenic effects of an alpha(v) integrin antagonist. Neurosurgery 48:151–157
Maglott A, Bartik P, Cosgun S, Klotz P, Ronde P, Fuhrmann G, Takeda K, Martin S, Dontenwill M (2006) The small alpha5beta1 integrin antagonist, SJ749, reduces proliferation and clonogenicity of human astrocytoma cells. Cancer Res 66:6002–6007
Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA (2001) Malignant glioma: genetics and biology of a grave matter. Genes Dev 15:1311–1333
Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, Compton D, McClain J, Aldrich TH, Papadopoulos N et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60
Marin-Padilla M (1985) Early vascularization of the embryonic cerebral cortex: Golgi and electron microscopic studies. J Comp Neurol 241:237–249
Markovic DS, Vinnakota K, Chirasani S, Synowitz M, Raguet H, Stock K, Sliwa M, Lehmann S, Kalin R, van Rooijen N et al (2009) Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc Natl Acad Sci U S A 106:12530–12535
Martin TA, Harding KG, Jiang WG (1999) Regulation of angiogenesis and endothelial cell motility by matrix-bound fibroblasts. Angiogenesis 3:69–76
Maruno M, Kovach JS, Kelly PJ, Yanagihara T (1997) Distribution of endogenous tumour necrosis factor alpha in gliomas. J Clin Pathol 50:559–562
Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103:295–309
Massague J, Gomis RR (2006) The logic of TGFbeta signaling. FEBS Lett 580:2811–2820
McCarty JH (2005) Cell biology of the neurovascular unit: implications for drug delivery across the blood-brain barrier. Assay Drug Dev Technol 3:89–95
McCarty JH (2009a) Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol 16:209–214
McCarty JH (2009b) Integrin-mediated regulation of neurovascular development, physiology and disease. Cell Adh Migr 3:211–215
McCarty JH, Lacy-Hulbert A, Charest A, Bronson RT, Crowley D, Housman D, Savill J, Roes J, Hynes RO (2005) Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132:165–176
McCarty JH, Monahan-Earley RA, Brown LF, Keller M, Gerhardt H, Rubin K, Shani M, Dvorak HF, Wolburg H, Bader BL et al (2002) Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol Cell Biol 22:7667–7677
Milner R, Campbell IL (2002) The integrin family of cell adhesion molecules has multiple functions within the CNS. J Neurosci Res 69:286–291
Mitjans F, Meyer T, Fittschen C, Goodman S, Jonczyk A, Marshall JF, Reyes G, Piulats J (2000) In vivo therapy of malignant melanoma by means of antagonists of alphav integrins. Int J Cancer 87:716–723
Mobley AK, McCarty JH (2011) beta8 integrin is essential for neuroblast migration in the rostral migratory stream. Glia 59:1579–1587
Mobley AK, Tchaicha JH, Shin J, Hossain MG, McCarty JH (2009) {beta}8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci 122:1842–1851
Moriyama T, Kataoka H, Kawano H, Yokogami K, Nakano S, Goya T, Uchino H, Koono M, Wakisaka S (1998) Comparative analysis of expression of hepatocyte growth factor and its receptor, c-met, in gliomas, meningiomas and schwannomas in humans. Cancer Lett 124:149–155
Morris PG, Abrey LE (2010) Novel targeted agents for platelet-derived growth factor receptor and c-KIT in malignant gliomas. Target Oncol 5:193–200
Moser TL, Kenan DJ, Ashley TA, Roy JA, Goodman MD, Misra UK, Cheek DJ, Pizzo SV (2001) Endothelial cell surface F1-F0 ATP synthase is active in ATP synthesis and is inhibited by angiostatin. Proc Natl Acad Sci U S A 98:6656–6661
Moses HL, Serra R (1996) Regulation of differentiation by TGF-beta. Curr Opin Genet Dev 6:581–586
Mrugala MM, Crew LK, Fink JR, Spence AM (2012) Carboplatin and bevacizumab for recurrent malignant glioma. Oncol Lett 4:1082–1086
Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL (2002) The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157:493–507
Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S, Sukhatme VP (1997) The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol Cell Biol 17:5629–5639
Murphy PM, Tiffany HL (1991) Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 253:1280–1283
Nagane M, Nishikawa R, Narita Y, Kobayashi H, Takano S, Shinoura N, Aoki T, Sugiyama K, Kuratsu J, Muragaki Y et al (2012) Phase II study of single-agent bevacizumab in Japanese patients with recurrent malignant glioma. Jpn J Clin Oncol 42:887–895
Nakamura Y, Morishita R, Higaki J, Kida I, Aoki M, Moriguchi A, Yamada K, Hayashi S, Yo Y, Matsumoto K et al (1995) Expression of local hepatocyte growth factor system in vascular tissues. Biochem Biophys Res Commun 215:483–488
Neuwelt EA (2004) Mechanisms of disease: the blood-brain barrier. Neurosurgery 54:131–140, discussion 141–132
Nguyen HL, Lee YJ, Shin J, Lee E, Park SO, McCarty JH, Oh SP (2011) TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest 91:1554–1563
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M (2006) Angiogenesis in cancer. Vasc Health Risk Manag 2:213–219
Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444:1032–1037
Norden AD, Young GS, Setayesh K, Muzikansky A, Klufas R, Ross GL, Ciampa AS, Ebbeling LG, Levy B, Drappatz J et al (2008) Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 70:779–787
O’Donnell PH, Undevia SD, Stadler WM, Karrison TM, Nicholas MK, Janisch L, Ratain MJ (2012) A phase I study of continuous infusion cilengitide in patients with solid tumors. Invest New Drugs 30:604–610
O’Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J (1997) Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell 88:277–285
O’Reilly MS, Holmgren L, Shing Y, Chen C, Rosenthal RA, Moses M, Lane WS, Cao Y, Sage EH, Folkman J (1994) Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 79:315–328
Ohgaki H (2005) Genetic pathways to glioblastomas. Neuropathology 25:1–7
Okamura K, Morimoto A, Hamanaka R, Ono M, Kohno K, Uchida Y, Kuwano M (1992) A model system for tumor angiogenesis: involvement of transforming growth factor-alpha in tube formation of human microvascular endothelial cells induced by esophageal cancer cells. Biochem Biophys Res Commun 186:1471–1479
Onishi M, Ichikawa T, Kurozumi K, Date I (2011) Angiogenesis and invasion in glioma. Brain Tumor Pathol 28:13–24
Onishi M, Ichikawa T, Kurozumi K, Fujii K, Yoshida K, Inoue S, Michiue H, Chiocca EA, Kaur B, Date I (2013) Bimodal anti-glioma mechanisms of cilengitide demonstrated by novel invasive glioma models. Neuropathology 33(2):162–174
Orlova VV, Liu Z, Goumans MJ, ten Dijke P (2011) Controlling angiogenesis by two unique TGF-beta type I receptor signaling pathways. Histol Histopathol 26:1219–1230
Pan LH, Beppu T, Kurose A, Yamauchi K, Sugawara A, Suzuki M, Ogawa A, Sawai T (2002) Neoplastic cells and proliferating endothelial cells express connective tissue growth factor (CTGF) in glioblastoma. Neurol Res 24:677–683
Pardridge WM (2002) Drug and gene targeting to the brain with molecular Trojan horses. Nat Rev Drug Discov 1:131–139
Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL et al (2008) ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood 111:633–642
Parsons JT, Horwitz AR, Schwartz MA (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11:633–643
Peeters M, Strickland AH, Lichinitser M, Suresh AV, Manikhas G, Shapiro J, Rogowski W, Huang X, Wu B, Warner D et al (2013) A randomised, double-blind, placebo-controlled phase 2 study of trebananib (AMG 386) in combination with FOLFIRI in patients with previously treated metastatic colorectal carcinoma. Br J Cancer 108:503–511
Penuelas S, Anido J, Prieto-Sanchez RM, Folch G, Barba I, Cuartas I, Garcia-Dorado D, Poca MA, Sahuquillo J, Baselga J et al (2009) TGF-beta increases glioma-initiating cell self-renewal through the induction of LIF in human glioblastoma. Cancer Cell 15:315–327
Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9:157–173
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146:873–887
Proctor JM, Zang K, Wang D, Wang R, Reichardt LF (2005) Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci 25:9940–9948
Radford IR (2002) Imatinib. Novartis. Curr Opin Investig Drugs 3:492–499
Rao S, Sengupta R, Choe EJ, Woerner BM, Jackson E, Sun T, Leonard J, Piwnica-Worms D, Rubin JB (2012) CXCL12 mediates trophic interactions between endothelial and tumor cells in glioblastoma. PLoS One 7:e33005
Read TA, Hegedus B, Wechsler-Reya R, Gutmann DH (2006) The neurobiology of neurooncology. Ann Neurol 60:3–11
Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE II, Cokgor I, McLendon RE, Pegram CN, Provenzale JM et al (2002) Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol 20:1389–1397
Rege TA, Fears CY, Gladson CL (2005) Endogenous inhibitors of angiogenesis in malignant gliomas: nature’s antiangiogenic therapy. Neuro Oncol 7:106–121
Rehn M, Veikkola T, Kukk-Valdre E, Nakamura H, Ilmonen M, Lombardo C, Pihlajaniemi T, Alitalo K, Vuori K (2001) Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci U S A 98:1024–1029
Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T (2000) Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet 26:109–113
Rempel SA, Dudas S, Ge S, Gutierrez JA (2000) Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res 6:102–111
Reyes SB, Narayanan AS, Lee HS, Tchaicha JH, Aldape KD, Lang FF, Tolias KF, McCarty JH (2013) alphavbeta 8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Mol Biol Cell 24(4):474–482
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM et al (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468:824–828
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM et al (2011) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468:824–828
Rich JN, Bigner DD (2004) Development of novel targeted therapies in the treatment of malignant glioma. Nat Rev Drug Discov 3:430–446
Rich JN, Zhang M, Datto MB, Bigner DD, Wang XF (1999) Transforming growth factor-beta-mediated p15(INK4B) induction and growth inhibition in astrocytes is SMAD3-dependent and a pathway prominently altered in human glioma cell lines. J Biol Chem 274:35053–35058
Rodriguez FJ, Orr BA, Ligon KL, Eberhart CG (2012) Neoplastic cells are a rare component in human glioblastoma microvasculature. Oncotarget 3:98–106
Rong Y, Durden DL, Van Meir EG, Brat DJ (2006) ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol 65:529–539
Rosen EM, Laterra J, Joseph A, Jin L, Fuchs A, Way D, Witte M, Weinand M, Goldberg ID (1996) Scatter factor expression and regulation in human glial tumors. Int J Cancer 67:248–255
Rundhaug JE (2005) Matrix metalloproteinases and angiogenesis. J Cell Mol Med 9:267–285
Ryuto M, Ono M, Izumi H, Yoshida S, Weich HA, Kohno K, Kuwano M (1996) Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J Biol Chem 271:28220–28228
Sahebjam S, Garoufalis E, Guiot MC, Muanza T, Del Maestro R, Petrecca K, Sharma R, Kavan P (2013) Bevacizumab use for recurrent high-grade glioma at McGill University Hospital. Can J Neurol Sci 40:241–246
Salmaggi A, Boiardi A, Gelati M, Russo A, Calatozzolo C, Ciusani E, Sciacca FL, Ottolina A, Parati EA, La Porta C et al (2006) Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia 54:850–860
Sanai N, Alvarez-Buylla A, Berger MS (2005) Neural stem cells and the origin of gliomas. N Engl J Med 353:811–822
Scaringi C, Minniti G, Caporello P, Enrici RM (2012) Integrin inhibitor cilengitide for the treatment of glioblastoma: a brief overview of current clinical results. Anticancer Res 32:4213–4223
Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, Thon N, Goetz C, Kretzschmar HA, Tonn JC et al (2008) Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol 18:378–386
Scott BJ, Quant EC, McNamara MB, Ryg PA, Batchelor TT, Wen PY (2010) Bevacizumab salvage therapy following progression in high-grade glioma patients treated with VEGF receptor tyrosine kinase inhibitors. Neuro Oncol 12:603–607
Scully S, Francescone R, Faibish M, Bentley B, Taylor SL, Oh D, Schapiro R, Moral L, Yan W, Shao R (2012) Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J Neurosci 32:12950–12960
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Sethi N, Dai X, Winter CG, Kang Y (2011) Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19:192–205
Shapiro LQ, Beal K, Goenka A, Karimi S, Iwamoto FM, Yamada Y, Zhang Z, Lassman AB, Abrey LE, Gutin PH (2013) Patterns of failure after concurrent bevacizumab and hypofractionated stereotactic radiation therapy for recurrent high-grade glioma. Int J Radiat Oncol Biol Phys 85:636–642
Shattil SJ, Kim C, Ginsberg MH (2010) The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol 11:288–300
Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN (2007a) Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene 26:4084–4094
Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS, Rich JN (2007b) A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog 46:488–496
Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC (2004) Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res 64:4783–4789
Shih AH, Holland EC (2004) Developmental neurobiology and the origin of brain tumors. J Neurooncol 70:125–136
Siegelin MD, Raskett CM, Gilbert CA, Ross AH, Altieri DC (2010) Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci Lett 478:165–170
Siekmann AF, Lawson ND (2007) Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445:781–784
Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23:9254–9262
Slowik MR, De Luca LG, Fiers W, Pober JS (1993) Tumor necrosis factor activates human endothelial cells through the p55 tumor necrosis factor receptor but the p75 receptor contributes to activation at low tumor necrosis factor concentration. Am J Pathol 143:1724–1730
Snuderl M, Batista A, Kirkpatrick ND, Ruiz de Almodovar C, Riedemann L, Walsh EC, Anolik R, Huang Y, Martin JD, Kamoun W et al (2013) Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152:1065–1076
Soda Y, Marumoto T, Friedmann-Morvinski D, Soda M, Liu F, Michiue H, Pastorino S, Yang M, Hoffman RM, Kesari S et al (2011) Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A 108:4274–4280
Stallcup WB, Huang FJ (2008) A role for the NG2 proteoglycan in glioma progression. Cell Adh Migr 2:192–201
Stefanik DF, Rizkalla LR, Soi A, Goldblatt SA, Rizkalla WM (1991) Acidic and basic fibroblast growth factors are present in glioblastoma multiforme. Cancer Res 51:5760–5765
Stenman JM, Rajagopal J, Carroll TJ, Ishibashi M, McMahon J, McMahon AP (2008) Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322:1247–1250
Steward WP, Thomas AL (2000) Marimastat: the clinical development of a matrix metalloproteinase inhibitor. Expert Opin Investig Drugs 9:2913–2922
Stockhausen MT, Kristoffersen K, Poulsen HS (2010) The functional role of Notch signaling in human gliomas. Neuro Oncol 12:199–211
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U et al (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Su H, Kim H, Pawlikowska L, Kitamura H, Shen F, Cambier S, Markovics J, Lawton MT, Sidney S, Bollen AW et al (2010) Reduced expression of integrin alphavbeta8 is associated with brain arteriovenous malformation pathogenesis. Am J Pathol 176:1018–1027
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD (1996) Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87:1171–1180
Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA (2003) Talin binding to integrin beta tails: a final common step in integrin activation. Science 302:103–106
Taga T, Suzuki A, Gonzalez-Gomez I, Gilles FH, Stins M, Shimada H, Barsky L, Weinberg KI, Laug WE (2002) alpha v-Integrin antagonist EMD 121974 induces apoptosis in brain tumor cells growing on vitronectin and tenascin. Int J Cancer 98:690–697
Takagi J, Petre BM, Walz T, Springer TA (2002) Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110:599–611
Tarui T, Miles LA, Takada Y (2001) Specific interaction of angiostatin with integrin alpha(v)beta(3) in endothelial cells. J Biol Chem 276:39562–39568
Tate MC, Aghi MK (2009) Biology of angiogenesis and invasion in glioma. Neurotherapeutics 6:447–457
Taylor J, Gerstner ER (2013) Anti-angiogenic therapy in high-grade glioma (treatment and toxicity). Curr Treat Options Neurol 15(3):328–337
Tchaicha JH, Reyes SB, Shin J, Hossain MG, Lang FF, McCarty JH (2011) Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res 71:6371–6381
Thiery JP (2003) Cell adhesion in development: a complex signaling network. Curr Opin Genet Dev 13:365–371
Treasurywala S, Berens ME (1998) Migration arrest in glioma cells is dependent on the alphav integrin subunit. Glia 24:236–243
Tsai JC, Goldman CK, Gillespie GY (1995) Vascular endothelial growth factor in human glioma cell lines: induced secretion by EGF, PDGF-BB, and bFGF. J Neurosurg 82:864–873
Tucker GC (2006) Integrins: molecular targets in cancer therapy. Curr Oncol Rep 8:96–103
Uhm JH, Dooley NP, Kyritsis AP, Rao JS, Gladson CL (1999a) Vitronectin, a glioma-derived extracellular matrix protein, protects tumor cells from apoptotic death. Clin Cancer Res 5:1587–1594
Uhm JH, Gladson CL, Rao JS (1999b) The role of integrins in the malignant phenotype of gliomas. Front Biosci 4:D188–D199
Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC (2002) Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res 62:5551–5558
Valter MM, Wiestler OD, Pietsche T (1999) Differential control of VEGF synthesis and secretion in human glioma cells by IL-1 and EGF. Int J Dev Neurosci 17:565–577
Van Meir E, Ceska M, Effenberger F, Walz A, Grouzmann E, Desbaillets I, Frei K, Fontana A, de Tribolet N (1992) Interleukin-8 is produced in neoplastic and infectious diseases of the human central nervous system. Cancer Res 52:4297–4305
Van Meir E, Sawamura Y, Diserens AC, Hamou MF, de Tribolet N (1990) Human glioblastoma cells release interleukin 6 in vivo and in vitro. Cancer Res 50:6683–6688
Venstrom K, Reichardt L (1995) Beta 8 integrins mediate interactions of chick sensory neurons with laminin-1, collagen IV, and fibronectin. Mol Biol Cell 6:419–431
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110
Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J (2002) A structural mechanism of integrin alpha(IIb)beta(3) “inside-out” activation as regulated by its cytoplasmic face. Cell 110:587–597
Vredenburgh JJ, Desjardins A, Herndon JE II, Marcello J, Reardon DA, Quinn JA, Rich JN, Sathornsumetee S, Gururangan S, Sampson J et al (2007) Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol 25:4722–4729
Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V (2011) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468:829–833
Wang X, Zhou Y, Kim HP, Song R, Zarnegar R, Ryter SW, Choi AM (2004) Hepatocyte growth factor protects against hypoxia/reoxygenation-induced apoptosis in endothelial cells. J Biol Chem 279:5237–5243
Ware ML, Berger MS, Binder DK (2003) Molecular biology of glioma tumorigenesis. Histol Histopathol 18:207–216
Wechsler-Reya R, Scott MP (2001) The developmental biology of brain tumors. Annu Rev Neurosci 24:385–428
Wickstrom SA, Alitalo K, Keski-Oja J (2002) Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res 62:5580–5589
Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E et al (2004) Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 6:553–563
Wojta J, Kaun C, Breuss JM, Koshelnick Y, Beckmann R, Hattey E, Mildner M, Weninger W, Nakamura T, Tschachler E et al (1999) Hepatocyte growth factor increases expression of vascular endothelial growth factor and plasminogen activator inhibitor-1 in human keratinocytes and the vascular endothelial growth factor receptor flk-1 in human endothelial cells. Lab Invest 79:427–438
Woods SA, McGlade CJ, Guha A (2002) Phosphatidylinositol 3′-kinase and MAPK/ERK kinase 1/2 differentially regulate expression of vascular endothelial growth factor in human malignant astrocytoma cells. Neuro Oncol 4:242–252
Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T (2002) Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell 1:157–168
Xie D, Yin D, Wang HJ, Liu GT, Elashoff R, Black K, Koeffler HP (2004) Levels of expression of CYR61 and CTGF are prognostic for tumor progression and survival of individuals with gliomas. Clin Cancer Res 10:2072–2081
Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA (2001) Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 294:339–345
Zacchigna S, Lambrechts D, Carmeliet P (2008) Neurovascular signalling defects in neurodegeneration. Nat Rev Neurosci 9:169–181
Zagzag D, Friedlander DR, Dosik J, Chikramane S, Chan W, Greco MA, Allen JC, Dorovini-Zis K, Grumet M (1996) Tenascin-C expression by angiogenic vessels in human astrocytomas and by human brain endothelial cells in vitro. Cancer Res 56:182–189
Zagzag D, Shiff B, Jallo GI, Greco MA, Blanco C, Cohen H, Hukin J, Allen JC, Friedlander DR (2002) Tenascin-C promotes microvascular cell migration and phosphorylation of focal adhesion kinase. Cancer Res 62:2660–2668
Zeng Q, Li S, Chepeha DB, Giordano TJ, Li J, Zhang H, Polverini PJ, Nor J, Kitajewski J, Wang CY (2005) Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 8:13–23
Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z et al (2008) p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455:1129–1133
Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF (2002) beta8 integrins are required for vascular morphogenesis in mouse embryos. Development 129:2891–2903
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28:202–208
Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50
Acknowledgments
This research was supported by grant funds awarded to J.H.M. from the National Institutes of Neurological Disease and Stroke (R01NS059876 and R01NS078402) and the National Cancer Institute (P50CA127001). Due to space constraints, we apologize to colleagues in the field for not citing their relevant studies.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Wien
About this chapter
Cite this chapter
Cheerathodi, M., McCarty, J.H. (2014). Angiogenesis in Gliomas. In: Sedo, A., Mentlein, R. (eds) Glioma Cell Biology. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1431-5_7
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1431-5_7
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1430-8
Online ISBN: 978-3-7091-1431-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)