
Abstract
Hypothermia is currently the only approved therapy for global cerebral ischemia (GCI) after cardiac arrest; however, it unfortunately has multiple adverse effects. As a noninvasive procedure, photobiomodulation (PBM) therapy has emerged as a potential novel treatment for brain injury. PBM involves the use of low-level laser light therapy to influence cell behavior. In this study, we evaluated the therapeutic effects of PBM treatment with an 808-nm diode laser initiated 6 h after GCI. It was noted that PBM dose-dependently protected against GCI-induced neuronal death in the vulnerable hippocampal CA1 subregion. Functional assessments demonstrated that PBM markedly preserved both short-term (a week) and long-term (6 months) spatial learning and memory function following GCI. Further mechanistic studies revealed that PBM post-treatment (a) preserved healthy mitochondrial dynamics and suppressed substantial mitochondrial fragmentation of CA1 neurons, by reducing the detrimental Drp1 GTPase activity and its interactions with adaptor proteins Mff and Fis1 and by balancing mitochondrial targeting fission and fusion protein levels; (b) reduced mitochondrial oxidative damage and excessive mitophagy and restored mitochondrial overall health status and preserved mitochondrial function; and (c) suppressed mitochondria-dependent apoptosome formation/caspase-3/9 apoptosis-processing activities. Additionally, we validated, in an in vitro ischemia model, that cytochrome c oxidase served as a key PBM target for mitochondrial function preservation and neuroprotection. Our findings suggest that PBM serves as a promising therapeutic strategy for the functional recovery after GCI, with mechanisms involving PBM’s preservation on mitochondrial dynamics and functions and the inhibition of delayed apoptotic neuronal death in GCI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiac arrest is a leading cause of death and disability in the USA [1]. Approximately half of the survivors of cardiac arrest experience impairments in memory, attention, and executive functioning [2,3,4]. These cognitive deficits are thought to be primarily due to global cerebral ischemia (GCI) that occurs after cardiac arrest, which can induce delayed neuronal cell death in the vulnerable hippocampal CA1 region [5,6,7,8,9]. Similar to humans, experimental GCI induced in animal models has been shown to result in selective and delayed neuronal death of pyramidal neurons in the hippocampal CA1 region as well as cognitive impairments, and thus, experimental GCI in animals has become a valuable model for the study of GCI [8,9,10,11]. Unfortunately, currently, treatment options for cardiac arrest-induced neurological impairments are limited. Therapeutic hypothermia has been reported in some studies to have neurological benefit in cardiac arrest [12], but a recent meta-analysis failed to find any significant benefit on survival or neurological outcome [13, 14]. Thus, new therapeutic strategies for protection of the brain from GCI after cardiac arrest are critically needed.
Interestingly, there is considerable evidence suggesting that mitochondria damage and dysfunction may play a significant role in the neuronal cell death and neurological impairment that follows GCI [15,16,17], and thus, could be a viable therapeutic target in GCI. The mitochondria play a key role in cells to produce energy in the form of ATP [18], but in pathological situations, such as ischemia and other damaging situations, the mitochondria can generate damaging reactive oxygen species (ROS) and can release cytochrome c from the intermembrane space of mitochondria to induce apoptosis [19, 20]. Recent work has demonstrated that cytochrome c is maintained in the cristae folds of the inner mitochondrial membrane by optic atrophy 1 (OPA1), a GTPase dynamin-related protein [21]. It has also been demonstrated that cristae fold tightness depends upon the balance of short and long oligomers of OPA1 and that disruption of OPA1 oligomers leads to disruption of cristae folds and cytochrome c release [21, 22].
Further work has shown that the mitochondria undergo cycles of fission and fusion and that OPA1 plays an important role in the mitochondrial fusion process [23]. Furthermore, GCI has been shown to increase mitochondria fragmentation (fission), which corresponded with a loss of OPA1 complexes [16]. Knockdown of OPA1 was shown to lead to rapid release of cytochrome c and increased mitochondria fragmentation in the brain, indicating a critical role of OPA1 in mitochondrial fusion, cytochrome C release, and apoptosis [22, 24]. In addition to OPA1, further work has revealed that Dynamin-related protein 1 (Drp1) functions as a mitochondrial fission protein [25,26,27], and has an important role in apoptosis by promoting Bcl-2-associated x protein (Bax) oligomerization, which facilitates mitochondrial outer membrane permeabilization and cell death [25, 28].
Due to the increasing evidence of a critical role of mitochondria dysfunction in cell death after GCI, in the current study, we examined the effect of a noninvasive procedure, photobiomodulation (PBM) therapy as a potential novel treatment for brain injury following GCI. The results show that transcranial PBM treatment preserved healthy mitochondrial dynamics and suppressed substantial mitochondrial fragmentation of CA1 neurons and significantly reduces both GCI-induced apoptosis in the vulnerable hippocampal CA1 region and the associated memory deficits.
Materials and Methods
Global Cerebral Ischemia Model and Photobiomodulation Treatment
Adult male Sprague-Dawley (SD) rats (250–300 g body weight, Charles River Laboratories) were used to conduct this study. The four-vessel occlusion rat model of GCI and reperfusion was performed as previously described [29, 30]. Briefly, under anesthesia with sodium pentobarbital (Diamondback Drugs, 40 mg/kg, intraperitoneal injection), the animal was placed prone in a stereotaxic frame with ear bars. Both alar foraminas of the first cervical vertebrae were exposed and the vertebral arteries were permanently electrocauterized using an electrocautery needle connected to an electrocoagulation device. The incision was closed with tissue glue and the animal was laid on its back after release from the stereotaxic platform. Then, the bilateral common carotid arteries (CCAs) were exposed and isolated, and a silastic ligature was placed loosely around each artery without interrupting the blood flow. The incision was closed with wound clips, and the animals were allowed to recover from the anesthetic before they were placed back into cages. Twenty-four hours afterwards, the animals were anesthetized under light anesthesia using a nose cone, and CCAs were exposed and occluded with aneurysm clips for 15 min to induce ischemia. Isofluorane was discontinued 1–2 min after the initiation of ischemia. Successful cerebral ischemia was ensured by monitoring the loss of righting reflex, dilated pupils, and the unresponsiveness to light during GCI. Following the procedure, arterial blood flow was confirmed before the wound was closed with tissue glue. Body temperature of the animals was maintained within the range 36.5–37.5 °C during and after ischemia using a thermal blanket and infrared lamp. Sham GCI animals underwent identical surgical procedures as ischemic animals except that the CCAs were not occluded. All animal surgery protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the local university and were carried out in compliance with National Institutes of Health guidelines.
For PBM treatment, a diode laser (continuous wave at 808 nm, model 808M100, Dragon Lasers) was applied transcranially by focusing the beam at 1 cm2 round spot covering the shaved scalp, as described in our previous work [31]. Daily PBM treatment was initiated 6 h after GCI as shown in the schematic diagram (Fig. 1a). The PBM power density was adjusted to 8.0 mW/cm2 at the hippocampus tissue level (without apparent local temperature increase). A laser power meter (#FM33-056, Coherent Inc., USA) was used to adjust and monitor the power output density.

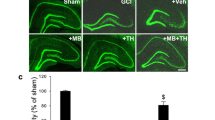
Dose-dependent neuroprotective effect of PBM on GCI-induced hippocampal CA1 injury. a Schematic diagram of the of PBM treatment protocol. Arrowheads indicate PBM-treated time points after ischemic reperfusion. b Typical NeuN and TUNEL staining of the CA1 subregion 14 days after GCI. c, d Quantitative data showing the number of surviving neurons and TUNEL-positive cells per 250 μm length of medial CA1 region. Mean ± SE values are shown; n = 8–12 per group. *P < 0.05 versus sham PBM (0.0 J) group, #P < 0.05 versus one-dose PBM-treated (at 6 h, 1.0 J) group, $P < 0.05 versus two-dose PBM-treated (at 6 h and 1 day, total 2.0 J) group. R: reperfusion
Histological Analysis
Routine histological examination of brain tissues was conducted with NeuN and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, as described previously [32, 33]. Briefly, rats were anesthetized with sodium pentobarbital and flush-perfused transcardially with ice-cold saline (0.9% NaCI), followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB), pH 7.4. The rat brains were extracted immediately following perfusion, post-fixed in PFA fixative overnight, and cryoprotected in 30% sucrose in PB at 4 °C for 3 days. The brains were embedded in OCT compound (Tissue-Tek, Ted Pella, Inc.), and free-floating coronal parallel series of sections (25 μm thick) were obtained on a Leica CM1950 Cryostat (Leica Microsystems). Double immunofluorescence labeling for NeuN (neuron-specific marker) and TUNEL (DNA fragmentation marker) was carried out on coronal sections collected through the entire dorsal hippocampus to investigate the neuroprotective effects of PBM. Briefly, brain sections were washed in PB, permeabilized with 0.4% Triton X-100 in PB for 1 h, and blocked with 10% normal donkey serum for 1 h followed by incubation with a mouse anti-NeuN primary antibody (1:500; Millipore) overnight at 4 °C. After washing in 0.1 M PB, the sections were then incubated with Alexa Fluor 488-conjugated donkey anti-mouse IgG (1:1000, Invitrogen) at room temperature for 1 h. The sections were further incubated in terminal deoxynucleotidyl transferase (TdT) reaction buffer, TdT reaction mixture, and the enzyme solution was provided in the Click-iT® Plus TUNEL assay kit (Thermo Fisher Scientific) according to the instructions by the manufacturer. Finally, sections were washed, mounted on Fisherbrand Superfrost Plus Microscope Slides (Fisher Scientific) coverslipped in VECTASHIELD® Antifade Mounting Medium (Vector Laboratories), and sealed with nail polish. Images were captured on a Zeiss LSM700 Meta confocal laser scanning microscope (Carl Zeiss Inc., Germany), and cell counting and quantification analysis were performed using the NIH ImageJ software (NIH ImageJ program, version 1.49) by investigators blinded to the experimental groups. The number of healthy neurons in the hippocampal CA1 pyramidal layer (e.g., cells displaying round soma and distinct nuclei similar to that observed in nonischemic sham-operated controls) and the number of TUNEL-positive cells per 250 μm length of medial CA1 layer were counted bilaterally in 3–5 representative sections per animal. Average counts from both the left and right hippocampus on each of the examined sections were calculated to provide a single value for each animal. Cell counts from the right and left hippocampi on the examined sections were averaged to provide a single value for each brain. Data are presented as mean ± standard error (SE) from 8 to 12 (n = 8–12) independent animals in each group. Statistical analysis of the data was carried out as described below.
Barnes Maze Task
The Barnes maze task was used to test hippocampus-dependent spatial reference learning and memory as described previously by our group [31, 34]. Briefly, tests of the escape latency (time taken to locate the recessed escape chamber) were performed on the circular maze platform with 18 holes around the perimeter, on days 182, 183, and 184 after GCI reperfusion. During the 3-min trial tests, latency and trace of each animal from the center of the platform to the escape box were recorded via an overhead video camera operated by ANY-maze video tracking software (Stoelting Co., Wood Dale, IL). Probe tests were carried out on day 185 after GCI, when the escape chamber was removed, and the time spent in the target quadrant (quadrant occupancy) was recorded in a 90-s period. All the recorded data were analyzed and compared between groups using the ANY-maze software. The maze platform was thoroughly cleaned with 70% ethanol and dried with a blower fan between each test.
Morris Water Maze Test
Morris water maze testing was conducted to evaluate spatial learning and memory as described in our previous studies [29, 32]. Briefly, animals were placed in the water facing the wall in random in one of the four quadrants ((I, II, III, and IV), located equidistant from one another around the rim of the pool. For each trial, animals were allowed to search the submerged platform (2 cm beneath the water surface) within a maximum time set to 90 s. If the rat located the platform in 90 s, it was allowed a 20-s rest period on the platform. If the platform was not located during 90 s of swimming, the rat was gently guided on the platform and allowed a 20-s rest. The procedure was repeated from each of the four start locations, within a 4-h interval. The escape latency, representing the average of the four trails to reach the platform, was recorded. After each testing, animals were manually dried with a cloth towel and placed in warming cages for 10 min before returning to the home cage. The training tests were administered on days 7, 8, and 9 after GCI. A probe test was performed 4 h after the last training session to examine spatial reference memory. During the probe trial, the platform was removed from the tank and the animal was allowed to swim freely for 90 s. Each rat was placed in the pool at the same random start location. The time spent in the quadrant that previously contained the platform was used to evaluate the level of the spatial reference memory on the given task. All behavioral tracks from the trials were recorded and analyzed using video tracking software, and the resultant data were statistically analyzed as described below.
Immunofluorescent Labeling and Confocal Microscopy
Coronal brain sections were prepared as described above. Immunofluorescence staining was performed following standard protocol as described previously [33]. Briefly, sections were washed three times and blocked in 10% normal donkey serum followed by overnight primary antibody incubation at 4 °C. The following primary antibodies were used in the current study: anti-cleaved caspase-3/9 (1:50, Cell Signaling), anti-NeuN (1:500, Millipore), anti-Tom20 and Annexin V (1:200, Proteintech Group), and anti-8-OHDG (1:100, Abcam). Sections were washed and incubated with appropriate Alexa Fluor donkey anti-mouse or rabbit secondary antibodies (1:1000, Invitrogen) for 1 h at room temperature. Finally, the sections were washed, mounted, and coverslipped in VECTASHIELD Mounting Medium with 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) and sealed. To detect variations in mitochondrial membrane potential (MMP), 100 μL of MitoTracker Red CMXRos (50 ng/mL, Life Technologies) or saline was administered via tail vein injection 5 min before brain collection, as described previously [35]. The coronal brain sections (25 μm thick) were washed 3 times, mounted, and sealed in mounting medium with DAPI as mentioned above. All the fluorescence images were captured on a LSM700 confocal laser microscope (Carl Zeiss). The MitoTracker fluorescence intensity associated with MMP was quantitatively evaluated by using the ImageJ software, and the signals were normalized as percentage change compared to the sham GCI control group. Data were calculated as mean ± SE from four to five independent animals in each group.
Brain Sample Preparation, Immunoprecipitation, and Western Blotting
As described previously, both hippocampi were rapidly removed from animals under deep anesthesia [36]. The hippocampal CA1 regions were quickly microdissected on an ice pad and immediately frozen in liquid nitrogen. Brain homogenates and subcellular fractionations, including mitochondrial fractions and cytosolic fractions, were prepared according to our previous protocol [33]. Protein concentrations were determined using a modified Lowry Protein Assay kit (Pierce, Rockford, IL). For immunoprecipitation (IP) and Western blot analyses, equal amounts of CA1 proteins (200 μg) were diluted in HEPES buffer and incubated with specific primary antibody overnight at 4 °C as described [33]. The precipitated proteins were examined by Western blotting with detection antibody. For Western blotting, samples were mixed with Laemmli loading buffer, boiled, and separated on 4–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Separated proteins were transferred onto polyvinylidene fluoride (PVDF) membrane (Millipore), blocked in 2% BSA blocking solution for 1 h, followed by incubation with appropriate primary antibody antibodies at 4 °C overnight. The membrane was washed to remove unbound antibody, followed by incubation with HRP-conjugated secondary antibody for 1 h at room temperature. Blots were then washed 3 more times and bound proteins coupled to HRP/luminol-based chemiluminescent signals were visualized using a CCD digital imaging system. Semiquantitative analyses of the corresponding band densities were performed using ImageJ software with appropriate normalization to loading controls (COX4 or β-actin). A mean ± SE was calculated from the data for statistical comparison. The following primary antibodies were used: Drp1 and OPA1 from BD Biosciences; phosphor-Drp1 Ser 616 and Ser 637 antibodies from Cell Signaling Technology; antibodies to COX4, Mfn1, Mff, Fis1, Bcl-2, LC3, and Apaf-1 from Proteintech Group, Inc.; and antibodies to caspase-9, Bax, cytochrome c, TRX2, and SOD2 from Thermo Fisher Scientific.
Duo-Link II Proximity Ligation Assay
The Duo-Link II in situ proximity ligation assay (PLA) was carried out with the Duo-Link in situ detection reagent kit (Sigma-Aldrich) according to the manufacturer’s instructions, as described in our recent studies [32, 37]. Briefly, coronal parallel series of sections prepared as described above were blocked in 10% donkey serum for 1 h and followed by incubation with mouse and rabbit primary antibodies overnight at 4 °C. After washing, Duo-Link PLA Rabbit MINUS and PLA Mouse PLUS proximity secondary probes were added to the sections and incubated for 1 h at 37 °C. Sections were washed twice, and ligation mix was added to the samples and incubated for 30 min at 37 °C for ligation and circularization. Amplification and detection through rolling circle amplification was conducted after washing the sections according to the manufacturer’s protocol. Sections were then mounted on the slides and sealed in mounting medium with DAPI, and the PLA signals as distinct dots were visualized and captured under a LSM700 confocal microscope at ×40 magnification. The red spots representing protein-protein interaction were separated and analyzed using NIH ImageJ software.
Electron Microscopic Analysis
Changes in the ultrastructural features of mitochondria in hippocampal CA1 pyramidal neurons were evaluated using a transmission electron microscope as described previously by our laboratory [38]. Briefly, the brain hippocampal CA1 region were diced and fixed overnight in 2.5% glutaraldehyde solution at 4 °C. Samples were post-fixed in 2.5% glutaraldehyde solution containing 2.0% paraformaldehyde for 4 h. After three washes in 0.1 M PB, the tissues were then post-fixed with 2% osmium tetroxide for 30 min prior to impregnation in increasing concentrations of ethanol solutions. Subsequently, tissue segments were washed in propylene oxide and embedded in Epon 812. Ultrathin sections of 70 nm were then cut using a diamond knife on a Leica ultramicrotome and mounted on 200-mesh copper grids. After counterstaining with lead citrate and uranyl acetate, the copper grids were washed and the ultrastructure was examined on a transmission electron microscope (Hitachi-7650). Representative images of the mitochondria ultrastructure in hippocampal CA1 pyramidal neurons from four to five animals in each group are shown.
Drp1 GTPase Activity Assay
Equal amount of protein samples (200 μg) of hippocampal CA1 from each group was immunoprecipitated with 5 μg of anti-Drp1 antibody and 30 μL of protein A/G agarose (Santa Cruz). The beads were washed four times with GTPase wash buffer (50 mM Tris-HCl, pH 7.5, 1.0 M NaCl, 10 mM MgCl2, 4 mM DTT, 1 mM PMSF) and incubated with 0.5 mM GTP in reaction buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 20 mM EDTA, and 5 mM MgCl2) at 30 °C for 1 h. The released free phosphate was measured using a PiColorLock Gold Colorimetric Assay Kit (#303-0030, Novus Biologicals) according to the kit booklet protocol. Absorbance was recorded at 660 nm on a microplate spectrophotometer (Bio-Rad, Benchmark Plus). A standard curve was prepared using the 0.1-mM Pi stock provided with the kit for quantification. Each sample was measured in triplicate using the kit, and values were calculated and presented as percentage changes compared to the sham GCI group.
Quantification of Cytochrome c Oxidase Activity and ATP Production
To examine the effect of PBM on mitochondrial function, assays of cytochrome c oxidase (CCO) activity and ATP production were carried out as described previously [39]. Briefly, ATP concentration was determined using an ENLITEN® rLuciferase/Luciferin kit (FF2021, Promega) as described. Cytosolic protein samples (30 μg) were suspended in 100 μL of reconstituted rL/L reagent buffer. The reactions were measured in a microplate luminometer (PE Applied Biosystems), and values were calculated from the ATP standard curve. CCO activity was measured with a Complex IV Rodent Enzyme Activity Microplate Assay Kit (ab109911, Abcam) using an equal amount of protein sample from the mitochondrial fractions. The activities of immunocaptured CCO proteins within the 96-well microplate were determined colorimetrically by measuring the degree of cytochrome c oxidation at 550 nm absorbance on a microplate spectrophotometer (Bio-Rad). The values of ATP concentration and relative CCO activity were expressed as percentage changes versus the sham GCI group for graphical depiction.
Measurement of Mitochondrial Total Antioxidant Capacity
Total antioxidant capacity of the mitochondrial fractions from the hippocampal CA1 subregion was measured using an antioxidant assay kit (#709001, Cayman Chemical) according to the vendor’s instructions. The assay measures total antioxidant ability in the sample to inhibit the oxidation of 2,2′-azino-di(3-ethylbenzthiazoline-6-sulfonic acid (ABTS) by metmyoglobin. As described previously [31], the reactions were initiated by mixing 10 μL of mitochondrial proteins or trolox standards with 40 μL of the metmyoglobin working solution according to the manufacturer’s protocol. The absorbance at 450 nm was recorded on a spectrophotometer (Bio-Rad) after 5 min incubation, and the standard was plotted. Total antioxidant capacities of the samples were then determined using the trolox standard curve, and the values were reported as percentage changes compared to the sham GCI control.
Caspase Activity Assay
Caspase-3 and caspase-9 activities were measured by incubation of the protein homogenate with the fluorometric substrates Ac-DEVD-AMC and Ac-LEHD-AMC, respectively, as described previously by our laboratory [35]. In brief, the reaction was initiated by mixing the hippocampal proteins and specific substrate in protease assay buffer and incubated at 37 °C. One hour later, the fluorescence of free AMC, proteolytically cleaved by the corresponding caspase, was measured on a spectrophotometer (Perkin Elmer) with excitation at 360 nm and emission at 460 nm. Relative caspase activity in each sample was determined as differences in fluorescent units and presented as fold changes compared to the sham GCI group.
Neuronal Culture and Oxygen-Glucose Deprivation Model
Primary hippocampal neurons were prepared from SD rats at embryonic day 18 (E18) as described previously [40]. Briefly, dissociated cells were suspended in plating medium and plated on poly-D-lysine (Sigma)-coated 24-well culture plates at a density of 4.50 × 105 cells/mL. Cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum in a humidified incubator in an atmosphere of 5% CO2 at 37 °C. On day 2 post-plating, half of the plating medium was removed and replaced with maintenance medium (Neurobasal medium, 2% B-27 supplement, and 0.5 μM L-glutamine). Subsequent media changes were performed every 2–3 days, and cells were used on day 9 in vitro. To achieve oxygen-glucose deprivation (OGD) challenge, cells were transferred to glucose-free media and culture plates were placed in a humidified chamber and maintained at 37 °C in 0% O2/95% N2/5% CO2 for 3.0 h. Thereafter, extracellular solution was replaced with maintenance medium and cultures were returned to the incubator. For control OGD treatment, cultures were transferred to standard neurobasal media, introduced into the humidified chamber, and maintained at 37 °C for 3.0 h in a CO2 incubator. For drug treatment, the complex IV/CCO specific inhibitor potassium cyanide (KCN 50 μM, a low concentration to avoid cell death [41]) or vehicle was applied in the culture media immediately after OGD. PBM treatment (2 min at 8 mW/cm2) was initiated 30 min after KCN and continued every 6 h for a total of 4 doses (Fig. 7a).
Mitochondrial Health Assay
The HCS Mitochondrial Health Kit (H10295, Thermo Fisher Scientific) was used to measure mitotoxicity and cytotoxicity simultaneously in the same cell. Briefly, cell staining solution was prepared by adding 10.5 μL of the MitoHealth stain solution and 2.1 μL of Image-iT® DEAD Green™ viability stain to 6 mL complete medium according to the manufacturer’s protocol. The neuronal cultures were incubated with the cell staining solution for 30 min under culture conditions, followed by adding the counterstain/fixation solution for 15 min at room temperature. The red MitoHealth stain corresponding to MMP and DEAD Green (cytotoxicity) were captured under a LSM700 Meta confocal laser microscope (Carl Zeiss).
Neuronal Viability Measurement
Neuronal viability was assessed 24 h after OGD by double labeling with fluorescein diacetate (FD, Sigma) and propidium iodide (PI, Sigma). Briefly, cultures were incubated with FD (10 μg/mL) and PI (5 μg/mL) for 20 min. The cultures were then washed and images were captured using a confocal laser microscope. Neuronal viability was determined by calculating the ratio of FD-labeled cells over PI-labeled cells. Cell counting was performed by investigators blinded to the experimental treatment. The release of lactate dehydrogenase (LDH) was measured using a Pierce LDH Cytotoxicity Assay Kit (88953, Thermo Scientific) based on the manufacturer’s instructions. Briefly, the cells were treated according to the manufacturer’s protocol, and the assay was performed by transferring the culture media into new 96-well plate and adding the kit reagents. The reactions were stopped 30 min after incubation at room temperature, and LDH activity was determined by absorbance at 490 nm using a Microplate Spectrophotometer (Bio-Rad Benchmark Plus). The levels of LDH release were normalized to protein concentration and data were presented as percentage change versus controls.
Experimental Design and Statistical Analysis
For the in vivo studies, adult male SD rats were randomly divided into four groups: sham GCI control group without ischemia, GCI group, GCI with PBM treatment group, and a GCI with sham PMB treatment group (underwent identical procedures with the PBM treatment group except that the laser device was not turned on).
Additional groups with vehicle or a selective inhibitor of mitochondrial fission protein Drp1, Mdivi-1, was administrated at 1 mg/kg/day with 3-day release Alzet osmotic mini-pumps (DURECT Corporation) as described previously [42]. Eight to 12 animals in each group were used for the behavioral tests and histological analysis. Four to six independent rats per group were used in the other in vivo experiments. For the in vitro studies, primary hippocampal cultures were prepared from mixed-sex E18 SD rat. The cultures were randomly assigned to the “sham” control group, OGD group, OGD with PBM group, and OGD with sham PMB treatment group. Values for OGD analysis were pooled from at least four independent cultures. Detailed sample sizes (n) and analyses are provided within each figure legend. Randomization was used to assign subjects to different groups and to collect and analyze data. Statistical analysis was performed with the SigmaStat software (version 3.5, Systat Software Inc.) and ANY-maze software (version 5.1, Stoelting Co.) using either one-way or two-way analysis of variance (ANOVA). After ANOVA, Student-Newman-Keuls or Dunnett’s test was adopted for post hoc analyses to determine differences between groups or compare with the control group. All results are presented as mean ± SE. Statistically significant difference was accepted at the 95% confidence (P < 0.05).
Results
PBM Post-Treatment Significantly Protects Hippocampal CA1 Neurons from GCI Injury in a Dose-Dependent Manner
As shown in Fig. 1a, PBM post-treatment was initiated 6 h after GCI followed by 1–3 more doses each day, and histology was conducted 2 weeks after GCI. As can be seen in Fig. 1b, c, NeuN staining (green) revealed that GCI induced severe neuronal loss of the CA1 subregion compared with the sham GCI control and PBM-treated GCI rats. Intriguingly, the density of CA1 pyramidal neurons was remarkably increased in the animals with PBM treatments at doses of 2.0 and 3.0 J (Fig. 1b). Further cell counting analyses indicated that PBM post-treatment (power density, 8.0 mW/cm2; each hippocampus received doses, 1.0, 2.0, 3.0, and 4.0 J) significantly and dose-dependently increased the number of surviving neurons (Fig. 1a, c). High PBM doses (3.0 and 4.0 J) saved around 78–80% neurons as in sham animals, while lower PBM doses (1.0 and 2.0 J) were shown as less protective. In addition, as seen in Fig. 1b, d, the number of apoptotic cells based on TUNEL labeling increased dramatically in GCI animals without PBM treatment. Moderate TUNEL staining was found in 2.0-J PBM-treated animals, and the lowest staining was found in 3.0- and 4.0-J PBM-treated groups, compared to GCI control and 1.0-J PBM-treated groups. No significant differences of NeuN and TUNEL staining were found between 3.0- and 4.0-J PBM-treated GCI groups, and therefore, this 3.0-J treatment profile was selected for the following experiments. These results demonstrated strong effects of PBM in preserving hippocampal CA1 neurons and decreasing the number of TUNEL-positive cells, suggesting that PBM neuroprotection is very efficient against GCI-induced delayed neuronal cell death.
PBM Significantly Attenuates the Hippocampal-Dependent Short-Term and Long-Term Behavioral Deficits Following GCI
The vulnerable hippocampal CA1 pyramidal neurons play a pivotal role in spatial learning and memory. To investigate whether PBM’s neuroprotective effect also led to functional improvement, we first examined and compared the short-term spatial learning ability of animals from each group using the Morris water maze test. As shown in Fig. 2a, b, the escape latency became progressively shorter in all experimental groups. On each trial day (days 7, 8, and 9), the GCI group animals with sham PBM treatment spent more time to find the hidden platform as compared to sham GCI animals. However, PBM-treated animals displayed a significant shorter time in latency to find the platform (Fig. 2b (a)) and a significant longer time spent in the platform quadrant (Fig. 2b(b)), as compared to non-PBM ischemic control animals.

Effect of PBM on the short-term and long-term behavioral deficits tested 1 week and 6 months after GCI. a, b Morris water maze test was performed to examine the short-term spatial learning and memory ability on days 7, 8, and 9 after GCI. Representative traces showing the animals to explore the hidden platform and quadrant occupancy (a, d: sham GCI; b, e: GCI + sham PBM; and c, f: GCI + PBM). c, d Barnes maze task was performed to test the long-term learning and memory deficits 6 months after GCI. Representative escaping traces of sham GCI rats (a, e), GCI rats (b, f), GCI + sham PBM (c, g), and GCI + PBM (d, h) rats at day 184 (a–d) and day 185 (e–h) after GCI, respectively. b, d The escape latency (a, time taken to locate the submerged platform or escape hole) and quadrant occupancy (b, time spent in the target quadrant) were recorded and statistically analyzed. Data are expressed as means ± SE from 8 to 12 animals in each group. *P < 0.05 versus sham GCI group, #P < 0.05 versus GCI or GCI + sham PBM group
Previous studies have shown that there is a long-lasting impairment in learning and memory in rat models of GCI. In this study, we also assessed the effect of PBM on long-term functional outcome at 6 months after GCI, using the Barnes maze task. As shown in Fig. 2c, d, in the training session on days 182, 183, and 184 after GCI, GCI control and sham PBM-treated animals spent more time in escape latency to locate the hidden escape box, compared to sham control rats. By contrast, PBM treatment significantly decreased the escape latency to find the escape box in the training trials. In addition, in the probe test 185 days after GCI, the GCI group animals and sham PBM-treated animals spent less time in the target quadrant where the escape box was located, whereas the PBM-treated GCI animals spent significantly longer time in quadrant occupancy (Fig. 2c (e–h), d (b)). Taken together, PBM has the ability to significantly preserve both short-term and long-term spatial learning and reference memory following GCI.
PBM Post-Treatment Significantly Suppresses GCI-Induced Mitochondrial Fragmentation in Hippocampal CA1 Neurons
We next investigated the effect of PBM post-treatment on the degree of mitochondrial network fragmentation following GCI. As shown in Fig. 3a (a–d), representative confocal immunofluorescence staining of Tom20 demonstrated remarkable mitochondrial network fragmentation in the hippocampal CA1 pyramidal neurons of GCI animals when compared to the sham GCI control. Further comparative quantification of mitochondrial segments was carried out after the processing of Tom20 fluorescent staining using the NIH ImageJ program. As indicated in Fig. 3a (a–l), b (a, b), relative to the higher densities of mitochondrial total particles and small particles (size < 1.5 μm) in the GCI control and sham PBM-treated GCI groups, PBM-treated ischemic neurons displayed a significantly lower degree of mitochondrial network fragmentation. Furthermore, percent changes of continuous mitochondrial structures (size > 2 μm) were analyzed and compared among groups. As shown in Fig. 3a (m–p), b (c), PBM-treated animals exhibited a significant recovery of the large continuous mitochondrial structures, as compared to non-PBM ischemic controls, demonstrating an efficacious preservation of healthy mitochondrial structures similar to that observed in sham GCI animals. These results suggest the strong capability of PBM in favorably preserving mitochondrial dynamics following GCI stress.

Effect of PBM on GCI-induced changes in mitochondrial dynamics of hippocampal CA1 pyramidal neurons. a Confocal microscopy images of immunofluorescence staining for Tom20 (green), and the nuclear counterstaining with DAPI (blue), of CA1 neurons 3 days after GCI (a–d). Scale bar: 5 μm. (e–p) Tom20 fluorescent in the acquired images was thresholded, filtered (median, 2.0 pixels), and binarized to analyze mitochondrial segments using ImageJ (NIH image program, version 1.49). b The counts of total and small mitochondrial fragmentation were obtained by normalization of the number of total and small mitochondrial particles (size < 1.5 mm) to total mitochondrial areas, respectively. The areas of large particles (size > 2 mm), from image views in each group, were normalized to the total mitochondrial areas to obtain the percentage changes of continuous mitochondrial structures (c). Data are presented as means ± SE, n = 5–6 in each group. *P < 0.05 versus sham GCI control, #P < 0.05 versus GCI or GCI + sham PBM group
PBM Effectively Attenuates the Activity and Mitochondrial Location of Drp1 GTPase Following GCI
We hypothesized that Drp1 GTPase, a key mitochondrial fission protein, may play a crucial role for GCI-induced excessive mitochondrial fragmentation and neuronal impairment. It has been demonstrated that there are two critical sites on Drp1 phosphorylation, serine 616 (S616), which is required for Drp1 activity and mitochondrial fission, and S637, which serves to inhibit Drp1 activity and suppress the fission process [43, 44]. The results of the time course study in Fig. 4a revealed that mitochondrial-located Drp1-S616 and Drp1-S637 levels were gradually increased and decreased from 6 h to 3 days after ischemic reperfusion, respectively. Intriguingly, PBM treatment effectively attenuated the ratio of Drp1-S616 to Drp1-S637 as shown from Western blot analysis using mitochondrial protein samples from the hippocampal CA1 subregion at 3 days of reperfusion (Fig. 4b). In addition, the levels of mitochondrial-located total Drp1, at ischemic reperfusion day 3 after GCI, as well as the Drp1 GTPase activity induced by GCI, were significantly inhibited following PBM treatment (Fig. 4c, d). To evaluate the potential role of Drp1 GTPase induction in GCI-induced mitochondrial fragmentation and the delayed neuronal death in the hippocampal CA1 region, we administered a specific Drp1 GTPase inhibitor, Mdivi-1, via 3-day release osmotic mini-pumps immediately after GCI. As shown in Fig. 4e, f, Mdivi-1 prevented GCI-induced mitochondrial fragmentation in CA1 pyramidal neurons, an effect that was correlated with a significant protection of the hippocampus CA1 region from GCI-induced delayed neuronal cell death. This finding suggests that Drp1 GTPase activation is critical for GCI induction of mitochondrial fragmentation and that PBM’s ability to attenuate excessive mitochondrial fission may be a critical mechanism for its beneficial effects after GCI.

Effect of PBM on Drp1 GTPase—a key mitochondrial fission protein, and the effect of Mdivi-1 on GCI-induced mitochondrial fragmentation and neuronal loss in hippocampal CA1. a–c Western blotting analyses of Drp1 phosphorylation (Ser616, Ser637) and mitochondrial location of total Drp1, at ischemic reperfusion day 3 after GCI, using mitochondrial protein samples from CA1 subregion. d Drp1 GTPase activity in hippocampal CA1 protein samples was quantified using a PiColorLock Gold kit. e Total mitochondrial fragmentation in CA1 pyramidal neurons from sham, vehicle (Veh), and Mdivi-1-treated animals was examined 3 days after GCI. f Typical staining of NeuN in vehicle (a) and Mdivi-1-treated (b) animals at day 14 after GCI, and the quantitative analyses of the numbers of surviving neurons in hippocampal CA1 layer (c). Dashed line represents percent value in sham control. Scale bar: 50 μm. Data are presented as means ± SE, n = 4–5 in a–e, n = 8–12 in f. *P < 0.05 versus sham control, #P < 0.05 versus GCI or GCI + sham PBM group. $P < 0.05 versus vehicle-treated group
PBM Promotes Mitochondrial Targeting of Opa1 and Mfn1 and Inhibits Drp1 Binding with Mff and Fis1 in Ischemic Hippocampal CA1 Subregion
We next examined whether PBM regulates Opa1 and Mfn1, key mitochondrial fusion proteins. As shown in Fig. 5a, representative Western blot of the time course analysis showed a gradual decreased mitochondrial expression of Opa1 and Mfn1 from 6 h to 3 days after GCI reperfusion. In contrast, PBM induced a highly significant increase in Opa1 and Mfn1 levels within mitochondrial fractions at 3 days after GCI (Fig. 5b). Additionally, Western blot and quantitative analyses of mitochondrial fractions from the hippocampal CA1 region indicated that the temporally induced expression of Mff and Fis1, key mitochondrial membrane adaptor proteins for fission, could be efficaciously inhibited following PBM treatment after GCI reperfusion (Fig. 5c, d). Further co-IP experiments using hippocampal CA1 protein samples at the 3-day time point after GCI and data analysis demonstrated that the interactions of Drp1-Fis1 and Drp1-Mff were remarkably increased in non-PBM-treated GCI animals and significantly attenuated by PBM (Fig. 5e, f). The protein-protein complex formations were confirmed by Duo-Link II in situ PLA immunostaining and quantitative analyses. As indicated in Fig. 5g, representative confocal microscopy images (a–i) and quantitative analyses (m) of the red spots representing the interactions of Drp1-Fis1 and Drp1-Mff were significantly higher in the non-PBM-treated GCI group than those in the sham control and PBM-treated GCI animals, further demonstrating the effectiveness of PBM in regulating the mitochondrial fission process.

Effect of PBM on mitochondrial location of key fusion proteins (Opa1 and Mfn1), and the effect on mitochondrial membrane adaptor proteins (Mff and Fis1) and their interactions with Drp1. a–d Western blot analyses of mitochondrial targeting fusion proteins (OPA1 and Mfn1) and fission proteins (Mff and Fis1) in hippocampal CA1 subregion 3 days after GCI. e, f Hippocampal CA1 protein samples at 3 days after GCI were subjected to immunoprecipitation (IP) analysis of the interaction of Drp1 with Fis1 and Mff. g Duo-Link II in situ PLA immunostaining (a–i) and quantitative analyses (m) of Drp1-Fis1 and Drp1-Mff bindings in hippocampal CA1 at reperfusion day 3. Red spots representing the interactions were separated and analyzed using NIH ImageJ software. Scale bar: 20 μm. Data are shown as means ± SE from 4 to 5 independent rats per group. *P < 0.05 versus sham group, #P < 0.05 versus GCI or GCI + sham PBM group
PBM Improves Mitochondrial Overall Health Status and Mitochondrial Function in Hippocampal CA1 Neurons Following GCI
Additional experiments were conducted to determine whether PBM regulates mitochondrial health status in the hippocampus CA1 subregion following GCI and whether mitochondrial morphology and function are preserved in the same neurons after ischemia. As shown in Fig. 6a, representative Western blots demonstrated the upregulated expression of mitochondrial antioxidants SOD2 and TRX2 in the CA1 protein samples from PBM-treated animals, as compared with PBM untreated groups 3 days after GCI. Indeed, using mitochondrial protein samples from the CA1 subregion and an antioxidant assay kit, we detected a strong increase in the total mitochondrial antioxidant capacity at 3 days of reperfusion (Fig. 6b). Consistent with these observations, PBM untreated ischemic rats exhibited a greater level of mitochondrial oxidation as evidenced by the colocalization of oxidized DNA marker 8-OHDG with mitochondrial marker Tom20, whereas the deleterious effect in GCI-induced oxidized mtDNA was ameliorated with PBM treatment (Fig. 6c). We also detected decreased levels of LC3II from mitochondrial fractions of PBM-treated animals compared to sham PBM in GCI controls (Fig. 6d), suggesting a potential inhibitory effect of PBM on GCI-induced excessive mitophagy in maintaining mitochondrial health and homeostasis. We further performed electron microscopy (EM) analysis to test whether PBM was able to preserve mitochondria ultrastructure of hippocampal CA1 pyramidal neurons. Figure 6e, middle panel, shows that the neuronal ultrastructure without PBM was markedly impaired at 3 days after GCI, as evidenced by nuclear membrane distortion and hyperchromasia (blue arrow), and the mitochondria display a fragmented structure or vacuolized with broken or disintegrated cristae (green arrow). However, PBM preserved the ultrastructure of CA1 neurons, which displayed a regular nucleus and intact ultrastructure with distinct cristae in most mitochondria (Fig. 6e (c)). MMP is critical for maintaining the physiological function of healthy mitochondria. Figure 6f demonstrated that GCI-induced collapse of MMP or the depolarization of mitochondrial membrane, reflected by the MitoTracker® dye staining and analyses, was markedly abolished by PBM in CA1 neurons. Remarkably, measurements of CCO activity and ATP contents revealed impaired levels in GCI controls and significant improvements in PBM-treated animals (Fig. 6g), confirming a vital role of PBM in modulating mitochondrial function following GCI insult.

Effect of PBM on the changes of mitochondrial health status and mitochondrial function in hippocampal CA1 following GCI. a Mitochondrial protein fractions from hippocampal CA1 were collected 3 days after GCI and subjected to Western blot analyses using the indicated antibodies. b Mitochondrial antioxidant capacity was measured with an antioxidant assay kit using mitochondrial protein samples from CA1 subregion. c Typical confocal microscopy images showing TOM20 (green) and 8-OHDG (red) double labeling for the detection of oxidized mtDNA at 3 days after GCI. Scale bar: 10 mm. d Mitochondrial fractions of CA1 protein 3 days after GCI were subjected to Western blotting using LC3 antibody. e Representative electron microscopy (EM) images of hippocampal CA1 pyramidal neurons showing mitochondrial ultrastructure (arrowheads). Scale bar: 500 nm. f Brain sections stained with MitoTracker Red was collected to detect variations in mitochondrial membrane potential (MMP). Representative confocal microscopy images counterstained with DAPI were shown (a–f), and MitoTracker fluorescence intensity associated with MMP was quantitatively analyzed and compared. Scale bar: 20 mm. g CCO activity and ATP level reflecting mitochondrial function was assessed with assay kits using the mitochondrial fractions and cytosolic protein samples, respectively. Values are shown as means ± SE (n = 4–5 per group).*P < 0.05 versus sham group, #P < 0.05 versus GCI or GCI + sham PBM group
PBM Post-Treatment Significantly Inhibits the Mitochondria-Dependent Apoptosome Pathway in Hippocampal CA1 Neurons Following GCI
Bcl-2 and Bax are pivotal regulatory proteins for mitochondria-mediated apoptosis process. Western blot analyses, as presented in Fig. 7a, exhibited a significantly preserved Bcl-2 level and reduced accumulation of active Bax in mitochondrial fractions from CA1 subregion compared with GCI and sham PBM controls, with a lower Bax/Bcl-2 ratio maintained to a level comparable with sham controls. Consequently, PBM treatment significantly inhibited cytosolic content of cytochrome c (cyt c), as shown in Fig. 7b, implying an effective PBM protection on the release of cyt c from the mitochondria into the cytosol through the permeability transition pore (PTP). The formation of Apaf-1/caspase-9 apoptosome, downstream of cyt c, was then examined by co-IP experiment and in situ PLA immunoassay, respectively. Figure 7c, d demonstrated that GCI-induced Apaf-1/caspase-9 component interaction in the cytosols could be dramatically suppressed by PBM treatment. Furthermore, caspase-3 and caspase-9 activities, determined by specific fluorometric substrate assays using 3-day cytosolic proteins from the CA1 subregion, exhibited low caspase-processing activities in PBM animals compared with non-PBM GCI controls (Fig. 7e). Finally, as shown in the representative confocal microscopy images in Fig. 7f, PBM displayed a remarkable ability in inhibiting the immunoreactivity of Annexin V at the early stage of apoptosis (i: a–c, day 2 after GCI), as well as of the active forms of caspase-3 and caspase-9 (ii–iii: day 3 after GCI) in hippocampus CA1 neurons.

Effect of PBM on the mitochondria-mediated caspase activation pathway in hippocampal CA1 subregion following GCI. a, b Mitochondrial location of Bax and Bcl2 proteins and the cytosolic content of cytochrome c (cyt c) protein in hippocampal CA1 was examined by Western blotting analyses. COX4 was used as a negative control for protein purity of the cytosol without mitochondria fraction. c, d At 3 days after GCI, the effect of PBM on the formation of Apaf-1/caspase-9 apoptosome was examined by Co-IP and in situ PLA immunoassay, respectively. e The changes of caspase-3 and caspase-9 activities were determined by specific fluorometric substrate assays using 3-day cytosolic proteins from hippocampal CA1 subregion. f Representative confocal microscopy images of the staining of Annexin V (i, day 2 after GCI), and the active forms of caspase-3 and caspase-9 (ii and iii, Cle-casp3/9, day 3 after GCI) in hippocampus CA1 subregion. Scale bar: 20 mm. Data are presented as means ± SE from 4 to 6 animals in each group.*P < 0.05 versus sham group, #P < 0.05 versus GCI or GCI + sham PBM group
Evidence of Mitochondrial CCO in Mediating PBM’s Neuroprotection in Primary Hippocampal Neurons Subjected to OGD and Reperfusion
To further mechanistically determine the potential mechanism underlying PBM’s beneficial effects, the CCO-specific inhibitor KCN (50 μM, a nonlethal dose [41]) or vehicle was applied to the media in hippocampal neuron cultures immediately after OGD, and PBM treatment was conducted 30 min after KCN and continued every 6 h for an additional 3 more doses (see diagram in Fig. 8a). Mitochondrial dysfunction was first tested using a mitochondrial health assay kit, and representative images of red MitoHealth stain and DEAD Green corresponding to MMP and cytotoxicity, respectively, are shown in Fig. 8b. Mitochondrial function was impaired in neurons subjected to OGD and 24 h reperfusion, as evidenced by the shifting of red and green signaling in OGD- and KCN-treated neurons. It was noted that PBM significantly reduced the extent of mitochondrial dysfunction as a recovery of MMP and decreased cellular counts of DEAD Green were observed, an effect that was remarkably attenuated with KCN treatment (Fig. 8b, c). To validate the role of CCO in mediating the enhancement of neuronal survival, hippocampal neurons were double-labeled with PI (red) and FD (green) 24 h after OGD, and data analyses of neuronal viability were carried out. Our data showed that after OGD insult, PBM treatment significantly protected against hippocampal neuronal injury (Fig. 8d–f (a, b, d)). While a low dose of KCN treatment did not add to OGD-induced cell death, the application of a nonlethal dose of KCN could effectively reverse PBM’s neuroprotective effects (Fig. 8d–f (b–e)). These results strongly suggest a key role of targeting mitochondria for PBM’s beneficial neuroprotective effects in GCI.

Effect of CCO inhibition on PBM neuroprotection in primary hippocampal neurons subjected to OGD followed by reoxygenation. a Diagram showing the OGD model and treatment protocol of KCN (50 μM, immediately after OGD) and PBM (8 mW/cm2, every 6 h after OGD). b, c Representative fluorescence microscopic images of hippocampal neurons stained with a HCS Mitochondrial Health Kit (ThermoFisher), and the analyses of MMP intensity (arbitrary unit) and the density of DEAD Green-positive cells. d–f Representative images of hippocampal neurons double-labeled with PI (red) and FD (green), and data analyses of the ratio of FD/PI and LDH release in the indicated groups. Scale bar: 20 mm. Data are presented as means ± SE from 4 to 6 cultures per group. For each culture, 4–5 vision fields in each group were used for calculations.*P < 0.05 versus control group (a), #P < 0.05 between KCN and PBM + KCN groups. KCN, potassium cyanide; OGD, oxygen-glucose deprivation; PI, propidium iodide; FD, fluorescein diacetate; LDH, lactate dehydrogenase
Discussion
To the best of our knowledge, the current study is the first to demonstrate that post-treatment with PBM exerts profound neuroprotective and functional improvement effects in an experimental animal model of global ischemic brain injury. Transient GCI due to cardiac arrest is known to cause significant atrophy and death of neurons in the hippocampal CA1 subregion and irreversible long-lasting cognitive injury [45]. In addition, hippocampal neuronal death after ischemia can also increase the incidence of ischemic-type dementia [46]. A major novel finding of our current study is the significant functional improvements induced by PBM post-treatment in GCI, which could provide a solid groundwork for potential preclinical studies of PBM as a simple and convenient noninvasive therapy against cognitive deficits induced by cardiac arrest.
Of significant importance, the beneficial functional improvement effect of PBM after GCI was long lasting, as examination at 30 and 180 days after GCI revealed that PBM-treated animals still had significantly higher performance in spatial learning and memory, as compared to control animals. It is well established that the type of neuronal death in the hippocampal CA1 region initiates at around 3 days after GCI insult and matures 7 days after GCI reperfusion, which is known as the delayed apoptotic neuronal cell death [12, 47, 48]. Our results suggest that the cognitive preservation effects of PBM post-treatment are likely due to the fact that PBM is highly efficient at preserving neuronal survival and preventing apoptotic cell death in the critical hippocampal CA1 region after GCI. Indeed, we found that PBM post-treatment saved approximately 78–80% of the hippocampal CA1 region neurons, as compared to sham animals. It should be mentioned that similar neuroprotective results were observed in female rats 2 weeks after ovariectomy and subjected to GCI (data not shown here), suggesting that there were no gender differences in PBM’s neuroprotection.
It is well known that the mitochondria are the powerhouses of cells and are responsible for most of the body’s energy production in the form of ATP [49, 50]. Mechanistically, PBM has been suggested to exert its beneficial effects in a variety of disorders via effects upon its proposed photoreceptor in the mitochondria, CCO, leading to greater oxygen consumption and ATP production [51,52,53,54]. Indeed, we found that PBM significantly enhanced both CCO activity and ATP levels in the hippocampal CA1 region after GCI. Furthermore, using an in vitro model for ischemia, hippocampal neuron cultures exposed to OGD, we mechanistically validated that the mitochondrial component CCO served as a potential target underlying PBM’s beneficial effects for mitochondrial function preservation and neuroprotection. Our study also revealed that PBM significantly improved mitochondrial overall health status and preserved mitochondrial morphology and mitochondrial function in the hippocampal CA1 region following GCI. This is evidenced by the fact that PBM induced a significant increase in total mitochondrial antioxidant capacity in the hippocampal CA1 region at 3 days after GCI. This effect may be due, in part, to our associated finding that PBM upregulated the expression of two key mitochondrial antioxidants, SOD2 and TRX2, in the hippocampal CA1 region after GCI. It is well known that ROS generation increases markedly after GCI and causes significant oxidative damage to DNA, protein, and lipids in neurons and other cell types [55, 56]. Therefore, PBM-induced enhancement of the antioxidant capability of mitochondria after GCI could help preserve mitochondrial function and cell survival in the face of increasing damaging ROS after cerebral ischemia. Our observations are consistent with previous in vitro reports showing PBM’s protective effects in cortical neurons against ischemia-like conditions such as glutamate, NMDA or kainite excitotoxicity, and OGD [57, 58].
In addition to regulating mitochondria antioxidant activity and ATP generation, PBM significantly attenuated mitochondria-dependent apoptosome signaling, an ischemia-induced intrinsic apoptotic pathway of apoptosis in the hippocampal CA1 region. It is now known that a ubiquitin-like protein, LC3, plays a key role in targeting damaged mitochondria to autophagosomes and exclusively activates the process of mitophagy in delayed neuronal death after brain ischemia [59]. Excessive mitophagy and the unbalanced fission that results from abnormal mitochondrial dynamics lead to excessive mitochondrial fragmentation, which impairs mitochondrial function [60, 61]. It is well accepted that mitochondrial dysfunction leads to programmed cell death, wherein Bax insertion into the mitochondrial outer membrane increases its permeability leading to the release of apoptosis-inducing factor cytochrome c [62, 63]. Interestingly, PBM treatment dramatically reduced mitochondrial fragmentation in the hippocampal CA1 region after GCI. To our knowledge, our study is the first to demonstrate that PBM regulates mitochondrial fragmentation in the GCI brain. The mitochondrial regulatory effect that we observed was associated with and likely due in part to (1) reduced Drp1 GTPase fission protein activity and its bindings with adaptor proteins (Mff and Fis1), (2) increased mitochondrial location of fusion proteins (Opa1 and Mfn1), and (3) decreased mitochondrial targeting of fission proteins (Mff and Fis1) in hippocampal CA1 neurons. The importance of mitochondrial-mediated apoptosis induced by mitochondrial fission is evidenced by our finding that administration of a specific Drp1 GTPase inhibitor, Mdivi-1, prevented GCI-induced mitochondrial fragmentation and induced significant protection of hippocampal CA1 neurons from delayed neuronal cell death. This finding agrees with other studies, which demonstrated that post-ischemic treatment with Mdivi-1 prevents OGD-induced apoptosis of hippocampal neurons in vitro [64] and protects against ischemic brain injury in a rat MCAO model of focal cerebral ischemia [65].
Near infrared (NIR) light has the features of minimal thermal effects, deep penetration, and minimal absorption through tissue media (see review [66]). Previous studies also show that the 808-nm NIR could penetrate the rabbit skull and brain to a depth of 2.5–3 cm if placed on the skin surface [67,68,69]. We indeed found mild to moderate cell damage in the striatum and cerebellum where these regions are also vulnerable to GCI insult [10, 70, 71]. Intriguingly, PBM could also remarkably protect these brain areas (data not shown). In fact, the laser beam covered at 1 cm2 round spot on the shaved scalp can also penetrate to these brain regions. It should be mentioned that there are some potential obstacles for the application of PBM in patients. Unlike animal models used in the preclinical works, NIR has limited penetration due to humans’ thicker skulls and larger brains. Unfortunately, due to financial problems, the company PhotoThera did not fully complete their latest clinical trial on PBM effectiveness titled NeuroThera Effectiveness and Safety Trial 3 (NEST 3), although studies from NEST 1 and NEST 2 gave encouraging results showing improvement in functional outcome after stroke [72, 73]. Therefore, preclinical studies to establish key parameters including the optimal wavelength, therapeutic time window, and multiple effective doses of PBM delivered to the affected brain layers are urgently needed to evaluate before translation to the clinic.
In conclusion, taken together, our data suggest that PBM post-treatment has robust beneficial effects upon mitochondrial health, fragmentation, and apoptosome signaling that is associated with significantly enhanced neuronal survival and preservation of cognitive function after GCI. Based on these beneficial effects, and the fact that PBM is a noninvasive and inexpensive form of therapy, PBM may be an excellent potential therapy to test in humans to ameliorate the poor cognitive outcomes that can occur after GCI.
References
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, Carnethon MR, Dai S et al (2011) Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 123(4):e18–e209. https://doi.org/10.1161/CIR.0b013e3182009701
Moulaert VR, Verbunt JA, van Heugten CM, Wade DT (2009) Cognitive impairments in survivors of out-of-hospital cardiac arrest: a systematic review. Resuscitation 80(3):297–305. https://doi.org/10.1016/j.resuscitation.2008.10.034
Sauve MJ, Doolittle N, Walker JA, Paul SM, Scheinman MM (1996) Factors associated with cognitive recovery after cardiopulmonary resuscitation. Am J Crit Care 5(2):127–139
Roine RO, Kajaste S, Kaste M (1993) Neuropsychological sequelae of cardiac arrest. JAMA 269(2):237–242
Wolman RL, Nussmeier NA, Aggarwal A, Kanchuger MS, Roach GW, Newman MF, Mangano CM, Marschall KE et al (1999) Cerebral injury after cardiac surgery: identification of a group at extraordinary risk. Multicenter Study of Perioperative Ischemia Research Group (McSPI) and the Ischemia Research Education Foundation (IREF) Investigators. Stroke 30(3):514–522
Brillman J (1993) Central nervous system complications in coronary artery bypass graft surgery. Neurol Clin 11(2):475–495
Swain JA, Anderson RV, Siegman MG (1993) Low-flow cardiopulmonary bypass and cerebral protection: a summary of investigations. Ann Thorac Surg 56(6):1490–1492
Pulsinelli WA, Brierley JB, Plum F (1982) Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol 11(5):491–498. https://doi.org/10.1002/ana.410110509
Kirino T (1982) Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res 239(1):57–69
Kirino T, Sano K (1984) Selective vulnerability in the gerbil hippocampus following transient ischemia. Acta Neuropathol 62(3):201–208
Chen J, Zhu RL, Nakayama M, Kawaguchi K, Jin K, Stetler RA, Simon RP, Graham SH (1996) Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J Neurochem 67(1):64–71
Harukuni I, Bhardwaj A (2006) Mechanisms of brain injury after global cerebral ischemia. Neurol Clin 24(1):1–21. https://doi.org/10.1016/j.ncl.2005.10.004
Kim YM, Yim HW, Jeong SH, Klem ML, Callaway CW (2012) Does therapeutic hypothermia benefit adult cardiac arrest patients presenting with non-shockable initial rhythms?: a systematic review and meta-analysis of randomized and non-randomized studies. Resuscitation 83(2):188–196. https://doi.org/10.1016/j.resuscitation.2011.07.031
Nielsen N, Friberg H, Gluud C, Herlitz J, Wetterslev J (2011) Hypothermia after cardiac arrest should be further evaluated—a systematic review of randomised trials with meta-analysis and trial sequential analysis. Int J Cardiol 151(3):333–341. https://doi.org/10.1016/j.ijcard.2010.06.008
Tucker D, Lu Y, Zhang Q (2017) From mitochondrial function to neuroprotection—an emerging role for methylene blue. Mol Neurobiol 55:5137–5153. https://doi.org/10.1007/s12035-017-0712-2
Kumar R, Bukowski MJ, Wider JM, Reynolds CA, Calo L, Lepore B, Tousignant R, Jones M et al (2016) Mitochondrial dynamics following global cerebral ischemia. Mol Cell Neurosci 76:68–75. https://doi.org/10.1016/j.mcn.2016.08.010
Chan PH (2004) Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res 29(11):1943–1949
Herst PM, Rowe MR, Carson GM, Berridge MV (2017) Functional mitochondria in health and disease. Front Endocrinol 8:296. https://doi.org/10.3389/fendo.2017.00296
Bakthavachalam P, Shanmugam PST (2017) Mitochondrial dysfunction—silent killer in cerebral ischemia. J Neurol Sci 375:417–423. https://doi.org/10.1016/j.jns.2017.02.043
Niizuma K, Yoshioka H, Chen H, Kim GS, Jung JE, Katsu M, Okami N, Chan PH (2010) Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim Biophys Acta 1802(1):92–99. https://doi.org/10.1016/j.bbadis.2009.09.002
Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS et al (2006) OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126(1):177–189. https://doi.org/10.1016/j.cell.2006.06.025
Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278(10):7743–7746. https://doi.org/10.1074/jbc.C200677200
Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L (2004) OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101(45):15927–15932. https://doi.org/10.1073/pnas.0407043101
Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C (2005) Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem 280(42):35742–35750. https://doi.org/10.1074/jbc.M505970200
Sharp WW (2015) Dynamin-related protein 1 as a therapeutic target in cardiac arrest. J Mol Med 93(3):243–252. https://doi.org/10.1007/s00109-015-1257-3
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12(8):2245–2256
Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM (1999) The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol 1(5):298–304. https://doi.org/10.1038/13014
Park JH, Ko J, Hwang J, Koh HC (2015) Dynamin-related protein 1 mediates mitochondria-dependent apoptosis in chlorpyrifos-treated SH-SY5Y cells. Neurotoxicology 51:145–157. https://doi.org/10.1016/j.neuro.2015.10.008
Zhang QG, Wang RM, Scott E, Han D, Dong Y, Tu JY, Yang F, Reddy Sareddy G et al (2013) Hypersensitivity of the hippocampal CA3 region to stress-induced neurodegeneration and amyloidogenesis in a rat model of surgical menopause. Brain 136(Pt 5):1432–1445. https://doi.org/10.1093/brain/awt046
Zhang QG, Han D, Wang RM, Dong Y, Yang F, Vadlamudi RK, Brann DW (2011) C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-alpha and the critical period hypothesis of estrogen neuroprotection. Proc Natl Acad Sci U S A 108(35):E617–E624. https://doi.org/10.1073/pnas.1104391108
Lu Y, Wang R, Dong Y, Tucker D, Zhao N, Ahmed ME, Zhu L, Liu TC et al (2017) Low-level laser therapy for beta amyloid toxicity in rat hippocampus. Neurobiol Aging 49:165–182. https://doi.org/10.1016/j.neurobiolaging.2016.10.003
Tu J, Zhang X, Zhu Y, Dai Y, Li N, Yang F, Zhang Q, Brann DW et al (2015) Cell-permeable peptide targeting the Nrf2-Keap1 interaction: a potential novel therapy for global cerebral ischemia. J Neurosci 35(44):14727–14739. https://doi.org/10.1523/JNEUROSCI.1304-15.2015
Zhang QG, Raz L, Wang R, Han D, De Sevilla L, Yang F, Vadlamudi RK, Brann DW (2009) Estrogen attenuates ischemic oxidative damage via an estrogen receptor alpha-mediated inhibition of NADPH oxidase activation. J Neurosci 29(44):13823–13836. https://doi.org/10.1523/JNEUROSCI.3574-09.2009
Lu Y, Dong Y, Tucker D, Wang R, Ahmed ME, Brann D, Zhang Q (2017) Treadmill exercise exerts neuroprotection and regulates microglial polarization and oxidative stress in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. J Alzheimers Dis 56(4):1469–1484. https://doi.org/10.3233/JAD-160869
Lu Q, Tucker D, Dong Y, Zhao N, Zhang Q (2016) Neuroprotective and functional improvement effects of methylene blue in global cerebral ischemia. Mol Neurobiol 53(8):5344–5355. https://doi.org/10.1007/s12035-015-9455-0
Bondarenko AL, Serova LD, Shabalin VN (1991) The role of the major histocompatibility complex antigens in the development of allergic diseases in the Korean population. Sovetskaia meditsina (4):26–28
Sareddy GR, Zhang Q, Wang R, Scott E, Zou Y, O’Connor JC, Chen Y, Dong Y et al (2015) Proline-, glutamic acid-, and leucine-rich protein 1 mediates estrogen rapid signaling and neuroprotection in the brain. Proc Natl Acad Sci U S A 112(48):E6673–E6682. https://doi.org/10.1073/pnas.1516729112
Zhu Y, Zhang Q, Zhang W, Li N, Dai Y, Tu J, Yang F, Brann DW et al (2017) Protective effect of 17beta-estradiol upon hippocampal spine density and cognitive function in an animal model of vascular dementia. Sci Rep 7:42660. https://doi.org/10.1038/srep42660
Xu Z, Guo X, Yang Y, Tucker D, Lu Y, Xin N, Zhang G, Yang L et al (2017) Low-level laser irradiation improves depression-like behaviors in mice. Mol Neurobiol 54(6):4551–4559. https://doi.org/10.1007/s12035-016-9983-2
Liu B, Li L, Zhang Q, Chang N, Wang D, Shan Y, Li L, Wang H et al (2010) Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic stroke. Stroke 41(5):1018–1026. https://doi.org/10.1161/STROKEAHA.110.579011
Li L, Prabhakaran K, Mills EM, Borowitz JL, Isom GE (2005) Enhancement of cyanide-induced mitochondrial dysfunction and cortical cell necrosis by uncoupling protein-2. Toxicol Sci 86(1):116–124. https://doi.org/10.1093/toxsci/kfi164
Zhang QG, Wang R, Tang H, Dong Y, Chan A, Sareddy GR, Vadlamudi RK, Brann DW (2014) Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol Cell Endocrinol 389(1–2):84–91. https://doi.org/10.1016/j.mce.2013.12.019
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105(41):15803–15808. https://doi.org/10.1073/pnas.0808249105
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8(10):939–944. https://doi.org/10.1038/sj.embor.7401062
Kiryk A, Pluta R, Figiel I, Mikosz M, Ulamek M, Niewiadomska G, Jablonski M, Kaczmarek L (2011) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219(1):1–7. https://doi.org/10.1016/j.bbr.2010.12.004
Ulamek-Koziol M, Pluta R, Bogucka-Kocka A, Januszewski S, Kocki J, Czuczwar SJ (2016) Brain ischemia with Alzheimer phenotype dysregulates Alzheimer’s disease-related proteins. Pharmacol Rep 68(3):582–591. https://doi.org/10.1016/j.pharep.2016.01.006
Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79(4):1431–1568
Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y (1995) Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke 26(8):1478–1489
Mishra P, Chan DC (2016) Metabolic regulation of mitochondrial dynamics. J Cell Biol 212(4):379–387. https://doi.org/10.1083/jcb.201511036
Stein LR, Imai S (2012) The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 23(9):420–428. https://doi.org/10.1016/j.tem.2012.06.005
de la Torre JC (2017) Treating cognitive impairment with transcranial low level laser therapy. J Photochem Photobiol B 168:149–155. https://doi.org/10.1016/j.jphotobiol.2017.02.008
Agrawal T, Gupta GK, Rai V, Carroll JD, Hamblin MR (2014) Pre-conditioning with low-level laser (light) therapy: light before the storm. Dose Response 12(4):619–649. https://doi.org/10.2203/dose-response.14-032.Agrawal
Gonzalez-Lima F, Barksdale BR, Rojas JC (2014) Mitochondrial respiration as a target for neuroprotection and cognitive enhancement. Biochem Pharmacol 88(4):584–593. https://doi.org/10.1016/j.bcp.2013.11.010
Hashmi JT, Huang YY, Osmani BZ, Sharma SK, Naeser MA, Hamblin MR (2010) Role of low-level laser therapy in neurorehabilitation. PM R 2(12 Suppl 2):S292–S305. https://doi.org/10.1016/j.pmrj.2010.10.013
Lee JC, Won MH (2014) Neuroprotection of antioxidant enzymes against transient global cerebral ischemia in gerbils. Anat Cell Biol 47(3):149–156. https://doi.org/10.5115/acb.2014.47.3.149
Friberg H, Wieloch T, Castilho RF (2002) Mitochondrial oxidative stress after global brain ischemia in rats. Neurosci Lett 334(2):111–114
Yu Z, Liu N, Zhao J, Li Y, McCarthy TJ, Tedford CE, Lo EH, Wang X (2015) Near infrared radiation rescues mitochondrial dysfunction in cortical neurons after oxygen-glucose deprivation. Metab Brain Dis 30(2):491–496. https://doi.org/10.1007/s11011-014-9515-6
Huang YY, Nagata K, Tedford CE, Hamblin MR (2014) Low-level laser therapy (810 nm) protects primary cortical neurons against excitotoxicity in vitro. J Biophotonics 7(8):656–664. https://doi.org/10.1002/jbio.201300125
Shi RY, Zhu SH, Li V, Gibson SB, Xu XS, Kong JM (2014) BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 20(12):1045–1055. https://doi.org/10.1111/cns.12325
Meyer JN, Leuthner TC, Luz AL (2017) Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391:42–53. https://doi.org/10.1016/j.tox.2017.07.019
Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X (2009) The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem 109(Suppl 1):153–159. https://doi.org/10.1111/j.1471-4159.2009.05867.x
Cartron PF, Bellot G, Oliver L, Grandier-Vazeille X, Manon S, Vallette FM (2008) Bax inserts into the mitochondrial outer membrane by different mechanisms. FEBS Lett 582(20):3045–3051. https://doi.org/10.1016/j.febslet.2008.07.047
Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol 159(6):923–929. https://doi.org/10.1083/jcb.200207071
Wang J, Wang P, Li S, Wang S, Li Y, Liang N, Wang M (2014) Mdivi-1 prevents apoptosis induced by ischemia-reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species-activated mitochondrial pathway. J Stroke Cerebrovasc Dis 23(6):1491–1499. https://doi.org/10.1016/j.jstrokecerebrovasdis.2013.12.021
Ma X, Xie Y, Chen Y, Han B, Li J, Qi S (2016) Post-ischemia mdivi-1 treatment protects against ischemia/reperfusion-induced brain injury in a rat model. Neurosci Lett 632:23–32. https://doi.org/10.1016/j.neulet.2016.08.026
Chung H, Dai T, Sharma SK, Huang YY, Carroll JD, Hamblin MR (2012) The nuts and bolts of low-level laser (light) therapy. Ann Biomed Eng 40(2):516–533. https://doi.org/10.1007/s10439-011-0454-7
Lapchak PA, Han MK, Salgado KF, Streeter J, Zivin JA (2008) Safety profile of transcranial near-infrared laser therapy administered in combination with thrombolytic therapy to embolized rabbits. Stroke 39(11):3073–3078. https://doi.org/10.1161/STROKEAHA.108.516393
Lapchak PA, Salgado KF, Chao CH, Zivin JA (2007) Transcranial near-infrared light therapy improves motor function following embolic strokes in rabbits: an extended therapeutic window study using continuous and pulse frequency delivery modes. Neuroscience 148(4):907–914. https://doi.org/10.1016/j.neuroscience.2007.07.002
Lapchak PA, Wei J, Zivin JA (2004) Transcranial infrared laser therapy improves clinical rating scores after embolic strokes in rabbits. Stroke 35(8):1985–1988. https://doi.org/10.1161/01.STR.0000131808.69640.b7
Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS, Narasimhan P, Chan PH (2011) NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab 31(3):868–880. https://doi.org/10.1038/jcbfm.2010.166
Taraszewska A, Zelman IB, Ogonowska W, Chrzanowska H (2002) The pattern of irreversible brain changes after cardiac arrest in humans. Folia Neuropathol 40(3):133–141
Hacke W, Schellinger PD, Albers GW, Bornstein NM, Dahlof BL, Fulton R, Kasner SE, Shuaib A et al (2014) Transcranial laser therapy in acute stroke treatment: results of neurothera effectiveness and safety trial 3, a phase III clinical end point device trial. Stroke 45(11):3187–3193. https://doi.org/10.1161/STROKEAHA.114.005795
Levine SR, Hill MD (2014) NeuroThera Effectiveness and Safety Trial 3: how do we align corporate and scientific integrity to complete and report pharma-sponsored trials properly? Stroke 45(11):3175–3177. https://doi.org/10.1161/STROKEAHA.114.006750
Funding
This study was supported by Research Grants NS086929 (to QZ) and NS088058 (to DW) from the National Institute of Neurological Disorders and Stroke, National Institutes of Health USA and by National Natural Science Foundation Grants of China: 30970664 and 31171354 (to RMW).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
All animal surgery protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of the local university and were carried out in compliance with National Institutes of Health guidelines.
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Wang, R., Dong, Y., Lu, Y. et al. Photobiomodulation for Global Cerebral Ischemia: Targeting Mitochondrial Dynamics and Functions. Mol Neurobiol 56, 1852–1869 (2019). https://doi.org/10.1007/s12035-018-1191-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1191-9