
Abstract
Despite improvements in cardiopulmonary resuscitation (CPR) quality, defibrillation technologies, and implementation of therapeutic hypothermia, less than 10 % of out-of-hospital cardiac arrest (OHCA) victims survive to hospital discharge. New resuscitation therapies have been slow to develop, in part, because the pathophysiologic mechanisms critical for resuscitation are not understood. During cardiac arrest, systemic cessation of blood flow results in whole body ischemia. CPR and the restoration of spontaneous circulation (ROSC), both result in immediate reperfusion injury of the heart that is characterized by severe contractile dysfunction. Unlike diseases of localized ischemia/reperfusion (IR) injury (myocardial infarction and stroke), global IR injury of organs results in profound organ dysfunction with far shorter ischemic times. The two most commonly injured organs following cardiac arrest resuscitation, the heart and brain, are critically dependent on mitochondrial function. New insights into mitochondrial dynamics and the role of the mitochondrial fission protein Dynamin-related protein 1 (Drp1) in apoptosis have made targeting these mechanisms attractive for IR therapy. In animal models, inhibiting Drp1 following IR injury or cardiac arrest confers protection to both the heart and brain. In this review, the relationship of the major mitochondrial fission protein Drp1 to ischemic changes in the heart and its targeting as a new therapeutic target following cardiac arrest are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cardiac arrest and ischemia reperfusion injury
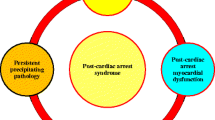
Sudden cardiac arrest (SCA) is the abrupt, unexpected, cessation of cardiac activity and loss of circulation. SCA is a leading cause of morbidity and mortality in the world. In the USA alone, there are approximately 400,000 out-of-hospital and 200,000 in-hospital cardiac arrests each year [1]. Although SCA was once viewed as irreversible, the clinical implementation of vasopressors, cardiopulmonary resuscitation (CPR), defibrillation, and the use of post-arrest therapeutic hypothermia has resulted in improvements in survival. Despite these advances, survival following cardiac arrest overall is poor with less than 10 % of out-of-hospital cardiac arrest (OHCA) victims surviving to hospital discharge [1]. Sixty percent of cardiac arrest victims never regain return of spontaneous circulation (ROSC) despite resuscitation attempts and of the remaining 40 % who are resuscitated; only 5 % are ever discharged from the hospital. Post-ROSC cardiovascular instability and hypoxic encephalopathy are two of the main contributors to death in this post-cardiac arrest syndrome [2, 3]. Therapies targeted at improving ROSC rates and at improving post-ROSC outcomes are needed.
Following cardiac arrest, survival is dependent upon a number of factors: reversing the inciting etiology, re-establishing adequate coronary perfusion pressure [4], and limiting systemic organ injury resulting from whole body ischemia/reperfusion (IR) injury (post-cardiac arrest syndrome) [3, 5]. During CPR and post-ROSC, global IR injury occurs resulting in myocardial dysfunction and encephalopathy. The heart and brain primarily rely on oxidative phosphorylation for their function and are particularly sensitive to oxygen deprivation. This reliance has made the function of mitochondria a target for therapy following IR injury and cardiac arrest [6, 7]. Mitochondria, however, are not just the “powerhouse of the cell” nor are their additional functions limited to production of reactive oxygen species (ROS), regulation of intracellular calcium, biosynthesis of iron sulfur clusters, and the mediation of apoptosis. They also serve as sensors of cellular oxygen tension and have been implicated in cellular signaling [8]. Thus, mitochondrial function following IR injury is likely to have important consequences for post-resuscitation cellular function.
Recently, it has been recognized that mitochondrial function is closely related to its morphology. Mitochondria are dynamic organelles undergoing continuous cycles of fission (division) and fusion (joining) [9]. The specific mechanisms governing mitochondrial fission and fusion are covered extensively elsewhere in this special edition and in previously published reports [8, 10]. The link between mitochondrial morphology and function is particularly evident in the cellular process of apoptosis. In order for apoptosis to occur, mitochondria must undergo fragmentation and cristae remodeling [10]. During this process, the mitochondrial fission protein, Dynamin-related protein 1 (Drp1), accumulates on the surface of the mitochondria promoting oligomerization of the Bcl-2 associated X protein (BAX). This then facilitates mitochondrial outer membrane permeabilization (MOMP) and cell death. Stress-induced Drp1 post-translational modification regulates this process. In cardiac ischemic injury, increased mitochondrial fission and Drp1 activation have been proposed as therapeutic targets [11, 12]. In this review, an overview of Drp1’s structure, function, and its post-translational regulation are discussed in the context of cardiac arrest and ischemic cellular injury.
Drp1 and its role in mitochondrial fission
Drp1, also known as human dynamin1-like protein (encoded by the human DNM1L gene), is an 82-kDa cytosolic protein that is a member of the Dynamin GTPase superfamily. Drp1 was discovered independently by several groups and described as Drp1 [13], DVLP [14], DLP1 [15], and Dymple [16]. In yeast, it is also known as Dnm1 [17]. Drp1 is found in most tissues, but its expression is highest in the brain and the heart [18]. In all of these studies, Drp1 was found to regulate mitochondrial scission, also known as fission. Drp1 upon activation translocates from the cytoplasm to the mitochondrial surface where it interacts with a number of binding partners including fission protein 1 (Fis1), mitochondrial fission factor (MFF), mitochondrial elongation factor (MEF) [10, 19]. Mitochondrial dynamics protein 49 (MiD49) and mitochondrial dynamics protein 51 (miD51) also serve to assist Drp1 in mitochondrial fission [20, 21]. In yeast, Dnm1 (homologue to Drp1) also interacts with the mitochondrial division protein 1 (Mdv1), but this protein has no mammalian homologues [22]. The exact interaction and necessity of each of these proteins is an active area of investigation. Fis1 does not appear to be specifically required for fission [19] although all of the above proteins may cooperatively act to assist Drp1’s fission activity [21]. Regardless of the exact mechanisms, it is clear that Drp1 and its accessory proteins encircle the mitochondria in localized areas and begin process of constricting it. Assembly sites of this process appear to occur at localized areas of contact between the mitochondria and the endoplasmic reticulum [23]. Experiments overexpressing dominant negative forms of Drp1 or inhibiting Drp1’s activity with the inhibitor Mdivi-1 have revealed that Drp1 is not only required for mitochondrial fission but also for apoptosis [24, 25].
Drp1 structure
Drp1 is composed of four domains including a GTPase domain, a middle domain, a variable domain (insert B domain), and a GTPase exchange domain (GED) (Fig. 1). The GTPase domain hydrolyzes GTP and is responsible for Drp1’s polymerization and recruitment to the mitochondrial membrane where it initiates mitochondrial fission. Overexpression of dominant negative mutants of Drp1’s GTPase domain blocks Drp1’s translocation to the mitochondria and its ability to initiate fission [18, 26, 27]. This inhibition of Drp1’s activity has no affect on secretory or endocytic pathways commonly regulated by Dynamins. Interestingly, although Drp1 lacks pleckstrin homology domains with which to bind lipids, the GTPase region of Drp1 has been implicated in Drp1 binding to and interacting with the mitochondrial lipid cardiolipin, though the functional significance of this is not yet understood [28, 29]. See Frohman article in this issue for further discussion of lipids and mitochondrial fission.

Drp1 structure and sites of post-translational modification
The middle domain of Drp1 is responsible for Drp1’s self-assembly and mutations in this region block Drp1’s GTPase activity [30]. A human case report of a spontaneous mutation of A395D in this domain has been described, resulting in lack of brain development and death [31]. Next is the variable or insert B domain, which is the least conserved region and has multiple sites for post-translational modification. Unlike other members of the Dynamin family, Drp1 lacks a pleckstrin homology domain in this region and does not bind directly to lipids (excluding cardiolipin affinity in the GTPase region described above). The variable region (insert B domain) has been shown to be dispensable with mitochondrial localization but seems to serve as an auto inhibitory region as Drp1 activity is enhanced by certain mutations in this region [32]. Finally, the GTPase exchange domain (GED) located at the C-terminal region of the peptide is known to fold back and interact with the middle and GTPase domain to facilitate Drp1’s GTPase activity [33].
Isoforms
There are six human and three murine isoforms of Drp1, which are regulated by alternative splicing [34]. Strack et al. recently demonstrated that Drp1-x01, a major isoform in immune cells, is mostly associated with microtubules rather than the cytosol and that its targeting to the mitochondria is regulated by cyclin-dependent kinase (CDK) [35]. Cortical neurons also express a unique splice form of Drp1 [34, 36]. Cell-specific isoform and post-translational modifications of Drp1 may thus contribute to some of the different post-translational modification findings for Drp1 in the literature. The role of Drp1 isoforms in the context of IR injury and apoptosis in the brain and the heart is unknown.
Post-translational regulation of Drp1
Drp1 has a number of sites where it is modified post-translationaly by phosphorylation, nitrosylation, small ubiquitin like modifier (SUMO) ylation, O-linked-N-acetyl-glucosamine glycosylation (O-GlcNAcylation), and ubiquitination (Fig. 1). These post-translational events are dependent upon the cell type, signaling pathway activated, and the Drp1 isoform involved. Each has different effects on Drp1’s activity. Drp1 is phosphorylated at a number of different sites including Serine 616, Serine 637, and Serine 693 with the first two sites of these being the best characterized. It has been proposed that Drp1’s activity is the result of a balance of Drp1’s phosphorylation on its Serine 616 site (promoting fission) and on its Serine 637 site (promoting fusion) [8]. Dephosphorylation of Drp1 at Serine 637 by calcineurin increases Drp1’s activity and mitochondrial fission [37], while phosphorylation at Serine 637 by protein kinase A (PKA) inhibits Drp1 and promotes fusion [38, 39]. However, modifications of Drp1 at Serine 637 have produced opposite effects in other cell types. For example, in renal podocytes, hyperglycemia promotes rho-associated coiled coil containing protein kinase 1 (ROCK1) phosphorylation of Drp1 at Serine 637 that results in mitochondrial fission [40]. In neurons, Drp1 phosphorylation at Serine 637 by CaMk1a similarly promotes mitochondrial fission [41].
Drp1 phosphorylation at Serine 616 also been demonstrated to have regulatory properties. Phosphorylation of Drp1 Serine 616 by cyclin B1-cyclin-dependent kinase (cyclin B1-CDK1), a mitosis initiator, activates Drp1 fission-promoting activity, coordinating mitochondrial division with cell division [42, 43]. Yu et al. discovered that elevated calcium (induced by hyperglycemia) was responsible for Erk1/2 phosphorylation of S616 that resulted in mitochondrial fission [44]. Finally, Drp1 phosphorylation occurs at Serine 693 in the GED region [45]. Phosphorylation at this site by GSK3β inhibits Drp1 GTPase activity and its activation of mitochondrial fission during apoptosis in HeLa and HEK293 cell lines.
Nitric oxide and the S-nitrosylation of proteins have been implicated in cardioprotection in ischemic heart disease and in cardiac arrest [46, 47]. S-nitrosylation of Drp1 has not been studied in the context of ischemic heart disease but has been implicated in Alzheimer’s disease. These findings however have been controversial and further study is warranted [48, 49].
Drp1 is also a substrate for SUMO1. Post-translational SUMOylation of Drp1 increases Drp1’s stability resulting in its cellular accumulation and increased mitochondrial fragmentation [50]. During apoptosis, Drp1 SUMOylation increases and is associated with its accumulation on the mitochondrial membrane in a Bax/Bak dependent manner. This process is regulated by the sumo protease SENP5 [51, 52]. Mitochondrial anchored protein ligase (MAPL) is another regulator of Drp1 SUMOylation [53]. In fact, Figueroa-Romero et al. demonstrated that Drp1 is a target for all three SUMO isoforms. Although Drp1 lacks SUMOylation consensus sequences, it harbors non-canonical conjugation sites within its insert B (variable) [54]. Drp1 SUMOylation is associated with the regulation of cellular apoptosis, but its role in ischemic cardiovascular disease is as yet unexplored.
O-linked-N-acetyl-glucosamine glycosylation (O-GlcNAcylation) of proteins at their serine and threonine residues regulates subsequent phosphorylation of these proteins at these same or even different residues and impacts protein functioning. Drp1 is O-GlcNAcylated at threonine residues 585 and 586 and if excessive can result in decreased phosphorylation of Drp1 at its serine 637 site [55]. Although these findings were in neonatal cardiac myocytes, the functional consequences beyond mitochondrial fragmentation were not studied.
Finally, Drp1 activity is regulated by ubiquitination. Conjugation of the 76 amino acid protein ubiquitin to proteins has been demonstrated to regulate a variety of cellular processes including protein degradation, membrane trafficking, and remodeling of mitochondrial membranes. Human membrane-associated RING-CH-V (MARCH-V), also identified as MITOL, is a transmembrane protein of the outer mitochondrial membrane that binds to ubiquitinated Drp1 and regulates mitochondrial division [56]. Drp1 can also be ubiquitinated by the E3 ligase Parkin thereby tagging it for proteasomal degradation [57]. Mutations of Parkin are one of the most common causes of familial Parkinson’s disease and abnormalities in mitochondrial fission and fusion contribute to its progression. In Drosophila, Parkin is known to ubiquitinate the mitofusins, but in mammals, Parkin has been shown to ubiquitinate Drp1, resulting in excessive accumulation of Drp1 and increased mitochondrial fission in Hela and 293 cells [57]. The exact role of Parkin regulation and mitochondrial dynamics in the context of cardiovascular disease is an area of active investigation (see Gustafsson review this issue on mitophagy).
Drp1 knockout animals
Drp1 has been knocked out in mice both globally and in a tissue-specific manner. Ishihara et al. created Drp1−/− mice using the Cre-loxP system with a deletion of exon 2 encoding the GTP-binding motif [58]. Embryos began to die between embryonic day 10.5 and 12.5. These animals were found to have a less developed cardiac system and nervous system and concluded Drp1 was essential for embryonic development. Wakabayahi et al. developed a brain-specific Drp1 knockout [59]. These mice began to die by embryonic day 11.5. Defects of the cerebellum were evident with particularly large mitochondria noted in the Purkinje cells. The neural tubes of these mice also failed to undergo regulated apoptosis resulting in abnormal formation. Ideda et al. recently reported the development of a cardiac-specific Drp1 knockout generated by crossing Drp1 fl/fl mice and αMHC-MerCreMer mice. The expression of Drp1 was then downregulated by tamoxifen injections for 5 days [60]. Drp1 protein was decreased in hearts of these animals and the animals all began to die between 8 and 13 weeks after tamoxifen injections. Significant hypertrophy, fibrosis, decreased fractional shortening, and increased diastolic pressures characterized the hearts of these animals. Increased mitochondrial size and mass were also observed and attributed to Drp1. These mitochondrial changes were not attributed to mitochondrial biogenesis since there were no increases in peroxisome proliferator-activated receptor gamma co-activator 1 alpha (PGC-1α) or mitochondrial transcription factor A (TFAM). Increased tissue ROS, myocardial necrosis, and apoptosis were further noted. These hearts were more sensitive to IR injury and had larger areas of infarct. Finally, Kageyama et al. created a cardiac-specific Drp1 knockout mouse (although not conditional). These mice died between post-natal day 9 and 11 and had significantly depressed ventricular function although they did not appear morphologically different than control littermates [61]. Mitochondria in these Drp1 knockout mice were enlarged and had defects in respiration.
Drp1 animal knockout studies have greatly aided the study of Drp1 but must be interpreted with caution when comparing to earlier studies that used Drp1 overexpression mutants. Entirely eliminating a cellular protein versus inhibiting a particular part of its function is likely to produce very different cellular effects. For example, Drp1, in addition to its role in mitochondrial fission, mediates peroxisome fission [62]. The mechanisms coordinating Drp1’s activity between the mitochondria and the peroxisome are unknown. Recent studies employing adenovirus Drp1 overexpression mutants have found Drp1 inhibition to be protective in the context of IR injury and cardiac hypertrophy [63, 64]. The consequences of Drp1 elimination versus the inhibition of its activity or the use of dominant negative mutants are only now beginning to be explored and must be interpreted with care given the multiple roles of Drp1 within the cell.
Pharmacological inhibitors of Drp1
To study the effects of Drp1 other than using overexpression of non-functional forms of Drp1, investigators began to screen compounds for fission-inhibiting capabilities. The first described by Cassidy-Stone et al. in 2008 found that Mdivi-1 at an IC of 10 μM in yeast and 50 μM in COS cells inhibited mitochondrial fission and made the cells more resistant to apoptosis [25]. Mdivi-1 inhibits Dnm1 GTPase activity specifically without affecting the GTPase activity of other dynamins. However, because recombinant Drp1 lacks GTPase activity, the authors were not able to directly test Mdivi-1’s capability of inhibiting it. Mdivi-1 has been found to be protective against myocardial [11, 12, 65], renal [40], and retinal IR injury [66]. A second inhibitor is in the form of a peptide known as P110 [67]. This compound blocks the binding of Drp1 to Fis1. P110 has been found to be protective in a model of neuronal toxicity and in myocardial IR injury [67, 68].
Drp1 in ischemia/reperfusion injury
Changes in mitochondrial morphology following IR injury and cardiac arrest are well known [6, 69]. The recognition that these changes might be related to mitochondrial fission was first realized in several different cell lines following exposure to ischemia or hypoxia. Drp1 involvement in IR injury of the kidney and the retina in addition to the heart has been reported [66, 70]. One of the first studies to investigate Drp1 following cardiac ischemia was by Ong et al. [11]. In this study, the authors found that overexpression of a dominant negative form of Drp1 or Mfn1 or Mfn2 in HL-1 cells exposed to 30 min of hypoxia decreased cellular apoptosis. They also observed that Mdivi-1 pretreatment was also associated with decreased infarct size in a murine model of myocardial infarction and protective of isolated adult cardiomyocytes. Zepeda et al. further demonstrated that adenovirus mediated overexpression of a dominant negative mutant of Drp1 in neonatal cardiac myocytes and in rat hearts is protective against IR injury [63].
Following IR injury, Drp1 undergoes a number of post-translational changes. Inhibition of some of these changes has been found to confer cellular protection. MiR-499 inhibition of IR-induced calcineurin activation and its subsequent dephosphorylation of Drp1 at Serine 637 protects neonatal cardiac myocytes and the intact heart from IR injury [71]. The serine/threonine kinase Pim1 similarly prevents Drp1 dephosphorylation at Serine 637 and its translocation to the mitochondria following IR injury in neonatal cardiac myocytes. Our lab demonstrated in neonatal cardiac myocytes that Drp1 translocates to the mitochondria within 30 min of ischemia and that inhibition of Drp1 increases cell survival, decreases IR-associated calcium and ROS, and preserves mitochondrial oxygen consumption and morphology [12]. Disatnik et al. reported that Drp1 translocated to the mitochondria following IR injury and that this could be blocked with the peptide inhibitor P110 in neonatal cardiac myocytes [68]. They further discovered that P110 administered following coronary artery ligation reduced infarct size and improved myocardial hemodynamics.
Recently, the role of Drp1 in IR injury has been studied in a conditional cardiac-specific Drp1 knockout mouse [60]. In contrast to expectations, Drp1 knockout increased myocardial infarction size in both the homozygote and heterozygote mice. This was in contrast to their findings that Mdivi-1 was protective following IR in the Drp1 heterozygote mice. The authors concluded that Drp1 is necessary for the normal health of the myocardium and that its inhibition is not protective following IR. They also concluded that the protective effects of Mdivi-1 are independent of Drp1. Although it is difficult to eliminate the possibility of off-target effects of a pharmacological inhibitor as Mdivi-1, Drp1 knockout animal studies must also be interpreted with caution. Although Drp1 plays an important role in the health of mitochondria, it may also have functions outside of the mitochondria (peroxisome biogenesis being one mentioned earlier in this review). Thus, Drp1’s elimination could have an impact on affecting other cellular functions. The authors of this study only investigated the effects of Mdivi-1 in heterozygote Drp1 knockdown mice, so it is possible that some of the protective effects may have been due to Drp1 inhibition of its GTPase activity while complete knockdown of Drp1 produces lethal toxicity unrelated to Drp1 GTPase activity. Testing the effects of Mdivi-1 in the complete Drp1 knockout mice to test whether it has cardioprotective properties might give insight into whether this agent is having off-target activities. This and multiple other studies that have knocked out Drp1 provide evidence that complete elimination of Drp1 over weeks is lethal and that Drp1 is essential for proper mitochondrial and cellular homeostasis. However, there is ample evidence to suggest that inhibition of Drp1’s GTPase activity for at least for several hours in the setting of IR injury is cardioprotective. Further studies inhibiting or blocking specific activities of Drp1 are now needed to further clarify the role of Drp1 in the myocardium.
Drp1 inhibition as a therapeutic target in cardiac arrest: the myocardium
Following cardiac arrest, there is cessation of myocardial blood flow, and this creates global cardiac ischemia. As the ischemia time is prolonged, return of spontaneous circulation becomes more difficult, and if resuscitation is achieved, myocardial dysfunction can be severe. These effects on myocardial function have been compared to those of myocardial stunning in which brief periods of coronary occlusion or global lack of perfusion result in ventricular wall motion abnormalities and contractile dysfunction [72, 73]. Although this dysfunction is severe, the contractile function returns to normal after several hours to days and there is no evidence of myocardial necrosis. For these reasons, studies applicable to cardiac arrest employ models of global cardiac ischemia/reperfusion injury that are relatively short in duration (less than 30 min) compared to myocardial infarction models.
We first investigated the role of Drp1 in myocardial dysfunction by examining its role in a Langendorff model of global cardiac ischemia [12]. We found that after 30 min of ischemia and 20 min of reperfusion, Drp1 was dephosphorylated at its Serine 637 site and increased in purified mitochondrial protein fractions. This accumulation was associated with mitochondrial swelling, increased mitochondrial ROS, and increased myocardial diastolic pressures. These alterations could be blocked by Drp1 inhibition either directly with the inhibitor Mdivi-1 or indirectly with the calcineurin inhibitor FK506 or induced hypothermia (both of which blocked Drp1 dephosphorylation at S637). In isolated neonatal cardiomyocytes, Drp1 inhibition was also associated with blocking IR-induced increases in cellular intracellular calcium, reduction in cellular oxygen consumption, and prevented mitochondrial fission. Drp1 inhibition was effective at preventing IR-induced changes whether performed prior to IR injury or immediately after (Fig. 2, only Mdivi-1 data shown).

Post-ischemic administration of Mdivi-1 improves diastolic function. Isolated Langendorff-mounted hearts underwent 20 min of perfusion, 30 min of ischemia, and an additional 20 min of reperfusion; 25 μM Mdivi-1 was administered at the onset of reperfusion. a Mdivi-1 post-ischemic administration preserved relatively lower diastolic pressure. b Pulse pressure was significantly improved compared to IR control (DMSO). N = 7. Figure republished from Sharp et al. FASEB J 2014: Jan;28(1):316–26
To study the role of Drp1 in in vivo cardiac arrest, we employed a model of murine cardiac arrest [74]. Similar to our perfused heart preparations, cardiac arrest resulted in Drp1S637 dephosphorylation and Drp1 accumulation in mitochondrial fractions associated with increased ROS, changes in mitochondrial morphology, and myocardial dysfunction. Drp1 inhibition with Mdivi-1 given at the time of cardiopulmonary resuscitation (CPR) preserved myocardial function and mitochondrial morphology, decreased ROS and myocardial lactate, and resulted in increased survival and improved neurological scores. These results demonstrate that Mdivi-1 is a promising therapy for Drp1 inhibition during cardiac arrest. Our proposed role for Drp1 in cardiac arrest is illustrated in Fig. 3. Although the effects of hypothermia on Drp1 activation were not examined in this study, hypothermia is well known to improve myocardial function and survival following cardiac arrest, and it is possible this protective property could be in part due to Drp1 inhibition as seen in our isolated perfused heart studies. Further studies examining the effects of other direct Drp1 inhibitors as P110 and indirect as FK506 will be needed to confirm the role of Drp1 in this setting. The development of Drp1 knockout mice is also a promising tool to study Drp1 function. However, it should be noted that Drp1 might have effects other than those directly related to mitochondrial fission and knocking down its protein levels may produce effects different than inhibition of its GTPase activity. In fact, this discrepancy between overexpression of non-functional Drp1 and its knockdown have already been described in several studies employing yeast.

Schematic overview of Drp1 in cardiac arrest. a Overview of role of Drp1 in cardiac arrest. b Mechanism of Drp1 activation in cardiac arrest (reproduced from Sharp et al. FASEB J 2014: Jan;28(1):316–26)
It is interesting to note the differences in mitochondrial morphology in the intact heart depending on the length of ischemia and reperfusion. In isolated perfused heart studies, we found that 30 min of ischemia followed by 20 min of reperfusion resulted in mitochondrial swelling. These results are consistent with those in other Langendorff heart model experiments as well as those in animal cardiac arrest studies with similar time frames [75]. However, in our murine cardiac arrest studies, smaller mitochondria were noted 2 h after IR injury, and these results are consistent with others examining heart mitochondria 2–24 h post-IR injury [11]. It is possible that mitochondria initially swell and enlarge following IR and then undergo fission and become smaller. Although this effect is yet to be confirmed in more comprehensive studies, it would be consistent with the description of fission in COS-7 cells which at first enlarge or swell prior to fission [24].
Drp1 inhibition as a therapeutic target in cardiac arrest: the brain
Neurological injury following cardiac arrest can be profound. Victims of cardiac arrest may remain comatose for hours if not days following even short cardiac arrest times. Longer cardiac arrest times are associated with brain swelling and global encephalopathy. Drp1 is highly expressed in the brain and has been investigated in the context of the neurological disorders Alzheimer’s, Huntington’s, and Parkinson’s diseases [34]. Mitochondrial targets for neurological disease are reviewed elsewhere [76]. The specific role of Drp1 in the brain following cardiac arrest has not been directly examined. In our studies, we have found that Drp1 inhibition is associated with improved neurological scores but did not directly examine the brain and the effects of Drp1 on post-arrest morphology. The cardioprotective properties of Drp1 in our study could have been indirect through its improvement of myocardial function or by directly affecting the nervous system. Mdivi-1 has been reported to be both injurious and protective. Wei-Zuo et al. found that Mdivi-1 worsened the infarct size when administered prior to a middle cerebral artery occlusion [77]. This contrasts with the findings of Wang et al. that found Mdivi-1 was protective of hippocampal neurons following IR injury [78]. Further studies to elucidate the role of Drp1 in this issue are needed.
Summary
Mitochondria are dynamic organelles with a complex system for regulating their shape and function. Drp1 as well as other regulators of mitochondrial form are encoded by the nuclear genome making it likely that changes in mitochondrial form and function will be tissue specific. Drp1 is essential for the proper development and functioning of the heart and brain, but its short-term inhibition in the context of IR injury has cardiac and neurological protective properties making it a promising target for cardiac arrest. The role of cardiac and neuronal Drp1 isoforms and their regulation by post-translational modification in the context of ischemic cellular injury are only now beginning and holds the promise for the development of pharmacological agents to improve cardiac and brain function following cardiac arrest.
References
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S et al (2014) Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation 129:e28–e292
Laver S, Farrow C, Turner D, Nolan J (2004) Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med 30:2126–2128
Neumar RW, Nolan JP, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC et al (2008) Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A consensus statement from the International Liaison Committee on Resuscitation (American Heart Association, Australian and New Zealand Council on Resuscitation, European Resuscitation Council, Heart and Stroke Foundation of Canada, InterAmerican Heart Foundation, Resuscitation Council of Asia, and the Resuscitation Council of Southern Africa); the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; and the Stroke Council. Circulation 118:2452–2483
Paradis NA, Martin GB, Rivers EP, Goetting MG, Appleton TJ, Feingold M, Nowak RM (1990) Coronary perfusion pressure and the return of spontaneous circulation in human cardiopulmonary resuscitation. JAMA 263:1106–1113
Nolan JP, Neumar RW, Adrie C, Aibiki M, Berg RA, Bottiger BW, Callaway C, Clark RS, Geocadin RG, Jauch EC et al (2008) Post-cardiac arrest syndrome: epidemiology, pathophysiology, treatment, and prognostication. A Scientific Statement from the International Liaison Committee on Resuscitation; the American Heart Association Emergency Cardiovascular Care Committee; the Council on Cardiovascular Surgery and Anesthesia; the Council on Cardiopulmonary, Perioperative, and Critical Care; the Council on Clinical Cardiology; the Council on Stroke. Resuscitation 79:350–379
Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL (2001) Mitochondrial dysfunction in cardiac disease: ischemia–reperfusion, aging, and heart failure. J Mol Cell Cardiol 33:1065–1089
Gazmuri RJ, Radhakrishnan J (2012) Protecting mitochondrial bioenergetic function during resuscitation from cardiac arrest. Crit Care Clin 28:245–270
Archer SL (2013) Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369:2236–2251
Kane LA, Youle RJ (2010) Mitochondrial fission and fusion and their roles in the heart. J Mol Med (Berl) 88:971–979
Kasahara A, Scorrano L (2014) Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol 24:761–770
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ (2010) Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–2022
Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, Morrow E, Ryan JJ, Archer SL (2014) Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J 28:316–326
Imoto M, Tachibana I, Urrutia R (1998) Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J Cell Sci 111(Pt 10):1341–1349
Shin HW, Shinotsuka C, Torii S, Murakami K, Nakayama K (1997) Identification and subcellular localization of a novel mammalian dynamin-related protein homologous to yeast Vps1p and Dnm1p. J Biochem 122:525–530
Yoon Y, Pitts KR, Dahan S, McNiven MA (1998) A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J Cell Biol 140:779–793
Kamimoto T, Nagai Y, Onogi H, Muro Y, Wakabayashi T, Hagiwara M (1998) Dymple, a novel dynamin-like high molecular weight GTPase lacking a proline-rich carboxyl-terminal domain in mammalian cells. J Biol Chem 273:1044–1051
Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM (1999) The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol 1:298–304
Smirnova E, Shurland DL, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143:351–358
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol 191:1141–1158
Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D, Ryan MT (2013) Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. J Biol Chem 288:27584–27593
Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667
Koirala S, Guo Q, Kalia R, Bui HT, Eckert DM, Frost A, Shaw JM (2013) Interchangeable adaptors regulate mitochondrial dynamin assembly for membrane scission. Proc Natl Acad Sci U S A 110:E1342–E1351
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334:358–362
Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1:515–525
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR et al (2008) Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 14:193–204
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12:2245–2256
van der Bliek AM (2000) A mitochondrial division apparatus takes shape. J Cell Biol 151:F1–F4
Montessuit S, Somasekharan SP, Terrones O, Lucken-Ardjomande S, Herzig S, Schwarzenbacher R, Manstein DJ, Bossy-Wetzel E, Basanez G, Meda P et al (2010) Membrane remodeling induced by the dynamin-related protein Drp1 stimulates Bax oligomerization. Cell 142:889–901
Macdonald PJ, Stepanyants N, Mehrotra N, Mears JA, Qi X, Sesaki H, Ramachandran R (2014) A dimeric equilibrium intermediate nucleates Drp1 reassembly on mitochondrial membranes for fission. Mol Biol Cell 25:1905–1915
Chang CR, Manlandro CM, Arnoult D, Stadler J, Posey AE, Hill RB, Blackstone C (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J Biol Chem 285:32494–32503
Waterham HR, Koster J, van Roermund CW, Mooyer PA, Wanders RJ, Leonard JV (2007) A lethal defect of mitochondrial and peroxisomal fission. N Engl J Med 356:1736–1741
Strack S, Cribbs JT (2012) Allosteric modulation of Drp1 mechanoenzyme assembly and mitochondrial fission by the variable domain. J Biol Chem 287:10990–11001
Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C (2004) Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem 279:35967–35974
Reddy PH, Reddy TP, Manczak M, Calkins MJ, Shirendeb U, Mao P (2011) Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res Rev 67:103–118
Strack S, Wilson TJ, Cribbs JT (2013) Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol 201:1037–1051
Uo T, Dworzak J, Kinoshita C, Inman DM, Kinoshita Y, Horner PJ, Morrison RS (2009) Drp1 levels constitutively regulate mitochondrial dynamics and cell survival in cortical neurons. Exp Neurol 218:274–285
Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 105:15803–15808
Cribbs JT, Strack S (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep 8:939–944
Chang CR, Blackstone C (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282:21583–21587
Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR (2012) Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15:186–200
Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M (2008) CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182:573–585
Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang YH, Thenappan T, Piao L, Zhang HJ, Pogoriler J et al (2012) Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110:1484–1497
Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem 282:11521–11529
Yu T, Jhun BS, Yoon Y (2011) High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxid Redox Signal 14:425–437
Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, Tu WY, Kao TC, Hsu CM, Cheng JT et al (2012) GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One 7:e49112
Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser DG, Munasinghe JP, Anderson SA, Chesley CF, Vanden Hoek TL, Gladwin MT (2009) Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation 120:897–905
Sun J, Murphy E (2010) Protein S-nitrosylation and cardioprotection. Circ Res 106:285–296
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324:102–105
Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, Masliah E, Schwarzenbacher R, Bossy-Wetzel E (2010) S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer's disease. J Alzheimers Dis 20(Suppl 2):S513–S526
Harder Z, Zunino R, McBride H (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol 14:340–345
Wasiak S, Zunino R, McBride HM (2007) Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J Cell Biol 177:439–450
Zunino R, Braschi E, Xu L, McBride HM (2009) Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem 284:17783–17795
Braschi E, Zunino R, McBride HM (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 10:748–754
Figueroa-Romero C, Iniguez-Lluhi JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23:3917–3927
Gawlowski T, Suarez J, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates JR 3rd, Hoshijima M, Dillmann W (2012) Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-beta-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287:30024–30034
Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S (2006) MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 7:1019–1022
Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C et al (2011) Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286:11649–11658
Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y et al (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat Cell Biol 11:958–966
Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, Iijima M, Sesaki H (2009) The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol 186:805–816
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M et al (2015) Endogenous drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–278
Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM et al (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33:2798–2813
Koch A, Thiemann M, Grabenbauer M, Yoon Y, McNiven MA, Schrader M (2003) Dynamin-like protein 1 is involved in peroxisomal fission. J Biol Chem 278:8597–8605
Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, Pedrozo Z, Chiong M, Sanchez G, Lavandero S (2014) Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J Cardiovasc Pharmacol 63:477–487
Pennanen C, Parra V, Lopez-Crisosto C, Morales PE, Del Campo A, Gutierrez T, Rivera-Mejias P, Kuzmicic J, Chiong M, Zorzano A et al (2014) Mitochondrial fission is required for cardiomyocyte hypertrophy mediated by a Ca2 + −calcineurin signaling pathway. J Cell Sci 127:2659–2671
Din S, Mason M, Volkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS et al (2013) Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc Natl Acad Sci U S A 110:5969–5974
Park SW, Kim KY, Lindsey JD, Dai Y, Heo H, Nguyen DH, Ellisman MH, Weinreb RN, Ju WK (2011) A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest Ophthalmol Vis Sci 52:2837–2843
Qi X, Qvit N, Su YC, Mochly-Rosen D (2013) A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci 126:789–802
Disatnik MH, Ferreira JC, Campos JC, Gomes KS, Dourado PM, Qi X, Mochly-Rosen D (2013) Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc 2:e000461
Yeh ST, Lee HL, Aune SE, Chen CL, Chen YR, Angelos MG (2009) Preservation of mitochondrial function with cardiopulmonary resuscitation in prolonged cardiac arrest in rats. J Mol Cell Cardiol 47:789–797
Brooks C, Wei Q, Cho SG, Dong Z (2009) Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest 119:1275–1285
Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP, Li YR, Li PF (2011) miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 17:71–78
Kern KB, Hilwig RW, Rhee KH, Berg RA (1996) Myocardial dysfunction after resuscitation from cardiac arrest: an example of global myocardial stunning. J Am Coll Cardiol 28:232–240
Kern KB (2002) Postresuscitation myocardial dysfunction. Cardiol Clin 20:89–101
Sharp WW, Beiser DG, Fang YH, Han M, Piao L, Varughese J, Archer SL (2015) Inhibition of the mitochondrial fission protein dynamin-related protein 1 improves survival in a murine cardiac arrest model. Crit Care Med 43:e38–e47
Edoute Y, van der Merwe E, Sanan D, Kotze JC, Steinmann C, Lochner A (1983) Normothermic ischemic cardiac arrest of the isolated working rat heart. Effects of time and reperfusion on myocardial ultrastructure, mitochondrial oxidative function, and mechanical recovery. Circ Res 53:663–678
Perez-Pinzon MA, Stetler RA, Fiskum G (2012) Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab 32:1362–1376
Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH (2014) Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 86:103–115
Wang J, Wang P, Li S, Wang S, Li Y, Liang N, Wang M (2014) Mdivi-1 prevents apoptosis induced by ischemia-reperfusion injury in primary hippocampal cells via inhibition of reactive oxygen species-activated mitochondrial pathway. J Stroke Cerebrovasc Dis 23:1491–1499
Acknowledgments
This work was supported by grants from the National Institutes of Health HL103901-01A1 and R03 HL110826-01A1. The Department of Medicine, University of Chicago, provided additional funding for Dr. Sharp’s work.
Conflict of interest
Dr. Sharp has no significant conflicts of interest to report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharp, W.W. Dynamin-related protein 1 as a therapeutic target in cardiac arrest. J Mol Med 93, 243–252 (2015). https://doi.org/10.1007/s00109-015-1257-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-015-1257-3