
Abstract
High glycine (GLY) levels have been suggested to induce neurotoxic effects in the central nervous system of patients with nonketotic hyperglycinemia (NKH). Since the mechanisms involved in the neuropathophysiology of NKH are not totally established, we evaluated the effect of a single intracerebroventricular administration of GLY on the content of proteins involved in neuronal damage and inflammatory response, as well as on the phosphorylation of the MAPK p38, ERK1/2, and JNK in rat striatum and cerebral cortex. We also examined glial fibrillary acidic protein (GFAP) staining, a marker of glial reactivity. The parameters were analyzed 30 min or 24 h after GLY administration. GLY decreased Tau phosphorylation in striatum and cerebral cortex 30 min and 24 h after its administration. On the other hand, synaptophysin levels were decreased in striatum at 30 min and in cerebral cortex at 24 h after GLY injection. GLY also decreased the phosphorylation of p38, ERK1/2, and JNK 30 min after its administration in both brain structures. Moreover, GLY-induced decrease of p38 phosphorylation in striatum was attenuated by N-methyl-d-aspartate receptor antagonist MK-801. In contrast, synuclein, NF-κB, iκB, inducible nitric oxide synthase and nitrotyrosine content, and GFAP immunostaining were not altered by GLY infusion. It may be presumed that the decreased phosphorylation of MAPK associated with alterations of markers of neuronal injury induced by GLY may contribute to the neurological dysfunction observed in NKH.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
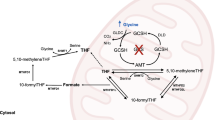
Nonketotic hyperglycinemia (NKH) is an autosomal recessive inborn error of metabolism caused by the deficient activity of glycine (GLY) cleavage system (GCS), which is the main catabolic pathway for GLY. This deficiency leads to the accumulation of GLY in cerebrospinal fluid (CSF), plasma, and tissues of affected patients, with a high CSF/plasma ratio. GCS is an intramitochondrial enzyme complex formed by P-, H-, T-, and L-protein. In the reaction, the P-protein decarboxylates GLY to release carbon dioxide and transfers the aminomethyl group to a lipoate on the H-protein. The H-protein interacts with T-protein that releases ammonia and forms methylenetetrahydrofolate. The last step consists in the reoxidation of reduced lipoate on H-protein by the L-protein.
Classical NKH is the most severe and frequent phenotype of this disorder and it is reported that 70–75% of patients with this phenotype have a mutation in GLDC gene that encodes the P-protein [1–3]. Symptoms of classical NKH appear neonatally with lethargy, myoclonic jerks, muscular hypotonia, and apnea that frequently lead to coma and death. Surviving individuals have profound neurological impairment and intractable seizures [3–5]. Cerebral MRI findings include progressive brain atrophy, hypoplasia of the corpus callosum, gliosis, and vacuolating myelinopathy [6–8].
The pathogenesis of the neurological dysfunction observed in NKH has been attributed to the accumulation of GLY in the brain of affected patients. In this context, it was showed that an increased CSF/plasma GLY ratio correlates with the severity of the disorder [2]. Moreover, previous reports suggested that GLY induces excitotoxic damage, since this molecule is a co-agonist of N-methyl-d-aspartate (NMDA) glutamate receptor [9–13]. This is in line with data showing that the NMDA receptor antagonist MK-801 prevents bioenergetics dysfunction caused by GLY in the brain of rats [14]. It was further demonstrated that GLY alters redox homeostasis and provokes glial reactivity in brain of rats [15–18]. Nevertheless, the exact pathomechanisms underlying the brain abnormalities observed in NKH are not yet fully established.
Regarding excitotoxicity, it is well known that this event leads to a cascade of deleterious intracellular effects that include calcium overload with consequent dysregulation of several signaling pathways, such as the mitogen-activated protein kinases (MAPK), oxidative stress, and energy metabolism impairment that may lead to neuronal damage [19–21]. Since GLY is suggested to cause excitotoxicity, in the present work, we evaluated the in vivo influence of GLY intracerebroventricular (ICV) administration on the phosphorylation of Tau protein and the MAPK p38, ERK1/2, and JNK, as well as on the content of synaptophysin and α-synuclein in the striatum and cerebral cortex of young rats. We also determined the content of NF-κB, iκB, inducible nitric oxide synthase (iNOS), and nitrotyrosine, proteins involved in inflammatory response, and glial fibrillary acidic protein (GFAP) staining, a marker of glial reactivity after GLY injection.
Material and Methods
Animals and Reagents
Thirty-day-old Wistar rats obtained from the Central Animal House of the Department of Biochemistry, ICBS, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil, were used. The animals were maintained on a 12:12 h light/dark cycle (lights on 07:00–19:00 h) in an air-conditioned constant temperature (22 ± 1 °C) colony room, with free access to water and 20% (w/w) protein commercial chow (SUPRA, Porto Alegre, RS, Brazil). All reagents used were of analytical grade and purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The experiments were approved by the local Animal Ethics Commission (Universidade Federal do Rio Grande do Sul) under the number 23787 and the National Animal Rights Regulations (Law 11.794/2008). The guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 80–23, revised 1996) and Directive 2010/63/EU were followed. All efforts were made to minimize the number of animals used and their suffering.
GLY Administration
The rats were deeply anesthetized with equitesine (3.33 mL kg−1, intraperitoneally-ip), which is a mixture of 0.25 M chloral hydrate, 88 mM magnesium sulfate heptahydrate, 10 mg mL−1 sodium thiopental, 5.8 M propylene glycol, and 2.0 M ethanol, and thereafter placed in a stereotaxic apparatus. Two small holes were drilled in the skull, and 2 μL of a 2.5-M GLY solution (5 μmol in a final volume of 2 μL) or NaCl (control) at the same volume and concentration (each solution was prepared in water and pH was adjusted to 7.4 with NaOH) was slowly injected bilaterally over 3 min into each lateral ventricle via a needle connected by a polyethylene tube to a 10-μL Hamilton syringe. The needle was left in place for another 1 min before being gently removed. The coordinates for injection were as follows: 0.6 mm posterior to the bregma, 1.1 mm lateral to the midline, and 3.2 mm ventral from dura [22]. The correct position of the needle was tested by injecting 0.5 μL of 4% methylene blue injection (prepared in saline solution) and carrying out histological analysis. In the experiment performed to evaluate the role of the NMDA glutamate receptor in GLY effects, the animals received a single injection of MK-801 (dizocilpine; 0.25 mg kg−1, ip) [23] 30 min before GLY injection.
Preparation of Samples and Western Blot Analysis
The rats were euthanized by decapitation 30 min or 24 h after GLY injection without anesthesia and had their brain rapidly excised on a Petri dish placed on ice. Striatum and cerebral cortex were homogenized in a RIPA buffer containing protease and phosphatase inhibitors (1 mM sodium orthovanadate, 1 mM aprotinin, and 1% protease inhibitor cocktail) and centrifuged at 10,000g for 10 min at 4 °C. Supernatant protein concentrations were determined by the method of Bradford [24], then denaturated in 4× Laemli buffer (250 mM Tris, 8% SDS, 40% glycerol, and 0.002% bromophenol blue, pH 6.7) and 10% 2-mercaptoethanol. These samples were then heated at 98 °C for 5 min and equal amounts of protein (30 μg/well) were fractionated by SDS-PAGE and electro-blotted onto nitrocellulose membranes. Protein loading and electro-blotting efficiency were verified through Ponceau S staining. Afterwards, the membrane was blocked with 5% albumin prepared with Tween-Tris-buffered saline (TTBS; 100 mM Tris-HCl, pH 7.6, containing 70 mM NaCl, and 0.1% Tween-20), incubated overnight at 4 °C with the primary antibodies diluted at 1:1000 in TTBS (anti-p38, anti-phospho-p38, anti-ERK1/2, anti-phospho-ERK1/2, anti-Tau, anti-phospho-Tau, anti-IκBα, and anti-synaptophysin were purchased from Cell Signaling Technology; anti-JNK and anti-phospho-JNK from R&D systems; anti-β-actin from Sigma-Aldrich Co.; and anti-α-synuclein, anti-NF-κB p105/p50, and anti-iNOS from Abcam), and washed with TTBS. The membrane was incubated with anti-IgG from mouse or rabbit (according to the species that originated the primary antibody) linked to a peroxidase for 2 h at room temperature (1:1000–10,000 dilution range) and washed with TTBS again. The immunoreactivity was detected by enhanced chemiluminescence using Millipore Immobilon™ Western chemiluminescent HRP substrate in a CCD camera. Densitometric analysis of the membranes or films was performed with the ImageJ software (Bethesda, MD). Blots were developed to be linear in the range used for densitometry.
Immunohistochemical Studies
After GLY administration, animals were anesthetized with an ip injection of a mixture of ketamine (80 mg kg−1) and xylazine (10 mg kg−1) until complete unresponsiveness to nociceptive stimuli and then transcardially perfused with 0.4% sodium citrate prepared in 0.9% saline and 4% paraformaldehyde (PFA) prepared in 0.1 M phosphate buffer, pH 7.4, to fix the brain. Fixed brains were removed, post-fixed by immersion in PFA during 24 h and then sectioned on a vibrating microtome to obtain 30–50 μm thick consecutive coronal series. Immunohistochemistry was performed in the striatum and cerebral cortex sections of rats euthanized 30 min after GLY administration. For each animal and staining procedure, three to six equivalent sections were immunostained. Free-floating sections were washed with PBS, submitted to antigen retrieval by boiling in 10 mM sodium citrate, pH 6.0. After 10 min cooling down, the sections were shaken for 20 min in sodium citrate and then washed three times with PBS for 10 min. The slices were then permeabilized with PBS plus 0.3% Triton X-100 (PBST) for 20 min and treated with blocking buffer (PBST and 5% bovine serum albumin) for 1 h. Afterwards, slices were incubated with the antibody anti-GFAP (Sigma-Aldrich Co., 1:300) diluted in PBST. After a 4 °C overnight incubation, sections were rinsed in PBS and incubated at room temperature for 2 h with a secondary antibody (1:500) conjugated to fluorescent probes (Molecular Probes). Sections were then washed, mounted using fluoroshield (Sigma-Aldrich Co.), and imaged in a FV300 Olympus confocal microscope provided with 488- and 546-nm lasers. Primary or secondary antibodies were omitted in negative controls [25].
Nitrotyrosine Content
Aliquots of cerebral cortex (50 μg) or striatum (45 μg) homogenates prepared in PBS buffer, pH 7.4, were added to an ELISA plate and incubated during 24 h for the proteins to adhere to the well. The plate was washed three times with TTBS and incubated overnight at 4 °C with anti-nitrotyrosine antibody (1:2000, Abcam). Afterwards, the plate was washed with TTBS again and incubated with rabbit IgG peroxidase-linked secondary antibody (1:1000) for 1 h. Two hundred microliters of tetramethylbenzidine (TMB) substrate solution was added, and after a 15-min incubation, the reaction was stopped with 50 μL of 12 M sulfuric acid. The colorimetric product was measured at 450 nm.
Data Normalization and Statistical Analysis
Sample protein content was quantified for data normalization according to the Bradford method [24]. Data were analyzed using the Student’s t test for unpaired samples or one-way analysis of variance (ANOVA) followed by the Duncan multiple range test when the F value was significant with the GraphPad 5.0 software. Only significant values are shown in the text. Differences between groups were rated significant at P < 0.05. All data presented here are the result of three or more independent experiments.
Results
GLY Administration Decreases Synaptophysin Content and Tau Protein Phosphorylation in Rat Brain
We investigated the effects of GLY ICV administration on the levels of synaptophysin, a membrane protein of pre-synaptic vesicles, in rat striatum and cerebral cortex. GLY significantly decreased synaptophysin levels in the striatum at 30 min [t (6) = 2.5, P < 0.05] (Fig.1) and in cerebral cortex at 24 h [t (6) = −2.503, P < 0.05] (Fig. 2) after the injection. In contrast, GLY did not alter the levels of synuclein, another synaptic protein, in the cerebral cortex and striatum 30 min after its injection (data not shown). The influence of GLY on the phosphorylation of Tau, a protein involved in the regulation of cell microtubule dynamics and stability, was also examined. It can be observed in Figs. 3 and 4 that GLY decreased Tau phosphorylation in the striatum and cerebral cortex 30 min [striatum, t (6) = 3.755; P < 0.01; cerebral cortex, t (6) = 3.340, P < 0.05] (Fig. 3) and 24 h [striatum, t (6) = 3.628; P < 0.05; cerebral cortex, t (6) = 5.152, P < 0.01] (Fig. 4) after its injection.

Effect of intracerebral administration of glycine (5 μmol) on the content of synaptophysin in striatum (a) and cerebral cortex (b) of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for four independent experiments (animals) normalized by β-actin content. *P < 0.05, compared to control (Student’s t test for unpaired samples)

Effect of intracerebral administration of glycine (5 μmol) on the content of synaptophysin in striatum (a) and cerebral cortex (b) of young rats. Rats were euthanized 24 h after glycine injection. Representative immunoblots are shown as mean ± SD for four independent experiments (animals) normalized by β-actin content. *P < 0.05, compared to control (Student’s t test for unpaired samples)

Effect of intracerebral administration of glycine (5 μmol) on Tau protein phosphorylation in striatum (a) and cerebral cortex (b) of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for four independent experiments (animals). *P < 0.05, **P < 0.01, compared to control (Student’s t test for unpaired samples)

Effect of intracerebral administration of glycine (5 μmol) on Tau protein phosphorylation in striatum (a) and cerebral cortex (b) of young rats. Rats were euthanized 24 h after glycine injection. Representative immunoblots are shown as mean ± SD for four independent experiments (animals). *P < 0.05, **P < 0.01, compared to control (Student’s t test for unpaired samples)
GLY Does Not Alter NF-κB, iκB, iNOS, and Nitrotyrosine Content in Rat Brain
In order to check whether high levels of GLY could elicit inflammation in brain, we assessed the influence of GLY on the content of NF-κB (Fig.5), iκB (Fig.5), iNOS (Fig.6), and nitrotyrosine (Fig.6). We found that GLY did not alter the levels of NF-κB and iκB in the striatum and of iNOS and nitrotyrosine in the cerebral cortex and striatum 30 min after its injection, suggesting that this amino acid does not cause neuroinflammation.

Effect of intracerebral administration of glycine (5 μmol) on the content of NF-κB and iκB in striatum of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for three to four independent experiments (animals). No significant alterations were verified

Effect of intracerebral administration of glycine (5 μmol) on the content of inducible nitric oxide synthase (iNOS) and nitrotyrosine in striatum (a, c) and cerebral cortex (b, d) of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for three to four independent experiments (animals). No significant alterations were verified
GLY Administration Decreases MAPK Phosphorylation in Rat Brain
We next examined the effects of GLY administration on the protein content of the MAPK p38, ERK1/2, and JNK. Our results demonstrate that GLY decreased the phosphorylation of p38, ERK1/2, and JNK in the striatum [p38, t (5) = 5.576; P < 0.01; ERK1/2, t (6) = 13.37; P < 0.001; JNK, t (5) = 4.903; P < 0.01] (Fig.7) and cerebral cortex [p38, t (6) = 3.768; P < 0.01; ERK1/2, t (5) = 2.968; P < 0.05; JNK, t (6) = 9.086; P < 0.001] (Fig.8) 30 min after GLY injection.

Effect of intracerebral administration of glycine (5 μmol) on the content of the mitogen-activated protein kinases (MAPK) p38 (a), ERK1/2 (b), and JNK (c) in striatum of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for three to four independent experiments (animals). **P < 0.01, ***P < 0.001, compared to control (Student’s t test for unpaired samples)

Effect of intracerebral administration of glycine (5 μmol) on the content of the mitogen-activated protein kinases (MAPK) p38 (a), ERK1/2 (b), and JNK (c) in cerebral cortex of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for three to four independent experiments (animals). *P < 0.05, **P < 0.01, ***P < 0.001, compared to control (Student’s t test for unpaired samples)
NMDA Receptor Antagonist MK-801 Prevents GLY-Induced Decrease of the Phosphorylation of p38 in Rat Striatum
We then evaluated the influence of the pre-treatment with the NMDA antagonist MK-801 on GLY-induced decrease of p38 phosphorylation once it was showed that GLY exerts excitotoxicity via NMDA receptor [9–13] and that MAPK activities may be influenced by this receptor [26, 27]. We verified that MK-801 prevented p38 phosphorylation decrease caused by GLY in striatum 30 min after its administration [F (2,8) = 6.191; P < 0.05] (Fig.9), suggesting that this MAPK is altered by GLY via NMDA receptor stimulation.

Effect of MK-801 (0.25 mg kg−1) on glycine (5 μmol)-induced p38 phosphorylation decrease in striatum of young rats. Rats were euthanized 30 min after glycine injection. Representative immunoblots are shown as mean ± SD for three independent experiments (animals). *P < 0.05, compared to control (Duncan multiple range test)
GLY Administration Does Not Alter GFAP Staining in Rat Brain
The effect of GLY administration on GFAP in the striatum and cerebral cortex was further investigated. We found that GLY did not alter this parameter in the brain regions evaluated 30 min after the injection (Fig. 10), implying that this amino acid does not cause glial reactivity.

Effect of intracerebral administration of glycine (5 μmol) on glial fibrillary acidic protein (GFAP) immunofluorescence staining in striatum (a) and cerebral cortex (b) of young rats. Rats were euthanized 30 min after glycine injection. Representative images were obtained from three independent experiments (animals) per group. GFAP is shown with magnification of 40× (scale bar of 200 μm)
Discussion
Patients affected by NKH usually present in the neonatal period lethargy, hypotonia, and seizures, whose pathophysiology is not yet established. However, previous data demonstrated that high levels of GLY cause excitotoxicity via NMDA receptor overstimulation, induce oxidative stress, and bioenergetics dysfunction in rat brain [2, 14, 17, 28, 29]. Aiming to better clarify the pathomechanisms involved in NKH, in the present study, we investigated the in vivo effects of GLY administration on proteins involved in neuronal damage, inflammatory response, and MAPK signaling pathways in the brain of rats.
We first evaluated the effect of GLY on the content of the synaptic proteins synaptophysin and α-synuclein and verified that this amino acid only decreased the levels of synaptophysin in the striatum and cerebral cortex. It is well known that synaptophysin is an abundant integral membrane protein of pre-synaptic vesicles essential for the formation and maintenance of synapses, regulation of neurotransmitter release, and synaptic plasticity [30–32], thus having a crucial role in cognitive function. Furthermore, previous studies showed that synaptophysin is downregulated in response to stress conditions, leading to impairment of synaptic integrity [33, 34] and that the lack of synaptophysin causes cognitive and learning deficits in knockout mice [35, 36]. GLY also decreased the phosphorylation of Tau, a protein involved in the outgrowth of neural processes, axonal transport, development of neuronal polarity, and maintenance of normal neuron morphology [37, 38]. Although we did not study the mechanisms involved in Tau phosphorylation decrease, it is conceivable that it occurred via oxidative stress once Zambrano et al. [39] showed that hydrogen peroxide decreases Tau phosphorylation in rat hippocampus cells. It should be emphasized here that previous findings from our group demonstrated that GLY increases reactive species production in vitro and in vivo in rat brain [15, 17, 18]. These observations imply that alterations in synaptophysin content and Tau phosphorylation may be involved in the neurological dysfunction of NKH.
Considering that Tau protein is a substrate for MAPK and that the activity of these kinases may be modulated by NMDA receptor overstimulation and/or reactive species [27, 40–42], we assessed the effect of GLY on the phosphorylation of p38, ERK1/2, and JNK. GLY decreased the phosphorylation of the three MAPK in rat brain, which is in accordance with our results showing a decrease in the phosphorylation of Tau protein caused by GLY. It is of note that dysregulation of MAPK signaling pathways was observed in different models of neurological disorders also characterized by excitotoxicity, oxidative stress, and bioenergetics dysfunction, such as Alzheimer’s disease [43], schizophrenia [44, 45], depression [46–48], and mental retardation [49].
The effect of the NMDA receptor antagonist MK-801 on GLY-induced decrease of p38 phosphorylation was then examined once the activation of this receptor may trigger alterations in the phosphorylation status of the MAPK [42, 50] and p38 is reported to mediate the pathogenesis of glutamate excitotoxicity [51, 52]. We indeed found that MK-801 prevented the decrease of p38 phosphorylation, indicating that NMDA receptor is involved in this effect. This is in line with the data reported by Wang et al. [47] showing that high calcium influx induced by chronic hypoxia provokes ERK (also a MAPK) dephosphorylation. The same report also verified that this alteration was prevented by the NMDA receptor antagonist memantine [47], reinforcing the view that MAPK activity is influenced by this receptor. It should be further considered here that NMDA receptor stimulation may be associated with a decrease in the phosphorylation of cytoskeletal proteins, such as Tau [38, 53, 54]. So, it can be assumed that GLY induces a decrease in p38 phosphorylation via NMDA receptor stimulation that leads to a reduced phosphorylation of Tau.
GLY did not change the levels of NF-κB, iκB, iNOS, and nitrotyrosine in the brain. Considering that the NF-κB pathway with consequent iNOS activation and nitrotyrosine formation is activated during pro-inflammatory responses in monocytes and macrophages [55, 56], our findings indicate that GLY does not elicit neuroinflammation in young rats. Moreover, the fact that GLY does not alter nitrotyrosine content corroborates the data reported by Seminotti et al. [57] demonstrating that a single GLY intrastriatal administration does not affect nitrate and nitrite content in rat striatum.
Brain abnormalities observed in patients affected by NKH consist of progressive cortical brain atrophy with gliosis [7, 58]. In this particular, it was previously demonstrated that GLY administration induces glial reactivity in brain of neonatal rats [18]. However, in the present study, no alterations were found in GFAP levels. Although these results seem controversial, it should be noted that the data from the literature showed induction of glial reactivity by GLY 5 days after its administration into the brain of newborn rats [18], while in the present study, we analyzed this parameter 30 min after the injection of GLY in brain of young rats. Taken together, these observations indicate that the newborn rat brain is more susceptible to GLY neurotoxic effects than the young rat brain.
In conclusion, this is the first report showing that GLY alters MAPK signaling pathways and causes neuronal damage. Our data also provide evidence that these alterations are elicited by GLY via NMDA receptor stimulation, thus reinforcing the view that the use of NMDA receptor antagonists is beneficial for patients affected by NKH [59–61].
References
Heindel W, Kugel H, Roth B (1993) Noninvasive detection of increased glycine content by proton MR spectroscopy in the brains of two infants with nonketotic hyperglycinemia. Am J Neuroradiol 14(3):629–635
Hamosh A, Johnston MV (2001) Non-ketotic hyperglycinemia. In: Scriver CR, Beaudet A, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, vol editors, 8th edn. McGraw-Hill, New York, pp. 2065–2078
Kure S, Korman SH, Kanno J, Narisawa A, Kubota M, Takayanagi T, Takayanagi M, Saito T et al (2006) Rapid diagnosis of glycine encephalopathy by 13C-glycine breath test. Ann Neurol 59(5):862–867. doi:10.1002/ana.20853
Applegarth DA, Toone JR (2006) Glycine encephalopathy (nonketotic hyperglycinemia): comments and speculations. Am J Med Genet A 140(2):186–188. doi:10.1002/ajmg.a.31030
Van Hove J, Coughlin C II, Scharer G (1993) Glycine encephalopathy. In: Pagon RA, Adam MP, Ardinger HH et al. (eds) Gene Reviews (R). Seattle (WA)
Mourmans J, Majoie CB, Barth PG, Duran M, Akkerman EM, Poll-The BT (2006) Sequential MR imaging changes in nonketotic hyperglycinemia. Am J Neuroradiol 27(1):208–211
Raghavendra S, Ashalatha R, Thomas SV, Kesavadas C (2007) Focal neuronal loss, reversible subcortical focal T2 hypointensity in seizures with a nonketotic hyperglycemic hyperosmolar state. Neuroradiology 49(4):299–305. doi:10.1007/s00234-006-0189-6
Shuman RM, Leech RW, Scott CR (1978) The neuropathology of the nonketotic and ketotic hyperglycinemias: three cases. Neurology 28(2):139–146
Hara H, Sukamoto T, Kogure K (1993) Mechanism and pathogenesis of ischemia-induced neuronal damage. Prog Neurobiol 40(6):645–670. doi:10.1016/0301-0082(93)90009-H
Kure S, Tada K, Narisawa K (1997) Nonketotic hyperglycinemia: biochemical, molecular, and neurological aspects. J Hum Genet 42(1):13–22. doi:10.1007/BF02766917
Applegarth DA, Toone JR (2001) Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Mol Genet Metab 74(1–2):139–146. doi:10.1006/mgme.2001.3224
Kono Y, Shigetomi E, Inoue K, Kato F (2007) Facilitation of spontaneous glycine release by anoxia potentiates NMDA receptor current in the hypoglossal motor neurons of the rat. Eur J Neurosci 25(6):1748–1756. doi:10.1111/j.1460-9568.2007.05426.x
Katsuki H, Watanabe Y, Fujimoto S, Kume T, Akaike A (2007) Contribution of endogenous glycine and d-serine to excitotoxic and ischemic cell death in rat cerebrocortical slice cultures. Life Sci 81(9):740–749. doi:10.1016/j.lfs.2007.07.001
Moura AP, Grings M, Dos Santos Parmeggiani B, Marcowich GF, Tonin AM, Viegas CM, Zanatta A, Ribeiro CA et al (2013) Glycine intracerebroventricular administration disrupts mitochondrial energy homeostasis in cerebral cortex and striatum of young rats. Neurotox Res 24(4):502–511. doi:10.1007/s12640-013-9396-1
Leipnitz G, Solano AF, Seminotti B, Amaral AU, Fernandes CG, Beskow AP, Dutra Filho CS, Wajner M (2009) Glycine provokes lipid oxidative damage and reduces the antioxidant defenses in brain cortex of young rats. Cell Mol Neurobiol 29(2):253–261. doi:10.1007/s10571-008-9318-6
Busanello EN, Moura AP, Viegas CM, Zanatta A, da Costa FG, Schuck PF, Wajner M (2010) Neurochemical evidence that glycine induces bioenergetical dysfunction. Neurochem Int 56(8):948–954. doi:10.1016/j.neuint.2010.04.002
Seminotti B, Knebel LA, Fernandes CG, Amaral AU, da Rosa MS, Eichler P, Leipnitz G, Wajner M (2011) Glycine intrastriatal administration induces lipid and protein oxidative damage and alters the enzymatic antioxidant defenses in rat brain. Life Sci 89(7–8):276–281. doi:10.1016/j.lfs.2011.06.013
Moura AP, Parmeggiani B, Grings M, Alvorcem LM, Boldrini RM, Bumbel AP, Motta MM, Seminotti B et al (2015) Intracerebral glycine administration impairs energy and redox homeostasis and induces glial reactivity in cerebral cortex of newborn rats. Mol Neurobiol. doi:10.1007/s12035-015-9493-7
Yang SH, Sharrocks AD, Whitmarsh AJ (2013) MAP kinase signalling cascades and transcriptional regulation. Gene 513(1):1–13. doi:10.1016/j.gene.2012.10.033
Hara MR, Snyder SH (2007) Cell signaling and neuronal death. Annu Rev Pharmacol Toxicol 47:117–141. doi:10.1146/annurev.pharmtox.47.120505.105311
Prentice H, Modi JP, Wu JY (2015) Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxidative Med Cell Longev 2015:964518. doi:10.1155/2015/964518
Paxinos G, Watson C (1986) The rat brain in stereotaxic coordinates. Academic Press, San Diego
Ribeiro CAJ, Grando V, Dutra CS, Wannmacher CMD, Wajner M (2006) Evidence that quinolinic acid severely impairs energy metabolism through activation of NMDA receptors in striatum from developing rats. J Neurochem 99(6):1531–1542. doi:10.1111/j.1471-4159.2006.04199.x
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Olivera S, Fernandez A, Latini A, Rosillo JC, Casanova G, Wajner M, Cassina P, Barbeito L (2008) Astrocytic proliferation and mitochondrial dysfunction induced by accumulated glutaric acidemia I (GAI) metabolites: possible implications for GAI pathogenesis. Neurobiol Dis 32(3):528–534. doi:10.1016/j.nbd.2008.09.011
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802(4):396–405. doi:10.1016/j.bbadis.2009.12.009
Poddar R, Paul S (2013) Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J Neurochem 124(4):558–570. doi:10.1111/jnc.12102
Terek D, Koroglu OA, Gunes S, Yalaz M, Akisu M, Ucar SK, Gokben S, Coker M et al (2012) Diagnostic tools of metabolic and structural brain disturbances in neonatal non-ketotic hyperglycinemia. Pediatr Int 54(5):717–720. doi:10.1111/j.1442-200X.2012.03591.x
Demirel N, Bas AY, Zenciroglu A, Aydemir C, Kalkanoglu S, Coskun T (2008) Neonatal non-ketotic hyperglycinemia: report of five cases. Pediatr Int 50(1):121–123. doi:10.1111/j.1442-200X.2007.02513.x
Alder J, Kanki H, Valtorta F, Greengard P, Poo MM (1995) Overexpression of synaptophysin enhances neurotransmitter secretion at Xenopus neuromuscular synapses. J Neuros 15(1 Pt 2):511–519
Janz R, Sudhof TC, Hammer RE, Unni V, Siegelbaum SA, Bolshakov VY (1999) Essential roles in synaptic plasticity for synaptogyrin I and synaptophysin I. Neuron 24(3):687–700
Kim E, Sheng M (2004) PDZ domain proteins of synapses. Nat Rev Neurosci 5(10):771–781. doi:10.1038/nrn1517
Thome J, Pesold B, Baader M, Hu M, Gewirtz JC, Duman RS, Henn FA (2001) Stress differentially regulates synaptophysin and synaptotagmin expression in hippocampus. Biol Psychiatry 50(10):809–812
Xu H, He J, Richardson JS, Li XM (2004) The response of synaptophysin and microtubule-associated protein 1 to restraint stress in rat hippocampus and its modulation by venlafaxine. J Neurochem 91(6):1380–1388. doi:10.1111/j.1471-4159.2004.02827.x
Schmitt U, Tanimoto N, Seeliger M, Schaeffel F, Leube RE (2009) Detection of behavioral alterations and learning deficits in mice lacking synaptophysin. Neuroscience 162(2):234–243. doi:10.1016/j.neuroscience.2009.04.046
Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ (1997) Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol 56(8):933–944
Terry RD (1998) The cytoskeleton in Alzheimer disease. J Neural Transm Suppl 53:141–145
Kuszczyk M, Gordon-Krajcer W, Lazarewicz JW (2009) Homocysteine-induced acute excitotoxicity in cerebellar granule cells in vitro is accompanied by PP2A-mediated dephosphorylation of tau. Neurochem Int 55(1–3):174–180. doi:10.1016/j.neuint.2009.02.010
Zambrano CA, Egana JT, Nunez MT, Maccioni RB, Gonzalez-Billault C (2004) Oxidative stress promotes tau dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radic Biol Med 36(11):1393–1402. doi:10.1016/j.freeradbiomed.2004.03.007
Kumar P, Jha NK, Jha SK, Ramani K, Ambasta RK (2015) Tau phosphorylation, molecular chaperones, and ubiquitin E3 ligase: clinical relevance in Alzheimer’s disease. J Alzheimers Dis 43(2):341–361. doi:10.3233/JAD-140933
Qi H, Prabakaran S, Cantrelle FX, Chambraud B, Gunawardena J, Lippens G, Landrieu I (2016) Characterization of neuronal tau protein as a target of extracellular signal-regulated kinase. J Biol Chem 291(14):7742–7753. doi:10.1074/jbc.M115.700914
Poddar R, Paul S (2009) Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J Neurochem 110(3):1095–1106. doi:10.1111/j.1471-4159.2009.06207.x
Subramaniam S, Zirrgiebel U, von Bohlen Und Halbach O, Strelau J, Laliberte C, Kaplan DR, Unsicker K (2004) ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol 165(3):357–369. doi:10.1083/jcb.200403028
Kyosseva SV, Elbein AD, Hutton TL, Griffin ST, Mrak RE, Sturner WQ, Karson CN (2000) Increased levels of transcription factors Elk-1, cyclic adenosine monophosphate response element-binding protein, and activating transcription factor 2 in the cerebellar vermis of schizophrenic patients. Arch Gen Psychiatry 57(7):685–691
Kyosseva SV, Elbein AD, Griffin WS, Mrak RE, Lyon M, Karson CN (1999) Mitogen-activated protein kinases in schizophrenia. Biol Psychiatry 46(5):689–696
Dwivedi Y, Rizavi HS, Roberts RC, Conley RC, Tamminga CA, Pandey GN (2001) Reduced activation and expression of ERK1/2 MAP kinase in the post-mortem brain of depressed suicide subjects. J Neurochem 77(3):916–928
Wang Z, Gu J, Wang X, Xie K, Luan Q, Wan N, Zhang Q, Jiang H et al (2013) Antidepressant-like activity of resveratrol treatment in the forced swim test and tail suspension test in mice: the HPA axis, BDNF expression and phosphorylation of ERK. Pharmacol Biochem Behav 112:104–110. doi:10.1016/j.pbb.2013.10.007
Duman CH, Schlesinger L, Kodama M, Russell DS, Duman RS (2007) A role for MAP kinase signaling in behavioral models of depression and antidepressant treatment. Biol Psychiatry 61(5):661–670. doi:10.1016/j.biopsych.2006.05.047
Kim SH, Markham JA, Weiler IJ, Greenough WT (2008) Aberrant early-phase ERK inactivation impedes neuronal function in fragile X syndrome. Proc Natl Acad Sci U S A 105(11):4429–4434. doi:10.1073/pnas.0800257105
Wang J, Ming H, Chen R, Ju JM, Peng WD, Zhang GX, Liu CF (2015) CIH-induced neurocognitive impairments are associated with hippocampal Ca(2+) overload, apoptosis, and dephosphorylation of ERK1/2 and CREB that are mediated by overactivation of NMDARs. Brain Res 1625:64–72. doi:10.1016/j.brainres.2015.08.012
Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, Legos JJ et al (2001) Inhibition of p38 mitogen-activated protein kinase provides neuroprotection in cerebral focal ischemia. Med Res Rev 21(2):129–145
Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF et al (2001) SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther 296(2):312–321
De Montigny A, Elhiri I, Allyson J, Cyr M, Massicotte G (2013) NMDA reduces Tau phosphorylation in rat hippocampal slices by targeting NR2A receptors, GSK3beta, and PKC activities. Neural Plast 2013:261593. doi:10.1155/2013/261593
Fleming LM, Johnson GV (1995) Modulation of the phosphorylation state of tau in situ: the roles of calcium and cyclic AMP. Biochem J 309(Pt 1):41–47
Schroder NW, Opitz B, Lamping N, Michelsen KS, Zahringer U, Gobel UB, Schumann RR (2000) Involvement of lipopolysaccharide binding protein, CD14, and Toll-like receptors in the initiation of innate immune responses by Treponema glycolipids. J Immunol 165(5):2683–2693
Aktan F (2004) iNOS-mediated nitric oxide production and its regulation. Life Sci 75(6):639–653. doi:10.1016/j.lfs.2003.10.042
Seminotti B, Ribeiro RT, Amaral AU, da Rosa MS, Pereira CC, Leipnitz G, Koeller DM, Goodman S et al (2014) Acute lysine overload provokes protein oxidative damage and reduction of antioxidant defenses in the brain of infant glutaryl-CoA dehydrogenase deficient mice: a role for oxidative stress in GA I neuropathology. J Neurol Sci 344(1–2):105–113. doi:10.1016/j.jns.2014.06.034
Shoham S, Javitt DC, Heresco-Levy U (1999) High dose glycine nutrition affects glial cell morphology in rat hippocampus and cerebellum. International Journal Neuropsychopharmacology 2(1):35–40. doi:10.1017/S1461145799001285
Ohya Y, Ochi N, Mizutani N, Hayakawa C, Watanabe K (1991) Nonketotic hyperglycinemia: treatment with NMDA antagonist and consideration of neuropathogenesis. Pediatr Neurol 7(1):65–68
Korman SH, Wexler ID, Gutman A, Rolland MO, Kanno J, Kure S (2006) Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol 59(2):411–415. doi:10.1002/ana.20759
Schmitt B, Steinmann B, Gitzelmann R, Thun-Hohenstein L, Mascher H, Dumermuth G (1993) Nonketotic hyperglycinemia: clinical and electrophysiologic effects of dextromethorphan, an antagonist of the NMDA receptor. Neurology 43(2):421–424
Acknowledgements
The authors declare that there is no conflict of interest. This work was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Programa de Apoio a Núcleos de Excelência (PRONEX II), Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS), Pró-Reitoria de Pesquisa/Universidade Federal do Rio Grande do Sul (PROPESQ/UFRGS), Financiadora de estudos e projetos (FINEP), Rede Instituto Brasileiro de Neurociência (IBN-Net) # 01.06.0842-00, and Instituto Nacional de Ciência e Tecnologia em Excitotoxicidade e Neuroproteção (INCT-EN).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The experiments were approved by the local Animal Ethics Commission (Universidade Federal do Rio Grande do Sul) under the number 23787 and the National Animal Rights Regulations (Law 11.794/2008). The guidelines of National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication no. 80–23, revised 1996) and Directive 2010/63/EU were followed. All efforts were made to minimize the number of animals used and their suffering.
Additional information
Alana Pimentel Moura and Belisa Parmeggiani have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Moura, A.P., Parmeggiani, B., Gasparotto, J. et al. Glycine Administration Alters MAPK Signaling Pathways and Causes Neuronal Damage in Rat Brain: Putative Mechanisms Involved in the Neurological Dysfunction in Nonketotic Hyperglycinemia. Mol Neurobiol 55, 741–750 (2018). https://doi.org/10.1007/s12035-016-0319-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-0319-z