
Abstract
Nonketotic hyperglycinemia (NKH) is an inherited disorder of amino acid metabolism biochemically characterized by the accumulation of glycine (Gly) predominantly in the brain. Affected patients usually manifest with neurological symptoms including hypotonia, seizures, epilepsy, lethargy, and coma, the pathophysiology of which is still not completely understood. Treatment is limited and based on lowering Gly levels aiming to reduce overstimulation of N-methyl‐D‐aspartate (NMDA) receptors. Mounting in vitro and in vivo animal and human evidence have recently suggested that excitotoxicity, oxidative stress, and bioenergetics disruption induced by Gly are relevant mechanisms involved in the neuropathology of NKH. This brief review gives emphasis to the deleterious effects of Gly in the brain of patients and animal models of NKH that may offer perspectives for the development of novel adjuvant treatments for this disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nonketotic Hyperglycinemia
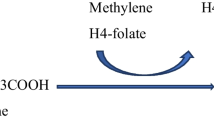
Nonketotic hyperglycinemia (NKH) (OMIM 605,899) is an autosomal recessive neurometabolic disorder caused by defects in the glycine cleavage system (GCS), which is well expressed in the liver, placenta, and brain (Van Hove et al. 2019). The GCS is composed of the P-protein, a pyridoxal phosphate‐dependent glycine decarboxylase encoded by the GLDC gene, the H‐protein, a lipoic acid-containing protein that carries hydrogen, encoded by the GCSH gene, the T‐protein, a tetrahydrofolate‐dependent protein encoded by the AMT gene, and the L‐protein, a lipoamide dehydrogenase encoded by the DLD gene. GLDC catalyzes the release of CO2 from glycine cleavage, which occurs in the presence of an accessory protein, GCSH, which receives the aminomethyl moiety. Next, the second one-carbon unit is transferred to tetrahydrofolate (THF) by AMT, forming 5,10-methylene THF. The following steps of folate one-carbon metabolism in the mitochondria culminate in supplying one-carbon units (e.g. formate) to the cytosol to be utilized in several pathways (e.g., methylation reactions and nucleotide biosynthesis) (Fig. 1). While classic NKH is caused by defects in the genes that encode protein components of the GCS, NKH in approximately 4% of patients is caused by defects in the synthesis of the cofactor lipoic acid (Baker et al. 2014; Swanson et al. 2015; Van Hove et al. 2019). Patients present with high levels of glycine (Gly) levels in plasma, brain, and cerebrospinal fluid (CSF), as well as a characteristically elevated CSF: plasma Gly ratio (Van Hove et al. 2019) (Fig. 1).
Two forms of NKH have been described pending the clinical manifestations: a severe form characterized by developmental progress impairment and intractable epilepsy, and an attenuated and treatable variant characterized by variable developmental progress and no epilepsy (Van Hove et al. 2019). Patients affected by severe NKH typically present soon after birth with lethargy progressing to coma, myoclonic epilepsy, and apnea requiring ventilation. They spontaneously regain ventilation but develop severe spasticity, developmental delay, and intractable epilepsy. Individuals with the mild form of NKH have mutations leading to low GCS residual activity. This form may appear in the neonatal period or infancy and is characterized by developmental delay, with convulsions responsive to usual anticonvulsant treatment or no seizures at all. Chorea and hyperactivity are also commonly seen (Van Hove et al. 2019).
Brain MRI shows that all patients with severe NKH have diffusion restriction in the posterior limb of the internal capsule, posterior tegmental tracts, anterior brain stem, and cerebellum in the first years of life. Older patients may present with supratentorial white matter abnormalities. Corpus callosum is found to be shortened or thin (Stence et al. 2019; Van Hove et al. 2019). Cerebellar cyst with hydrocephalus can be seen in a few individuals with the severe NKH form (Swanson et al. 2015), whereas cerebral volume abnormalities are also observed with a reduction in the size of the globus pallidus, hippocampus, thalamus, cerebral cortex, and cerebellum (Stence et al. 2019; Van Hove et al. 2019).
Plasma and CSF Gly levels, as well as the CSF: plasma Gly ratio, are higher in severe NKH as compared to the attenuated form (Swanson et al. 2015). According to this, patients affected by classic NKH have average CSF Gly levels of 228 µM (normal: <20 µM), 1,133 µM in plasma (normal values vary with age: 125–450 µM), and a CSF/plasma Gly ratio of 0.22 (normal: ≤0.02) (Swanson et al. 2015; Van Hove et al. 2019). Attenuated NKH patients have average CSF Gly levels of 99 µM, 822 µM in plasma, and a CSF/plasma Gly ratio of 0.13 (Van Hove et al.2019).
Determining the cerebrospinal fluid/plasma Gly concentration ratio is the main diagnostic method. A ratio value > 0.08 strongly indicates NKH diagnosis. The activity of hepatic GCS is also performed but diagnosis confirmation is primarily carried out by molecular genetic testing (Hamosh and Johnston 2001; Van Hove et al. 2019). Prenatal diagnosis may be carried out by determining the activity of GCS activity in chorionic villus (Hamosh and Johnston 2001).
Treatment is very limited and based on the administration of sodium benzoate to decrease plasma Gly levels, and N-methyl‐D‐aspartate (NMDA) receptor antagonists, including dextromethorphan and ketamine, aiming the reduction in Gly-induced overstimulation of this receptor. However, the latter therapeutic approach still has an unclear effect on NKH outcome (Swanson et al. 2015).
Glutamatergic Neurotransmission
Glutamate is the predominant excitatory neurotransmitter in the adult brain, virtually present in all CNS cells. It originates mainly from the transamination of α-ketoglutarate, a citric acid cycle (CAC) intermediate. When neuronal depolarization in glutamatergic neurons occurs, vesicles containing glutamate fuse to the membrane of presynaptic cells in a calcium-dependent process and release glutamate into the nerve terminals. Glutamate may activate ionotropic and metabotropic postsynaptic receptors, being NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate the major postsynaptic ionotropic receptors (Traynelis et al. 2010). Activation of the glutamatergic receptors triggers different pre- and post-synaptic responses that may be involved in physiological or pathological cell activity. NMDA receptors are activated by the obligatory binding of glutamate as agonist and Gly or D-serine as co-agonist for activation (Vyklicky et al. 2014). Three subgroups of NMDA receptors with variable subtypes were reported: GluN1, GluN2 (GluN2A-D), and GluN3 (GluN3A-B). GluN1 is essential in the composition of the receptor and combines usually with GluN2 dimers to form the common heterotetrameric NMDA receptor. However, different subunit compositions exist generating structural diversity. Noteworthy, GluN2 subunit binds glutamate, whereas GluN1 and GluN3 have the binding site for the co-agonist Gly (Paoletti et al. 2013).
Gly levels in glutamatergic synapses are regulated by Gly transporters. Four Gly transporters have been identified so far, and the so-called glycine transporters 1 (GlyT1) and 2 (GlyT2) are considered the principal proteins involved in the regulation of intracerebral brain Gly concentrations. They accumulate Gly within neurons and glial cells using the electrochemical gradient of Na+ as energy source. This transport also requires Cl− to be co-transported with Na+ and Gly (Piniella and Zafra 2023).
Besides ionotropic receptors, the so-called metabotropic receptors (mGluRs) can be also verified in glutamate-releasing and responsive neural cells. Eight types of mGluRs (mGluR1 to mGluR8) have been identified so far, consisting of G-protein coupled receptors. Presynaptic mGluR activation regulates glutamate release, whereas the activation of post-synaptic mGluR regulates the opening of ionotropic channels. After exerting its function in the synaptic cleft, glutamate is rapidly removed to avoid excitotoxicity by transporters called Excitatory Amino Acid Transporters (EAAT) 1 to 5 in humans. In rodents, the homologous transporters are named GLAST (EAAT1), GLT‐1 (EAAT2), EAAC1 (EAAT3), EAAT4, and EAAT5.
Although the distribution of these transporters is different across the brain structures, all of them transport glutamate against its concentration gradient using the driving force of one H+ and three Na+ ions (Beart and O’Shea 2007). Astrocytes are essential to regulate the extracellular levels of glutamate (DiNuzzo et al. 2014). After glutamate uptake, astrocytic glutamine synthetase converts it to glutamine in the cytosol. Glutamine is then released to the extracellular fluid and taken up by neurons. In neurons, glutamate is generated from glutamine by the enzyme glutaminase (Bak et al. 2006). The transport of glutamate and glutamine between these cells is named the “glutamate-glutamine cycle”.
The physiological glutamatergic neurotransmission has crucial functions, such as neuronal development and survival. However, the excessive activation of ionotropic glutamate receptors by high glutamate concentrations leads to an elevated influx of Ca2+ into the neurons, which may cause cell death and neurodegeneration. This pathological mechanism, called excitotoxicity, is involved in many neurodegenerative disorders (Lau and Tymianski 2010; Verma et al. 2022). During this process, excessive mitochondrial calcium influx may also occur, causing elevated reactive oxygen species (ROS) production, permeability transition, and cell death (Duchen 2012). Of note, NADPH oxidase is a relevant source of ROS following NMDA receptor activation (Duchen 2012). Furthermore, neuronal nitric oxide synthase (nNOS), which generates nitric oxide (NO), is also activated by NMDA receptors (Hardingham 2019). Additional enzymes that contribute to cell damage during excitotoxicity include calcium/calmodulin-dependent protein kinase II, protein kinase C, phospholipases, phosphatases, proteases, and endonucleases (Choi 2020).
Reactive Species and Oxidative Stress
Reactive species (RS) correspond to unstable molecules that may be produced in all cells, whereas free radicals are RS that possess unpaired electrons in their outer orbitals. Reactive oxygen species (ROS) are derived from molecular oxygen (O2) and are commonly generated physiologically in the mitochondrial electron transport chain (ETC) through the transference of electrons to O2. The reduction of O2 by one electron results in the formation of the free radical superoxide (O2•−), which is converted to hydrogen peroxide (H2O2) by the enzyme superoxide dismutase (SOD). Mitochondrial glutathione peroxidase (GPx) or peroxisomal catalase (CAT) metabolizes hydrogen peroxide to H2O. However, when in excess and in the presence of Fe2+ or Cu+, hydrogen peroxide can lead to the formation of hydroxyl radical (OH•), considered the most reactive and toxic ROS, by the Fenton reaction (Halliwell and Gutteridge 2015).
Reactive nitrogen species (RNS) are also generated in many cell types and include nitric oxide (NO•) and peroxynitrite (ONOO−) as the most relevant examples. Nitric oxide is produced by nitric oxide synthase (NOS), which converts L-arginine to nitric oxide and L-citrulline. So far, three isoforms of NOS have been identified: neuronal, endothelial, and inducible NOS. Noteworthy, nNOS is activated by NMDA receptors. Physiologically, nitric oxide is responsible for the regulation of blood pressure and contraction and is generated during inflammation by macrophages (Halliwell and Gutteridge 2015). In contrast, high levels of nitric oxide inhibit cytochrome c oxidase, blocking the ETC and leading to electron leakage and increased ROS production (Brown et al. 2010). Furthermore, peroxynitrite, which is the product of the reaction between nitric oxide and superoxide, is highly cytotoxic (Halliwell and Gutteridge 2015).
When RS are produced at high concentrations or when the antioxidant defenses are decreased, oxidative damage to biomolecules may occur, giving rise to the condition known as “oxidative stress”. Thus, oxidative stress is defined as an imbalance between pro-oxidants and antioxidants in favor of the former, leading to disturbances in redox signaling and biomolecule damage (Halliwell and Gutteridge 2015; Jones 2006; Sies 2014).
Mitochondrial Bioenergetics
Mitochondria are key organelles principally involved in cell bioenergetics. Mitochondrial CAC generates reducing equivalents (NADH and FADH2), which transfer electrons across the respiratory chain complexes to O2 ultimately forming H2O. The energy released during the electron transfer through the respiratory chain complexes is conserved by proton pumping from the mitochondrial matrix to the mitochondrial intermembrane space creating an electrochemical gradient that generates a protonmotive force. ATP synthase uses the protonmotive force to phosphorylate ADP and generate ATP. In turn, the enzyme creatine kinase (CK) is crucial for ATP cell transfer, especially in tissues with high mitochondrial mass, such as CNS, skeletal muscle, and heart. Various CK isoforms have been identified and are differentially distributed in mitochondria and cytosol in these tissues (Puurand et al. 2018). The mitochondrial CK isoform consumes ATP in mitochondria to produce creatine phosphate, which is shuttled to the cytosol, where ATP is regenerated by cytosolic CK isoforms (Puurand et al. 2018).
Pathophysiology of NKH: Insights from Patient and Animal Studies
Growing evidence has shown that Gly is neurotoxic through distinct deleterious mechanisms, including excitotoxicity caused by NMDA receptor overstimulation, oxidative stress, and bioenergetics impairment. While excitotoxicity was initially observed in patients and animal models, oxidative stress and bioenergetic impairment have been so far reported only in animal models of NKH. Three different animal models of NKH have been utilized: (1) in vitro experimental models consisting of tissue homogenates and enriched mitochondrial fractions from brain of wild type Wistar rats that were exposed to Gly (Leipnitz et al. 2009); (2) an in vivo model consisting of intracerebral administration of Gly in wild type Wistar rats (Seminotti et al. 2011); and (3) glycine decarboxylase-deficient (gldc−/−) zebrafish (Riche et al. 2018).
NMDA Receptor Overstimulation in the Pathogenesis of NKH
Different reports have suggested that excessive stimulation of NMDA receptors by Gly provokes excitotoxicity and represents a crucial event in the onset and progression of seizures and developmental delay in NKH patients (Ohya et al. 1991). It was first demonstrated that ketamine, an NMDA receptor antagonist, improved hyperirritability, voluntary movement, and electroencephalographic findings in a NKH patient (Ohya et al. 1991). Further studies also showed improvement in the symptomatology of some NKH patients, especially in those with a less severe phenotype, who were treated with ketamine and/or other NMDA receptor antagonists combined with sodium benzoate, a compound that reduces plasma Gly concentrations (Hamosh and Johnston 2001; Hennermann et al. 2012; Hoover-Fong et al. 2004; Van Hove et al. 2019). In contrast, other reports demonstrated variable effects of these drugs in some NKH patients (Hennermann et al. 2012; Nowak et al. 2022; Van Hove et al. 2019), indicating that this is still a matter of debate.
Regarding the experimental studies, overstimulation of NMDA receptors by Gly was observed in a zebrafish model of NKH (gldc−/−) (Riche et al. 2018). It was also seen that exposure of gldc−/− zebrafish embryos to the NMDA antagonist dextromethorphan rescued alterations in the swimming behavior of these animals. Moreover, overexpression of GlyT1, the transporter that removes Gly from the synaptic cleft, was shown to improve the motor deficit in the knockout NKH embryos (Riche et al. 2018). Finally, a recent study revealed that intracerebroventricular administration of Gly in neonatal rats reduced the levels of the NMDA receptor subunit 1 (NR1) in the cerebral cortex and striatum, and of GLAST transporter in the striatum of these animals (Parmeggiani et al. 2023). Overall, these data support excitotoxicity as a potentially contributing factor to the neurodegeneration in NKH.
Oxidative Stress in the Pathogenesis of NKH
Many in vitro and in vivo works have shown that oxidative stress is caused by Gly, representing a relevant pathomechanism in NKH neuropathology. In vitro reports demonstrated that Gly induces lipid peroxidation and decreases GSH levels, a major antioxidant defense, in the cerebral cortex, cerebellum, and striatum of developing rats (Leipnitz et al. 2009; Moura et al. 2014; Parmeggiani et al. 2019). Gly also reduces sulfhydryl content in a commercial GSH solution, possibly indicating a direct oxidative effect of this amino acid (Leipnitz et al. 2009). In addition, the antioxidants trolox (soluble alpha-tocopherol) and melatonin (MEL), which have efficient scavenging activity towards peroxyl and hydroxyl radicals, were shown to prevent Gly oxidative damage in rat cerebellum and cerebral cortex (Leipnitz et al. 2009; Moura et al. 2014). It was also observed that the NMDA receptor antagonist MK-801 mitigated the reduction in GSH levels (Moura et al. 2014), indicating NMDA receptor involvement in this Gly effect and reinforcing the data mentioned in the previous topic of our review ( NMDA Receptor Overstimulation in the Pathogenesis of NKH).
In vivo findings showed that Gly disrupts redox homeostasis in neonatal and adolescent rats, therefore reinforcing the deleterious effects of this amino acid revealed in the in vitro data. It was found that Gly intrastriatal administration in these animals induces lipid and protein oxidation, besides increasing the activities of antioxidant enzymes in the striatum. The later effects on the antioxidant system might have occurred through increased gene transcription of these enzymes, representing an adaptive mechanism to cope with oxidative stress induced by Gly. It was also found that Gly-induced lipid peroxidation in vivo was prevented by the NMDA receptor antagonist MK-801, suggesting the role of these receptors in this effect (Seminotti et al. 2011). Furthermore, when Gly was administered to adolescent rats intracerebroventricularly, it resulted in increased activities of striatal enzymatic antioxidant defenses and decreased hippocampal GSH levels (Parmeggiani et al. 2019). Interestingly, most of these effects were mitigated by the administration of the PPAR agonist bezafibrate (Parmeggiani et al. 2019).
Other findings showed that intracerebroventricular administration of Gly in neonatal rats induced reactive species generation, and lipid oxidation, and decreased GSH levels, besides disrupting the enzymatic antioxidant system in the cerebral cortex. Moreover, MEL pre-treatment diminished lipid oxidation and prevented the GSH level decrease caused by Gly (Moura et al. 2016; Parmeggiani et al. 2023).
Bioenergetic Dysfunction in the Pathogenesis of NKH
Gly was also shown to impair brain bioenergetics in vitro and in vivo. In vitro studies performed in rat cerebral cortex revealed that Gly impaired the CAC function by decreasing CO2 production from acetate and inhibiting citrate synthase activity, besides disrupting the electron transport chain (ETC) by inhibiting various complex activities. Gly also reduced the activities of mitochondrial CK, which is responsible for intracellular energy buffering and transference, and Na+,K+-ATPase, an enzyme crucial for maintaining cellular volume control and neuronal excitability (Busanello et al. 2010). Similarly, Gly in vivo intracerebroventricular administration was shown to decrease the activities of ETC complexes and CK in rat cerebral cortex and striatum, supporting the in vitro data. Furthermore, intracerebroventricular administration of Gly in rat striatum impaired CAC, as determined by reduced CO2 production and inhibition of citrate synthase and isocitrate dehydrogenase activities. Interestingly, the antioxidants N-acetylcysteine and creatine mitigated the reduction of the ETC complexes and CK activities caused by Gly administration in both the striatum and cerebral cortex, whereas MK-801 ameliorated the ETC disruption in the striatum (Moura et al. 2013). These observations indicate the involvement of glutamate receptors and redox homeostasis disruption in Gly-elicited disturbances of brain bioenergetics.
Concluding Remarks and Perspectives
In this brief review, we summarized relevant experimental in vitro and in vivo data in animal models and patients with NKH revealing potential deleterious effects of Gly on the glutamatergic system as well as on redox and bioenergetics homeostasis that may synergistically contribute to the neuropathology of NKH (Fig. 2). Additionally, a considerable number of clinical studies demonstrated that patients improved their clinical condition when treated with NMDA receptor antagonists, such as dextromethorphan, reinforcing that excitotoxicity via the overstimulation of these receptors probably represents an important pathomechanism induced by this amino acid (Van Hove et al. 2019).
These data indicate that the Gly-induced overstimulation of NMDA receptor causes a high influx of calcium into the cells, leading to increased production of ROS. The exact source of ROS involved in the Gly-induced effects has not yet been established but the activation of NADPH oxidase and neuronal nitric oxide synthase, which are sensitive to elevated calcium levels, are the main candidates (Görlach et al. 2015; Brennan et al. 2009; Halliwell and Gutteridge 2015). It is also conceivable that the inhibition of ETC, especially complexes I and III, caused by the mitochondrial accumulation of calcium may also be a source. However, it is also worth noting that some in vitro findings also suggested that Gly may bind to and directly inhibit energy metabolism enzymes. Therefore, the increased generation of ROS via Gly action may have caused bioenergetic impairment by provoking oxidative damage to the CAC enzymes and ETC complexes (Görlach et al. 2015). Although increased intracerebral concentrations of Gly may be excitotoxic because of its co-agonism at NMDA receptors, we cannot rule out that Gly-induced oxidative stress caused might potentially cause oxidative attack to glutamate receptors and transporters altering their functions and accentuating excitotoxicity (Michaelis 1998; Guan 2008).
Additionally, bioenergetic disruption caused by Gly may lead to further deleterious consequences, such as a decreased activity of membrane synaptic Na+/K+-ATPase, resulting in increased K+ extracellular concentrations and glutamate release to the synaptic cleft (secondary excitotoxicity) (Greene and Greenamyre 1996). Moreover, enzymes and proteins that modulate calcium concentrations, such as Ca2+-ATPases, Na+/Ca2+ exchangers, mitochondrial Ca2+ uniporter, and Ca2+-binding proteins may be affected by bioenergetic failure and dysregulation of calcium levels. Excessive calcium also leads to the activation of calpains that can degrade cytoskeletal proteins, membrane receptors, and enzymes. Glutamate-induced calcium influx may further activate cyclooxygenases and lipoxygenases, increasing ROS production and membrane lipid peroxidation, and impair the function of membrane ion-motive ATPases, as well as of glucose and glutamate transporters (Greene and Greenamyre 1996).
Other mitochondrial processes, such as mitochondrial quality control, may be affected by excitotoxicity and must be considered potential mechanisms involved in NKH. Reports have shown that excitotoxic events may impair mitochondrial dynamics (fusion and fission) and consequently mitochondrial morphology. In this sense, it was revealed that glutamate excitotoxicity leads to fragmented mitochondria in neurodegenerative diseases, mediated by nitric oxide-induced S-nitrosylation of dynamin-related protein 1, a mitochondrial fission protein (Nakamura et al. 2010) and mitofusin-2 degradation (Wang et al. 2015). Further data revealed that glutamate decreases mitochondrial size and movement in neurons (Tena-Morraja et al. 2023). Therefore, studies are needed to elucidate whether Gly affects mitochondrial dynamics and other quality control processes, such as biogenesis and mitophagy, aiming to provide additional mechanisms of NKH neuropathology that may target novel therapeutic interventions.
Regarding the cells mainly involved in bioenergetic modifications contributing to hyperglycinemia neurotoxicity, it seems that neurons are the major candidate since this cell has a high amount of NMDA receptors with high calcium permeability, low antioxidant defenses, and high dependence on oxidative phosphorylation (Halliwell and Gutteridge 2015). Interestingly, NMDAR receptors GluN1/GluN3A that are gated exclusively by Gly have been found in neurons (Stroebel et al. 2021). In these receptors, while Gly binding to GluN3 subunits triggers channel opening, Gly binding to the neighboring GluN1 subunits has an opposite effect causing auto-inhibition (Stroebel et al. 2021). So, it may be speculated that changes in the GluN1 and GluN3 subunits may alter NMDA responsiveness to Gly and thus have a role in NKH pathogenesis. On the other hand, NMDA receptors have also been found in non-neuronal cells, such as astrocytes and oligodendrocytes, also rendering them potential targets for Gly neurotoxicity (Liu et al. 2023). Consistent with this, previous investigations showed that the overstimulation of non-neuronal NMDA receptors mediates different pathological mechanisms, such as excitotoxicity, neuroinflammation, blood-brain barrier disruption, and myelin impairment (Liu et al. 2023).
On the other hand, since the benefits of treatment with NMDA receptors antagonists were observed especially after a long-term regimen, and particularly in attenuated rather than in the severe NKH variant of the disease (Hennermann et al. 2012), it is conceivable that other mechanisms such as oxidative stress and disruption of bioenergetics may play a key role in NKH pathophysiology. Consistent with this, studies performed in rodent brain supernatants, which are at least partially devoid of active NMDA receptor-mediated neurotransmission, and in media devoid of biological samples showed that Gly induced direct toxic effects (Leipnitz et al. 2009).
Although the data reviewed here provide important insights into the NKH pathophysiology, it should be emphasized that the animal models used so far have some limitations. Thus, the obtained results should be taken with caution with respect to the human condition. In this sense, more research, especially conducted in cells from affected patients, is necessary to confirm these potential pathomechanisms of brain damage in NKH. Finally, further studies evaluating the effects of novel NMDA receptor antagonists and mitochondria-targeted antioxidants seem important to improve the prognosis of these patients.

Glycine breakdown catalyzed by the glycine cleavage system (GCS), encompassing glycine decarboxylase (GLDC), GCS H-protein (GCSH), aminomethyltransferase (AMT), and DLD (dihydrolipoamide dehydrogenase). GLDC catalyzes the first step of glycine cleavage, causing the release of CO2 along with the transfer of the aminomethyl moiety to GCSH. In the next step, AMT transfers the second one-carbon unit (methylene) to tetrahydrofolate (THF), generating 5,10-methylene THF. GCS is regenerated when GCSH is oxidized to regenerate the disulfide bond in the active site by interaction with the DLD. Deficient activity of GCS causes nonketotic hyperglycinemia, which is characterized by glycine accumulation in the brain of patients. MTHFD1, methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1; MTHFD2/L, methylenetetrahydrofolate dehydrogenase 2/ 2-like; MTHFD1L, monofunctional tetrahydrofolate synthase, mitochondrial; SHMT, serine hydroxymethyltransferase

Potential pathomechanisms involved in NKH neuropathology. It is suggested that high levels of glycine (Gly) disrupt glutamatergic neurotransmission (1) by binding to and overstimulating NMDA receptors leading to increased Ca2+ influx in the postsynaptic neuron. Elevated levels of Ca2+ activate phospholipases and nitric oxide synthase (NOS) with subsequent production of reactive oxygen and nitrogen species (ROS and RNS, respectively). Gly provokes lipid oxidative damage and disturbs the antioxidant system ultimately causing oxidative stress (2). Gly also impairs the citric acid cycle (CAC) and electron transport chain functioning and inhibits the activities of mitochondrial creatine kinase (Mit-CK) (3) and Na+,K+-ATPase
Data Availability
No datasets were generated or analysed during the current study.
References
Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98(3):641–653
Baker PR et al (2014) Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Br J Neurol 137(2):366–379
Beart PM, O’shea RD (2007) Transporters for L-glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150(1):5–17
Brennan AM et al (2009) NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci 12:857–863
Brown GC (2010) Nitric oxide and neuronal death. Nitric oxide: Biology and Chemistry 23(3):153–165
Busanello ENB et al (2010) Neurochemical evidence that glycine induces bioenergetical dysfunction. Neurochem Int 56:948–954
Choi DW, Excitotoxicity (2020) Still Hammering the Ischemic Brain in 2020. Front Neurosci 14:579953
Dinuzzo M et al (2014) Physiological bases of the K + and the glutamate/GABA hypotheses of epilepsy. Epilepsy Res 108(6):995–1012
Duchen MR (2012) Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers Arch European J Physiol 464(1):111–121
Görlach A et al (2015) Calcium and ROS: A mutual interplay. Redox Biol 6:260–271
Greene JG, Greenamyre JT (1996) Bioenergetics and glutamate excitotoxicity. Prog Neurobiol 48(6):613–634
Guan ZZ (2008) Cross-talk between oxidative stress and modifications of cholinergic and glutaminergic receptors in the pathogenesis of Alzheimer’s disease. Acta Pharmacol Sin 29(7):773–780
Halliwell B, Gutteridge JMC (2015) Free Radicals in Biology and Medicine. 5th Edition, Oxford University Press, New York
Hamosh A, Johnston MV (2001) Non-ketotic hyperglycinemia. In: Scriver CR, Beaudet A, Sly WS, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn. McGraw-Hill, New York, pp 2065–2078
Hardingham G (2019) NMDA receptor C-terminal signaling in development, plasticity, and disease. F1000Research 8: F1000 Faculty Rev-1547
Hennermann JB et al (2012) Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metabolic Disease 35(2):253–261
Hoover-Fong JE et al (2004) Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 63(10):847–1853
Jones DP (2006) Redefining oxidative stress. Antioxid Redox Signal 8(9–10):1865–1879
Lau A, Tymianski M (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch European J Physiol 460(2):525–542
Leipnitz G et al (2009) Glycine provokes lipid oxidative damage and reduces the antioxidant defenses in brain cortex of young rats. Cell Mol Neurobiol 29(2):253–261
Liu W et al (2023) The role of N-methyl-D-aspartate glutamate receptors in Alzheimer’s disease: From pathophysiology to therapeutic approaches. Prog Neurobiol 231:102534
Michaelis EK (1998) Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol 54(4):369–415
Moura AP et al (2014) Evidence that glycine induces lipid peroxidation and decreases glutathione concentrations in rat cerebellum. Mol Cell Biochem 395(1–2):125–134
Moura AP et al (2013) Glycine intracerebroventricular administration disrupts mitochondrial energy homeostasis in cerebral cortex and striatum of young rats. Neurotox Res 24(4):502–511
Moura AP et al (2016) Intracerebral Glycine Administration Impairs Energy and Redox Homeostasis and induces glial reactivity in cerebral cortex of newborn rats. Mol Neurobiol 53(9):5864–5875
Nakamura T et al (2010) S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 10(5):573–578
Nowak M, Chuchra P, Paprocka J (2022) Nonketotic hyperglycinemia: insight into current therapies. J Clin Med 11(11):3027
Ohya Y et al (1991) Nonketotic hyperglycinemia: treatment with NMDA antagonist and consideration of neuropathogenesis. Pediatr Neurol 7(1):65–68
Paoletti et al (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14(6):383–400
Parmeggiani B et al (2019) Bezafibrate prevents Glycine-Induced increase of antioxidant enzyme activities in Rat Striatum. Mol Neurobiol 56(1):29–38
Parmeggiani B et al (2023) Glycine disrupts myelin, glutamatergic neurotransmission, and redox homeostasis in a neonatal model for non ketotic hyperglycinemia. Biochimie 219:21–32
Piniella D, Zafra F (2023) Functional crosstalk of the glycine transporter GlyT1 and NMDA receptors. Neuropharmacol 232:109514
Puurand M et al (2018) Intracellular Energy-Transfer Networks and High-Resolution Respirometry: A Convenient Approach for Studying Their Function. Int J Mol Sci 19(10):2933
Riché R et al (2018) Glycine decarboxylase deficiency–induced motor dysfunction in zebrafish is rescued by counterbalancing glycine synaptic level. JCI Insight 3(21):e124642
Seminotti B et al (2011) Glycine intrastriatal administration induces lipid and protein oxidative damage and alters the enzymatic antioxidant defenses in rat brain. Life Sci 89:7–8
Sies H (2014) Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem 289:8735–8741
Stence NV et al (2019) Brain imaging in classic nonketotic hyperglycinemia: Quantitative analysis and relation to phenotype. J Inherit Metab Dis 42(3): 438–450
Stroebel D, Mony l, Paoletti P (2021) Glycine agonism in ionotropic glutamate receptors. Neuropharmacology 193:108631
Swanson MA et al (2015) Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol 78(4):606–618
Tena-Morraja P et al (2023) Synaptic activity regulates mitochondrial Iron metabolism to enhance neuronal bioenergetics. Int J Mol Sci 24(2):922
Traynelis SF et al (2010) Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol Rev 62(3):405–496
Van Hove JLK, Coughlin CII, Swanson M, Hennermann JB, Nonketotic Hyperglycinemia (2002) Nov 14 [updated 2019 May 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023
Verma M, Lizama BN, Chu CT (2022) Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener 11(1):3
Vyklicky V et al (2014) Structure, function, and pharmacology of NMDA receptor channels. Physiol Res 63(1):S191–203
Wang W et al (2015) MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J Biol Chem 290(1):168–182
Funding
Conselho Nacional de Desenvolvimento Científico e Tecnológico-CNPq #312141/2020-3, and National Institute of Brain Health, INSC #406020/2022-1.
Author information
Authors and Affiliations
Contributions
GL, JS da R, and MW wrote the manuscript. JS da R created the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Leipnitz, G., da Rosa, J.S. & Wajner, M. The Role of Excitotoxicity, Oxidative Stress and Bioenergetics Disruption in the Neuropathology of Nonketotic Hyperglycinemia. Neurotox Res 42, 32 (2024). https://doi.org/10.1007/s12640-024-00711-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12640-024-00711-5