
Abstract
GLIS1 has multiple roles in embryonic development and in deriving induced pluripotent stem cells by aiding signaling pathways and chromatin assembly. An inexpensive and simple method to produce human GLIS1 protein from Escherichia coli (E. coli) is demonstrated in this study. Various parameters such as codon usage bias, E. coli strains, media, induction conditions (such as inducer concentration, cell density, time, and temperature), and genetic constructs were investigated to obtain soluble expression of human GLIS1 protein. Using identified expression conditions and an appropriate genetic construct, the human GLIS1 protein was homogeneously purified (purity > 90%) under native conditions. Importantly, the purified protein has upheld a stable secondary structure, as demonstrated by circular dichroism spectroscopy. To the best of our knowledge, this is the first study to report the ideal expression conditions of human GLIS1 protein in E. coli to achieve soluble expression and purification under native conditions, upholding its stable secondary structure post-purification. The biological activity of the purified GLIS1 fusion protein was further assessed in MDA-MB-231 cells. This biologically active human GLIS1 protein potentiates new avenues to understand its molecular mechanisms in different cellular functions in various cancers and in the generation of induced pluripotent stem cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glis1 belongs to the Gli-similar protein family and broadly to the subfamily of Kruppel-like zinc-finger proteins [1, 2]. The Glis1 gene encodes a ~ 66 kDa protein (620 amino acids) rich in proline residues and has zinc-finger motifs, with the highest sequence homology to different members of Gli and Zic subfamilies of Kruppel-like proteins [3, 4]. The pivotal role of the nuclear translocation of the GLIS1 protein was due to the contribution of the zinc-finger region in its structure. The ability to bind to the consensus sequence 5′-GACCACCCAC-3′ of the Gli-binding site was also observed for the same [1]. It has a transactivation domain present at both the N- and C-terminus of the protein, and thus, it acts as a transcription factor that regulates gene expression during specific embryonic developmental stages [1]. A spatiotemporal expression of GLIS1 during embryonic development contributed to its abundant expression in unfertilized eggs, one- and two-cell embryos, and placenta [3, 5]. Also, its abundant expression in the kidney and low expression in the brain, colon, testis, thymus, and adipose tissue was observed in adults [3, 5].
The expression profile of GLIS1 protein in embryonic stem cells (ESCs) showed a low level of expression, with drastic inhibition in ESC proliferation upon its overexpression [6]. Its importance to derive fully reprogrammed induced pluripotent stem cells (iPSCs) with high reprogramming efficiency was demonstrated by many studies [6,7,8,9,10,11,12]. It was reported that OCT4, SOX2, and KLF4 combined with GLIS1 enhanced the generation of both mouse and human fully reprogrammed iPSCs, along with the successful generation of germline-competent chimeras from mouse iPSCs [6]. In this study, Maekawa et al. demonstrated the generation of iPSCs by substituting c-MYC with GLIS1 from the Yamanaka factor cocktail (OCT4, SOX2, KLF4, and c-MYC) [6]. The mouse and human iPSCs generated formed bona fide ESC-like colonies when compared to the Yamanaka factor cocktail. Specifically, GLIS1 enhanced the generation of fully reprogrammed iPSCs, unlike c-MYC, which promoted the formation of partially reprogrammed clones in a higher proportion. The chimeric mice generated with these mouse iPSCs showed decreased incidence of tumor formation, thus, GLIS1 emerging as a potential promising candidate in the cocktail of reprogramming factors, replacing the oncogene c-MYC [6]. The same combination (OCT4, SOX2, KLF4, and GLIS1) showed enhanced reprogramming efficiency with the non-pathogenic, self-replicating Venezuelan equine encephalitis RNA virus to derive integration-free human iPSCs [8]. Further, GLIS1, in different reprogramming factor combinations, also efficiently generated iPSCs from mouse and human somatic cells [9,10,11, 13]. GLIS1 has multifaceted roles such as the promotion of pro-reprogramming pathways (Wnt, Nanog, Myc, Lin28, Esrrb), induction of the expression of the FOXA2 transcription factor, inhibition of epithelial to mesenchymal transition [6], facilitation of the change in chromatin state, and activation of a pluripotency-associated gene like SOX2 [14]. It induces multilevel epigenetic and metabolic remodeling in stem cells, thus, facilitating the induction of pluripotency [15]. All these functions enhanced the reprogramming efficiency and enabled the generation of bona fide iPSCs. Apart from its function in cell reprogramming, embryonic development, and mesodermal cell differentiation during fetal development [6, 16, 17], its role has also been implicated in various ciliated organ diseases of lung, pancreas and kidney [17], breast cancer [18], and in the late onset of Parkinson’s disease [19].
iPSCs are important cell sources for basic understanding of embryonic development, various prospects of disease modeling, development of drugs, toxicity screening, and personalized medicine [20]. Various approaches such as integrating and non-integrating have been developed to generate iPSCs from different somatic cell sources [21,22,23,24,25]. However, the major constraints concerning the integrative approaches are the permanent genomic mutations leading to tumor formation, inefficient gene silencing, and transgene reactivation [25,26,27,28,29,30,31]. These problems affect their potential to differentiate properly and limit the applicability of iPSCs in patient-specific therapies [26,27,28,29,30, 32]. The non-integrative gene delivery approaches provided a platform to generate iPSCs with no or minimum genetic alterations [23, 24, 33, 34]. Among all the non-integrative approaches, the recombinant protein approach is the safest so far [23, 33,34,35,36]. It provides great control over the time of application, dosage as well as flexibility in designing and screening different reprogramming factor combinations. It also helps in assessing their respective roles at specific reprogramming stages [23, 35]. However, the limiting factor of this approach is the low reprogramming efficiency and kinetics [23, 25, 35] due to the presence of various reprogramming roadblocks [22, 37, 38]. Therefore, the inclusion of reprogramming factors such as GLIS1, especially in recombinant form, may promote the formation of bona fide iPSCs efficiently with a step towards the generation of integration-free iPSCs.
Recombinant proteins have accrued great utility in clinical research, agricultural, and industrial applications [39, 40]. Despite its hassle-free and inexpensive generation advantages in host systems like E. coli, the bottlenecks, such as codon usage bias, soluble expression, native purification, retaining secondary structure, and protein folding, remain inevitable. In this study, we are the first to purify and determine the secondary structure of the recombinant GLIS1 fusion protein overcoming these limitations in E. coli.
Materials and Methods
Construction of GLIS1 Fusion Gene Constructs
The GLIS1 protein-coding sequence was obtained, codon optimized, and evaluated as shown in Fig. 1 and as per our previously published studies [41, 42]. Briefly, codon optimization was carried out using the Gene Optimizer tool (Thermo Scientific), and the codon-optimization quality was evaluated using GenScript Rare Codon Analysis (GRCA) and Graphical Codon Usage Analyzer 2.0 (GCUA) online tools. Further, the codon-optimized sequence was cloned in a protein expression vector for the heterologous expression in E. coli, as illustrated in Fig. 1. The sequences of the fusion tags used in this study are listed in Table S1. DNA sequencing and restriction digestion analysis was employed to confirm the resulting genetic constructs, as shown in Figs. 1 and S1.

Illustration of the codon optimization workflow and methodology
Expression of pET-HTN-GLIS1 and pET-GLIS1-NTH in Different E. coli Host Strains and Media Conditions
The plasmids (pET-HTN-GLIS1 and pET-GLIS1-NTH) were transformed into E. coli BL21(DE3) and Rosetta strains using standard calcium chloride (CaCl2) transformation protocol using the heat-shock method. The antibiotic selection for BL21(DE3) was kanamycin, whereas for Rosetta, kanamycin and chloramphenicol were used. Single colonies obtained were inoculated in Luria–Bertani/Lysogeny Broth (LB) media for primary culture. The secondary culture was prepared in LB and Terrific Broth/Tartoff-Hobbs Broth (TB) media for screening the influence of media conditions on protein expression of N- and C-terminal GLIS1 in both BL21(DE3) and Rosetta strains. Following the first screening (Table S2), two clones of pET-GLIS1-NTH were expressed in LB and TB media. The induced cultures were incubated at 37 °C for different time points in a shaker incubator at 180 rpm. The induction parameters for the experiments are mentioned in Table S2 (second screening). After induction, the harvested cells were centrifuged and resuspended in lysis buffer (20 mM phosphate buffer, 150 mM NaCl, and 20 mM imidazole). Throughout the process, the buffer and the lysates were kept on ice. Following resuspension, the cells were subjected to ultrasonication. The biochemical analysis was performed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting methods.
Screening for Minimum Inducer Concentration and Cell Density for pET-GLIS1-NTH in BL21(DE3) E. coli Strain
Screening for the minimal inducer concentration was carried out by inducing pET-GLIS1-NTH plasmid clones grown in LB media (40 ml) with isopropyl β-d-1-thiogalactopyranoside (IPTG) (Sisco Research Laboratories) in a range from 0, 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 1, 1.5, and 2 mM concentration at OD600 ~ 0.5, at 37 °C for 4 h in a shaker incubator at 180 rpm. Screening for the optimal optical density was performed by inducing the cells grown in LB media (40 ml) with optimal IPTG concentration (0.05 mM) for OD600 range ~ 0.5, ~ 1, and ~ 1.5, and incubated at 37 °C for 4 h in a shaker incubator at 180 rpm. After induction, the harvested cells were centrifuged and resuspended in lysis buffer (20 mM phosphate buffer, 150 mM NaCl and 20 mM imidazole). Throughout the process, the buffer and the lysates were kept on ice. Following resuspension, the cells were subjected to ultrasonication, and further, the biochemical analysis was performed by SDS-PAGE and immunoblotting methods.
Screening for the Optimal Induction Temperature for Soluble Expression of pET-GLIS1-NTH in BL21(DE3) E. coli Strain
The soluble expression of GLIS1-NTH was screened by inducing the cells grown in LB media (40 ml). At OD600 ~ 0.5, the cultures were divided equally into four centrifuge tubes. Cultures were induced with 0.05 mM IPTG and incubated at four temperatures; 37, 30, 25, and 18 °C, and incubated for 4 h at 37 °C, 8 h at 30 °C, 16 h at 25 °C, and 32 h at 18 °C, respectively. After induction, the harvested cells were centrifuged and resuspended in lysis buffer (20 mM phosphate buffer, 150 mM NaCl and 20 mM imidazole, pH 7.2). Following resuspension, the cells were subjected to ultrasonication and further separated into lysate and supernatant fractions, respectively. The biochemical analysis of the fractions was analyzed by SDS-PAGE and immunoblotting methods.
Screening for Clonal Variation of pET-GLIS1-NTH in BL21(DE3) E. coli Strain
The plasmids were transformed into E. coli BL21(DE3) strain using standard CaCl2 transformation protocol as mentioned in previously published studies [41, 43]. Four clones were selected and grown in LB media (10 ml) supplemented with kanamycin. The secondary culture (40 ml) in LB media was prepared, and upon reaching OD600 ~ 0.5, the cells were induced with 0.05 mM IPTG and incubated at 37 °C for 4 h in the shaker incubator. After induction, the harvested cells were centrifuged and resuspended in lysis buffer (20 mM phosphate buffer, 150 mM NaCl, and 20 mM imidazole, pH 7.2). Following resuspension, the cells were subjected to ultrasonication and further separated into lysate and supernatant fractions. The analysis was performed by SDS-PAGE and immunoblotting methods.
Native Purification of GLIS1-NTH Protein Using Ni2+-NTA Affinity Column Chromatography
1.2 L LB media was inoculated for the expression of GLIS1-NTH in a 2 L flask. The culture was induced with 0.05 mM IPTG and incubated at 37 °C for 4 h. After induction, the harvested cells were centrifuged. The pellet weight of ~ 6.5 gm was resuspended in 40 ml equilibration/lysis buffer (20 mM phosphate buffer, 150 mM NaCl and 20 mM imidazole at pH 7.2). GLIS1-NTH was purified using the affinity chromatography technique. Following resuspension, the cells were subjected to ultrasonication. The lysate was centrifuged at 8000 rpm at 4 °C for 20 min. The supernatant obtained was loaded onto the equilibrated 20 ml column (Bio-Rad) containing charged nickel resin. The equilibration buffer used was 20 mM phosphate buffer, 150 mM NaCl, and 20 mM imidazole at pH 7.2. The column was incubated for 2 h at 4 °C with continuous shaking, and the purification step was carried out using purification buffers at pH 7.2 (W1: 20 mM phosphate buffer, 150 mM NaCl and 50 mM imidazole; W2: 20 mM phosphate buffer, 150 mM NaCl and 100 mM imidazole; W3: 20 mM phosphate buffer, 150 mM NaCl and 150 mM imidazole; Elution: 20 mM phosphate buffer, 150 mM NaCl, and 500 mM imidazole) with 10 min of incubation on ice. After purification, the eluted fractions were loaded onto pre-packed PD10 size-exclusion chromatography columns (10 ml; GE healthcare). Columns were equilibrated using PD10 equilibration buffer (20 mM phosphate buffer, pH 7.2) and eluted using PD10 elution buffer (20 mM phosphate buffer, pH 7.2). The protein was eluted from the column as per the manufacturer’s protocol. The eluted protein was supplemented with 5% glycerol, quantified with Bradford assay, and stored at − 80 °C until further analysis.
SDS-PAGE and Immunoblotting Techniques
The protein estimation for the samples was performed using the Bradford assay [44]. Bovine serum albumin (Bio-Rad) at varying concentrations was used to make the standard plot for protein estimation. Following protein estimation, SDS-PAGE was run with the respective samples and subjected to Coomassie staining and immunoblotting, described in our recent studies [41, 45]. Anti-His primary antibody (1:5000; Bio BioBharati, BB-AB0010) and anti-rabbit IgG secondary antibody (1:5000; Invitrogen, 31460) were used for immunoblotting analysis. 5% bovine serum albumin was used as diluent for both the primary and secondary antibodies. Immunoblots were developed using chemiluminescence substrate (Bio-Rad). The gel and blot images from were taken and analyzed using the molecular imager (ChemiDoc™ XRS+) installed with Image Lab™ software (Bio-Rad).
Far UV Circular Dichroism Spectroscopy
Secondary structure analysis of purified GLIS1-NTH protein was performed using far UV circular dichroism (CD) spectroscopy. The PD10 eluted fraction without glycerol was used for the analysis. The same parameters, as mentioned in our recent studies [41, 43], were used for data accumulation. The raw CD data were analyzed using an online tool [Beta Structure Selection (BeStSel)]. BeStSel algorithm is described in detail in the recently published study [46].
Cell Culture
Breast cancer cell line MDA-MB-231 was procured (from National Centre for Cell Science, Pune, India) and cultured in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen), 10% fetal bovine serum (FBS) (Invitrogen), and 1% penicillin/streptomycin (P/S) (Invitrogen). Cells were cultured at standard cell culture conditions (37 °C with 5% CO2 under humidified conditions). Cells were passaged upon attaining confluency of 70–80% with 0.25% trypsin–EDTA (Invitrogen) for further culture.
Cell Migration Assay
Cell migration assay was performed by plating a specific number of breast cancer cells MDA-MB-231 (0.85 × 105 cells/well of a 24-well plate) and BJ cells (0.50 × 105 cells/well of 24-well plate), and the seeded cells were incubated overnight. Cells at 80–90% confluency were scratched with a 10 μL sterile pipette tip. The medium was aspirated out, followed by a PBS wash. Scratched monolayers were treated with GLIS1-NTH (200 nM) or 5% glycerol buffer for 2 days in protein transduction media, and for BJ cells, the protein media was changed to fibroblast growth media (DMEM supplemented with 10% FBS and 1% P/S) after 4 h. Images were captured at different time intervals (0, 24 and 48 h) for MDA-MB-231 cells and BJ cells using an inverted bright-field microscope (ZOE Fluorescent Cell Imager, Bio-Rad, California, USA) at ×20 magnification. The migration rate at 24 h was analyzed and calculated using ImageJ software.
Statistical Analysis
A statistical test (unpaired t-test) was performed to analyze the results obtained from migration assay using Graphpad Prism 5 software. p values ≤ 0.05 were considered to be statistically significant.
Results
Codon Optimization and Design of GLIS1 Genetic Construct in pET Vector
Codon optimization tools deal with sequence-related parameters such as transcription, splicing, translation, and mRNA degradation, involved in various aspects of gene expression. The presence of rare codons deteriorates the protein expression due to tRNA insufficiency for these codons during translation [47], and therefore, codon optimization was performed of the coding sequence of the human GLIS1 gene for its expression in the E. coli host system. As per the GRCA tool, 11% rare codons with codon usage frequency of ≤ 30% (in red) were observed, as shown in Fig. S2 and Table S3, which were codon optimized. As per the GCUA tool, a total of 23 codons were observed to have a relative adaptiveness value of ≤ 30%, as shown in Fig. S3 (left; magenta). For example, the rare codons CTC and CGA present in the first 50 codons of the sequence were substituted with codons CTG and CGT (Fig. S3). Upon its codon optimization using the Gene Optimizer tool, the relative adaptiveness values of these codons enhanced from 26 and 30 to 100% for both these codons (Fig. S3). The optimized sequence showed parametric differences considering the original (non-optimized) sequence, as shown in Figs. S2, S3, and Table S3. In congruence with our previously published study of stem cell-specific transcription factor SOX2 [42] and OCT4 [48], an increase in the CAI (Codon adaptability index defines the measurement and the respective quantification of the similar codon usage bias of the DNA or RNA sequence with the reference set) value was observed for the optimized GLIS1 gene compared to the non-optimized sequence. The results also further confirmed the absence of rare codons after codon optimization.
The codon-optimized gene was then tagged with the fusion tags (NLS: for nuclear delivery, TAT: for cell permeability, and 8X His: for affinity purification) at either N- or C-terminal end of the gene of interest (Fig. S1) and further cloned into a pET28a(+) expression vector using restriction enzymes NcoI and XhoI as shown in Fig. 1 and Fig. S1, generating two genetic constructs, pET-HTN-GLIS1 and pET-GLIS1-NTH (Fig. S1). We performed preliminary confirmation for assessing the integrity of the cloned pET-HTN-GLIS1 and pET-GLIS1-NTH by restriction digestion, as shown in Fig. S4. Restriction enzymes used for digestion and their respective cut sites are mentioned in Table S4.
Optimization of Various Expression Parameters for Soluble Expression of the GLIS1 Fusion Protein
Heterologous expression of eukaryotic proteins in the prokaryotic system has been challenging due to various factors such as codon bias, expression host system, media composition, inducer concentration, optical density, temperature, time, expression construct, and so forth. Moreover, obtaining soluble expression of human proteins from the bacterial system is even more challenging. Thus, optimizing these parameters is essential for the successful soluble expression of heterologous genes in E. coli [49,50,51,52].
Screening for Strains and Media Conditions for the Expression of HTN-GLIS1 and GLIS1-NTH Fusion Proteins
To overcome these bottlenecks, we have initially screened to determine the best expression host strain (E. coli, in this study) and media condition for expressing pET-HTN-GLIS1 and pET-GLIS1-NTH. Two E. coli strains, BL21(DE3) and Rosetta (BL21 derivatives designed to improve the expression of eukaryotic proteins that contain rare codons rarely used in E. coli), were used in this study, which were used in the first screening process. These two strains were selected because BL21(DE3) has a strong T7 promoter system, lacks lon and Omp T proteases, and is compatible with the pET expression vector [53]. On the other hand, Rosetta is a highly engineered strain and a derivative of BL21(DE3) containing plasmid pRARE, used to express genes containing rare codons [50, 54]. Selecting these two strains would give us an idea of the presence of any rare codon in the sequence, compromising the expression of the gene. Although codon optimization was performed, Rosetta was also chosen as the initial expression analysis in BL21(DE3) alone resulted in low expression of the GLIS1 fusion protein in E. coli. Rosetta strain is reported to increase the success in the expression and purification of human-recombinant proteins containing rare codons [55]. Therefore, to rule out the possibility of the presence of any rare codons even after codon optimization, Rosetta was chosen for comparison.
In addition, two culture media, LB and TB (more nutritionally rich), were used in the first screening process. The cells were induced as per the induction parameters mentioned in Table S2 (first screening). Post-induction, an intense band at ~ 73 kDa GLIS1-NTH fusion protein, was observed in BL21(DE3)-transformed clones compared to Rosetta in both LB and TB media (Fig. 2A, top and bottom; 2B top and bottom; Table 1). However, HTN-GLIS1 showed no expression in any of the conditions (Fig. 2A, top and bottom; 2B, top and bottom). Interestingly, faint degradation was observed for HTN-GLIS1 in BL21(DE3) grown in TB media (Fig. 2B, bottom). No significant difference in the overall cell biomass was observed in BL21(DE3) transformed with GLIS1-NTH in both LB and TB at 37 °C (data not shown). The further assessment (i.e., the second screening; Table S2) of the difference in expression of the protein in BL21(DE3) in LB and TB media at different time points confirms maximum expression of the protein at 4 h in LB media compared to TB (Fig. 3, top and bottom).

Screening of E. coli strains and media conditions for expressing pET-HTN-GLIS1 and pET-GLIS1-NTH. A The gene was expressed in LB in BL21(DE3) and Rosetta strains, and the expressed protein samples were resolved in 10% SDS-PAGE. B The gene was expressed in TB in BL21(DE3) and Rosetta strains, and the expressed protein samples were resolved in 10% SDS-PAGE. The loading was normalized for both Coomassie and immunoblotting, and the amount of protein loaded in each well was 40 µg/well. M protein marker (kDa); UI uninduced; Ab antibody (n = 2)

Comparison and assessment of GLIS1-NTH in LB and TB at different time points. The genetic construct was transformed and expressed in BL21(DE3) in LB and TB media. Post-induction, the cultures were incubated at different time points to compare and assess the difference in the expression between the two media. Protein samples were resolved in 10% SDS-PAGE, and the loading was normalized for both Coomassie and immunoblotting and the amount of protein loaded in each well was 40 µg/well. M protein marker (kDa); UI uninduced; Ab antibody (n = 2)
Screening for Minimum IPTG Concentration and Appropriate OD Value for GLIS1-NTH Expression
After the screening, we screened for the minimum IPTG concentration and ideal optical density to express the GLIS1-NTH fusion protein. The bacterial culture was inoculated in LB media and induced with different IPTG concentrations, as mentioned in Table 1. Screening showed the highest expression with 0.05 mM IPTG concentration for GLIS1-NTH (Fig. 4A, top and bottom). In addition, lower IPTG concentrations such as 0.01 and 0.025 were also screened and compared to 0.05; however, lower than 0.05 mM concentration of GLIS1-NTH failed to induce high protein expression (Fig. S5, top and bottom). Next, to screen the optical density, we induced the bacterial culture with optimized IPTG concentration (0.05 mM) after reaching the desired OD600 values (Table 1). The results showed that OD600 ~ 0.5 showed maximum expression for GLIS1-NTH (Fig. 4B, top and bottom; Table 1) compared to OD600 ~ 1 and ~ 1.5.

Screening for maximum expression of GLIS1-NTH fusion protein at different IPTG concentrations and optimal optical density. A Expression of GLIS1-NTH was screened at increasing order of different IPTG concentrations. The protein samples were resolved in 10% SDS-PAGE for visualization. B Optical density at three different growth phases was assessed for the maximum expression of GLIS1-NTH. The loading was normalized for both Coomassie and immunoblotting, and the amount of protein loaded in each well was 40 µg/well. M protein marker (kDa); UI uninduced; Ab antibody (n = 2)
Assessment of Soluble Expression of GLIS1-NTH at Varying Temperatures
The soluble expression of recombinant GLIS1-NTH was evaluated by screening different induction temperatures, as shown in Table 1 and Fig. 5A. It was observed that at only 37 °C, a small fraction of the protein was observed in the soluble fraction compared to other temperatures. At 30 °C, although expression was observed in the lysate fraction, no expression was detected in the supernatant fraction. The overall expression of the protein at 25 °C and 18 °C temperatures was also compromised (Fig. 5A, top and bottom; Table 1).

Determination of appropriate temperature for soluble expression of GLIS1-NTH fusion protein. A four different temperatures (37, 30, 25, 18 °C) were screened to determine the appropriate temperature for the soluble expression of GLIS1-NTH. B Clonal variability at 37 °C of four different clones were assessed. Lysate concentration was quantified, and the same volume was loaded for supernatant. C Soluble expression analysis of the selected clone 3 expressed under optimal parameters. D Quantitative analysis of soluble protein expression using ImageJ online tool. All the protein samples for Coomassie and immunoblotting were resolved in 10% SDS-PAGE for visualization and analysis. The loading was normalized, and the amount of protein loaded in each well was 40 µg/well. M protein marker (kDa); L lysate; P pellet; S supernatant; UI uninduced; Ab antibody (n = 2)
Assessment of Clonal Variation in Soluble Protein Expression Under Optimized Culture Conditions
Randomly four clones were picked from the freshly transformed pET-GLIS1-NTH dish and were expressed as per the optimized parameters mentioned in Table 1. No significant difference in the overall expression of the clones was observed in both the lysate and supernatant fractions (Fig. 5B, top and bottom). The protein solubility of the selected clone showed GLIS1-NTH in both supernatant (S) and pellet (P; pellet (inclusion bodies)) fraction (Fig. 5C; top and bottom). The supernatant fraction was chosen for purification. The Lysate (L), P, and S band intensity of the immunoblot was quantified using Image J, and the arbitrary area value for each intensity peak was plotted as shown in Fig. 5D.
Recombinant GLIS1-NTH Fusion Protein Purification
GLIS1-NTH was expressed using identified optimal conditions, and the supernatant fraction was used for purification to purify under native conditions. 1.2 L culture-encoding recombinant GLIS1-NTH was expressed according to the optimized culture conditions (Table 1). The supernatant was loaded onto the pre-equilibrated Ni2+-NTA column. The purified GLIS1-NTH was observed corresponding to the expected molecular weight of ~ 73 kDa (Calculated molecular weight: 73.017 kDa) in Coomassie-stained SDS-PAGE gel (eluted fraction) and immunoblot (Fig. 6A, top and bottom). However, the loss of protein was observed in the flow through in both Coomassie and immunoblot (Fig. 6A, top and bottom). This presumably could be due to the overloading of the sample on the purification column or due to the low resin volume used for purification. A very faint-truncated GLIS1 fusion protein at around 45 kDa was also observed in Coomassie-stained SDS-PAGE gel (eluted fraction) and immunoblot (Fig. 6A, top and bottom). These truncations observed could be possibly due to (i) the presence of intragenic sequences that mimic E. coli ribosomal entry sites within the protein-coding sequence [56], and (ii) proteolysis at specific sensitive sites of some protein molecules during expression [57]. A total of nine 1 ml elution fractions were collected, and their absorbance at 280 nm was measured and plotted. The elution profile shows the maximum peak at the fourth elution (Fig. 6B). The total protein yield obtained was 1.5 mg/L of culture with a purity of > 90% (quantified using Image J software).

Purification of the GLIS1-NTH fusion protein. Affinity purification method was applied for purifying GLIS1-NTH protein from the supernatant fraction of the lysate. A purification of GLIS1-NTH protein and its purity were visualized using Coomassie and immunoblot using anti-His antibody. M protein marker (kDa); L lysate; S supernatant; FT flow through; W wash buffer; E eluted fraction; Ab antibody; (*), GLIS1-NTH protein truncations (n = 3). B elution profile analysis of the eluted proteins per ml measured at absorbance 280 nm
Secondary Structure Prediction of the Recombinant GLIS1-NTH Fusion Protein
The far ultraviolet CD spectroscopic technique is the most frequently used method to study the folding conformation/characteristics of desired proteins in which secondary structure is unknown [58, 59], like GLIS1. Therefore, secondary structure determination for purified recombinant GLIS1-NTH fusion protein was performed using far UV CD spectroscopy. The results showed a positive peak at ~ 195 nm and a negative peak at ~ 218 nm (Fig. 7A), which are corresponding to the peaks for β-sheets [58, 59]. The GLIS1-NTH structure was observed to be majorly composed of random coils (~ 46%) and β-sheets (~ 27%) and a substantial contribution of α-helix (~ 14%) and turns (~ 13%) determined using the BestSel method (Fig. 7B). These results confirm that recombinant GLIS1-NTH fusion protein has maintained its secondary structure and shows great promise of being biologically active.

Investigating the secondary structure of purified human GLIS1-NTH fusion protein using CD spectroscopy. A Representation of secondary structure peaks (positive or negative) corresponding to the wavelength. Analysis using the BeStSel web server, and the spectra were plotted with wavelength (nm) in X-axis and delta epsilon (M−1/cm) on Y-axis. B the bar graph represents the quantitative secondary structure percentage present in the purified human GLIS1-NTH fusion protein (n = 3)
Effect of the Exogenously Delivered Recombinant GLIS1 Fusion Protein on the Rate of Migration of MDA-MB-231 Cells
Recently, a study reported that overexpression of GLIS1 in breast cancer MDA-MB-231 cells contributed to the increase in the migration rate of these cells [18]. Therefore, to determine the functionality of the purified GLIS1-NTH fusion protein, the rate of migration of the breast cancer cells MDA-MB-231 was determined using migration assay. Upon protein transduction in MDA-MB-231 cells at every 24 h, the results showed that the migration rate of cells treated with purified GLIS1-NTH protein migrated faster compared to the vehicle control (Fig. 8A, B). This increase in the rate of migration was in congruence with the recent study [18]. The calculation for the migration rate was performed as reported earlier [60]. The migration assay was also performed for BJ cells, and the results showed no significant difference in the migration rate (Fig. S6), indicating that the observation was specific to breast cancer cells.

Effect of the exogenously delivered recombinant GLIS1-NTH fusion protein on the rate of migration of MDA-MB-231 cells. A cells were seeded in 24-well culture dishes. The respective wells were treated with protein or vehicle control every 24 h for 2 days. B graphical representation of the rate of migration of protein vs. vehicle control-treated cells (p ≤ 0.05) (n = 4)
Discussion
GLIS1 is a multifaceted protein, and in cell reprogramming, it has a prominent role in enhancing the generation of fully reprogrammed iPSCs [6]. To date, the viral form of GLIS1 has been used for generating iPSCs in combination with OCT4, SOX2, and KLF4. In this study, we sought to purify the recombinant human GLIS1 fusion protein from E. coli, which can substitute its genetic and viral counterpart to generate integration-free iPSCs.
In this study, expression host E. coli was used, which is the most versatile and well-understood system with high transformation efficiency, rapid doubling time, inexpensive culture conditions, and hassle-free culturing techniques [39]. Moreover, this expression host is extensively used to generate recombinant human proteins for which post-translational modifications are not essential for their bioactivity [61,62,63,64]. In addition to codon optimization, the expression of human-recombinant protein in this host system is also influenced by the media composition. It was reported that complex and semi-defined media enhanced cell biomass, thus, boosting protein production [65, 66]. Also, the position of fusion tags at either end of the gene influences the solubility of the protein, as reported previously [41]. Owing to these reports, we have compared the expression of pET-HTN-GLIS1 and pET-GLIS1-NTH in two E. coli strains BL21(DE3) and Rosetta, grown in LB and TB media. Interestingly, the recombinant expression of pET-GLIS1-NTH comparatively showed high expression in BL21(DE3) strain, whereas only a trace amount was detected in the Rosetta strain (in LB media). This implied that codon optimization was efficiently performed, and the low expression was due to reasons other than the presence of rare codons. In general, a similar observation of high expression in BL21(DE3) strain and low expression in Rosetta strain was also made by a study purifying Cas9 protein [67] and few other proteins from a set of 68 human proteins [55]. We speculate that the extra metabolic burden due to an additional plasmid pRARE and the presence of chloramphenicol might have resulted in the low GLIS1 gene expression in Rosetta, similar to what was reported earlier with other recombinant proteins [55, 67, 68]. A solution to this would be to induce the expression of the protein of interest at low temperature (preferably at 18 °C) to reduce the metabolic burden [67]; however, this did not yield any expression in our study. In agreement with earlier studies, our study shows that the inclusion of plasmids containing extra copies of low abundance of tRNA genes and the presence of chloramphenicol may cause negative metabolic side effects decreasing the expression levels of recombinant proteins, but this may not be the case for other recombinant proteins and detailed screening will be required. Also, pET-HTN-GLIS1 failed to express in either of the expression host system in both LB and TB, thus, confirming the influence of the fusion tags on protein expression [69, 70].
Also, the higher expression of GLIS1-NTH in the case of LB compared to TB was observed. We speculate that TB being a more nutritionally rich medium (composed of increased concentrations of peptone, yeast extract, and glycerol as a carbon source) might have contributed to the accumulation of acetate in the culture, presumably affecting the overall expression of GLIS1-NTH fusion protein compared to LB media. Earlier studies have reported that acetate acts as an inhibitor in biomass production and, thus, reduces recombinant protein production [71, 72]. Although we did not see any difference in the overall cell biomass between LB and TB at 37 °C, the expression of GLIS1-NTH was affected in the case of TB compared to LB. This could be due to overflow metabolism (the Crabtree effect) producing acetate because of the presence of excess carbon sources in the TB medium [73]. Other studies have also reported that E. coli, in the presence of excess carbon sources like glucose and glycerol, produces acetate as a by-product due to acidic fermentation [74, 75].
The other factors affecting the soluble expression of recombinant protein are inducer concentration, the optical density of cells, induction time, and temperature. Several studies have reported that reducing the inducer concentration, time, and temperature helps maximize the solubility of the protein, reduces the metabolic burden, and facilitates protein folding [50, 76, 77]. This step is crucial to avoid protein purification from inclusion bodies that contain either misfolded or partially folded proteins. Hence, it demands solubilization with strong detergents and refolding to the native state [76]. The bacterial growth phase also plays a critical role in the soluble expression of recombinant proteins. Several studies have reported that the maximum expression of the protein was achieved at the early to mid-log phase (OD600 0.1–0.5) [78, 79]. Higher cell densities lead to exhaustion of nutrients leading to nutrient deprivation, production of acetate, reduced dissolved oxygen, and increased carbon dioxide post-induction. These factors contribute to the decrease in the expression of recombinant proteins [50, 80,81,82]. We have optimized these expression parameters for maximizing the soluble expression of GLIS1-NTH in E. coli. Upon screening the inducer concentration, maximum expression was obtained at minimum IPTG concentration. Also, upon screening the optical density of cells, the results were in tandem with the fact that maximum expression of the GLIS1-NTH fusion protein was obtained at an early- to mid-log phase compared to the late-log phase. However, the reduction in temperature did not enhance the soluble expression; instead, it curbed the overall protein expression at lower temperatures. This might be due to the decrease in total cell mass, thereby expressing GLIS1-NTH at an undetectable range.
To the best of our knowledge, we are the first to optimize the induction parameters and use a simple and straightforward approach to purify GLIS1-NTH protein under native conditions. The presence of an affinity tag (polyhistidine) aided purification through the affinity chromatography technique. The cell-penetrating peptide (TAT) and NLS will help deliver the protein to the subcellular and subnuclear locations of the mammalian cell. Our previous reports have corroborated that tagging the proteins with TAT and NLS promoted their entry into the cell and nucleus, respectively [43, 45, 48]. Similar fusion strategies were employed in previous studies, including ours, for the efficient subcellular and subnuclear delivery of reprogramming factors such as OCT4, NANOG, SOX2, PDX1, and GATA4 in the form of recombinant proteins in mammalian cells [43, 45, 48, 69, 83,84,85,86]. Thus, purified GLIS1-NTH fusion protein can potentially translocate into the cell and nucleus as well. The biological activity of a protein depends on its structural conformation, and thus, retention of the secondary structure is imperative for its functionality. Secondary structure analysis of GLIS1-NTH showed its structure composition, majorly comprising of β-sheets and random coils and a substantial contribution of α-helix and turns; thus, it is most likely to be biologically active.
GLIS1 overexpression in breast cancer cells MDA-MB-231 contributed to the increase in the migration and invasion capacities of the cells, possibly through the upregulation of WNT5A [18] or by cooperating with CUX1, thus stimulating activity of TCF/β-catenin transcription factor and enhancing cell migration and invasion of breast cancer cells [87]. In our study, we also observed an increase in migration rate when the same MDA-MB-231 cells were treated with the GLIS1 fusion protein. Interestingly, no significant difference in migration rate was observed when BJ cells were treated with the GLIS1 fusion protein. This might be due to the fact that GLIS1 alone has no prominent effect on somatic cells, but in combination with other transcription factors like OCT4, SOX2, and KLF4, it pushes the cell fate towards the generation of iPSCs. However, a further detailed understanding of the role of GLIS1 protein in various cellular processes and signaling pathways in cancer cells as well as in somatic cells would be an interesting and important topic of research in the near future.
This study optimized the parameters for the maximum expression of a human-recombinant GLIS1 fusion protein, successfully purified the protein under native conditions, determined its secondary structure and biological activity. However, compared to our recently published studies [41,42,43, 45, 88], the total protein expression is less. This might be due to the large size of the protein ~ 73 kDa compared to our other purified proteins, which are ≤ 50 kDa. Many studies have previously reported the constraints of purifying large molecular weight proteins [89], and similarly, the compromised expression of GLIS1-NTH could be due to its large size. Thus, further optimization or other novel strategies should be employed to increase the total yield of the protein. The methodology generated and used in our study is not only inexpensive and facile but also highly reproducible. The immense prospect of this biological tool (GLIS1-NTH) in the recombinant form will bring forth multiple opportunities. It will allow the scientific community to understand its detailed function in different stage-specific developmental processes, its function, and downstream implications in various cancers and other disease models, molecular interactions with reprogramming factors and signaling pathways, nuances of the mechanism during cell-fate transitions, and substituting its viral/genetic counterpart in the generation of integration-free iPSCs. Importantly, this tool can be used to gauge the specific spatiotemporal expression of GLIS1 reprogramming factor in different combinations during the generation of integration-free iPSCs and understand its expression and function in different diseases in the near future.
References
Kim, Y. S., Lewandoski, M., Perantoni, A. O., Kurebayashi, S., Nakanishi, G., & Jetten, A. M. (2002). Identification of Glis1, a novel Gli-related, Krüppel-like zinc finger protein containing transactivation and repressor functions. Journal of Biological Chemistry, 277(34), 30901–30913.
Scoville, D. W., Kang, H. S., & Jetten, A. M. (2017). GLIS1-3: Emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investigation, 4, 1–11.
Jetten, A. M. (2018). GLIS1–3 transcription factors: Critical roles in the regulation of multiple physiological processes and diseases. Cellular and Molecular Life Sciences, 75(19), 3473–3494.
Kang, H. S., Zeruth, G., Lichti-Kaiser, K., Vasanth, S., Yin, Z., Kim, Y.-S., & Jetten, A. M. (2010). Gli-similar (Glis) Krüppel-like zinc finger proteins: Insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histology and Histopathology, 25(11), 1481–1496.
Nakashima, M., Tanese, N., Ito, M., Auerbach, W., Bai, C., Furukawa, T., Toyono, T., Akamine, A., & Joyner, A. L. (2002). A novel gene, GliH1, with homology to the Gli zinc finger domain is not required for mouse development. Mechanisms of Development, 119(1), 21–34.
Maekawa, M., Yamaguchi, K., Nakamura, T., Shibukawa, R., Kodanaka, I., Ichisaka, T., Kawamura, Y., Mochizuki, H., Goshima, N., & Yamanaka, S. (2011). Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature, 474(7350), 225–228.
Yoshioka, N., & Dowdy, S. F. (2017). Enhanced generation of iPSCs from older adult human cells by a synthetic five-factor self-replicative RNA. PLoS ONE, 12(7), 0182018.
Yoshioka, N., Gros, E., Li, H. R., Kumar, S., Deacon, D. C., Maron, C., Muotri, A. R., Chi, N. C., Fu, X. D., Benjamin, D. Y., & Dowdy, S. F. (2013). Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell, 13(2), 246–254.
Wang, L., Chen, Y., Guan, C., Zhao, Z., Li, Q., Yang, J., Mo, J., Wang, B., Wu, W., Yang, X., & Song, L. (2017). Using low-risk factors to generate non-integrated human induced pluripotent stem cells from urine-derived cells. Stem Cell Research Therapy, 8(1), 245.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Sasaki, A., Yamamoto, M., Nakamura, M., Sutou, K., Osafune, K., & Yamanaka, S. (2014). Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nature Communications, 5(1), 3678.
Lee, S.-Y., Noh, H. B., Kim, H.-T., Lee, K.-I., & Hwang, D.-Y. (2017). Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget, 8(44), 77041–77049.
Maekawa, M., & Yamanaka, S. (2011). Glis1, a unique pro-reprogramming factor, may facilitate clinical applications of iPSC technology. Cell Cycle, 10(21), 3613–3614.
Liu, J., Han, Q., Peng, T., Peng, M., Wei, B., Li, D., Yao, Y., Wang, Y., Zhao, G., Wang, X., & Fu, M. (2015). The oncogene c-Jun impedes somatic cell reprogramming. Nature Cell Biology, 17(7), 856–867.
Wang, B., Wu, L., Li, D., Liu, Y., Guo, J., Li, C., et al. (2019). Induction of pluripotent stem cells from mouse embryonic fibroblasts by Jdp2-Jhdm1b-Mkk6-Glis1-Nanog-Essrb-Sall4. Cell Reports, 27(12), 3473–3485.
Li, L., Chen, K., Wang, T., Wu, Y., Xing, G., Chen, M., et al. (2020). Glis1 facilitates induction of pluripotency via an epigenome–metabolome–epigenome signalling cascade. Nature Metabolism, 2(9), 882–892.
Luo, R., Zhang, X., Wang, L., Zhang, L., Li, G., & Zheng, Z. (2021). GLIS1, a potential candidate gene affect fat deposition in sheep tail. Molecular Biology Reports, 48(5), 4925–4931.
Yasuoka, Y., Matsumoto, M., Yagi, K., & Okazaki, Y. (2020). Evolutionary history of GLIS genes illuminates their roles in cell reprogramming and ciliogenesis. Molecular Biology and Evolution, 37(1), 100–109.
Shimamoto, K., Tanimoto, K., Fukazawa, T., Nakamura, H., Kanai, A., Bono, H., Ono, H., Eguchi, H., & Hirohashi, N. (2020). GLIS1, a novel hypoxia-inducible transcription factor, promotes breast cancer cell motility via activation of WNT5A. Carcinogenesis, 41(9), 1184–1194.
Song, W., Chen, Y. P., Huang, R., Chen, K., Pan, P. L., Li, J., Yang, Y., & Shang, H. F. (2012). GLIS1: An increased risk factor for late-onset Parkinson’s disease in the Han Chinese population. European Neurology, 68(2), 89–92.
Singh, V. K., Kalsan, M., Kumar, N., Saini, A., & Chandra, R. (2015). Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Frontiers in Cell and Developmental Biology, 3, 2.
Hu, Q., Chen, R., Teesalu, T., Ruoslahti, E., & Clegg, D. O. (2014). Reprogramming human retinal pigmented epithelial cells to neurons using recombinant proteins. Stem Cells Translational Medicine, 3(12), 1526–1534.
Saha, B., Borgohain, P. M., Dey, C., & Thummer, R. P. (2018). iPS cell generation: Current and future challenges. Annals of Stem Cell Research & Therapy, 1(2), 1007.
Borgohain, M. P., Haridhasapavalan, K. K., Dey, C., Adhikari, P., & Thummer, R. P. (2019). An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Reviews and Reports, 15(2), 286–313.
Haridhasapavalan, K. K., Borgohain, M. P., Dey, C., Saha, B., Narayan, G., Kumar, S., & Thummer, R. P. (2019). An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene, 686, 146–159.
Dey, C., Raina, K., Thool, M., & Thummer, R. P. (2021). An overview of reprogramming approaches to derive integration-free induced pluripotent stem cells for prospective biomedical applications. In Recent advances in iPSC technology (1st ed., pp. 231–238). Elsevier Academic Press.
Okita, K., Ichisaka, T., & Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature, 448(7151), 313–317.
Ben-David, U., & Benvenisty, N. (2011). The tumorigenicity of human embryonic and induced pluripotent stem cells. Nature Reviews Cancer, 11(4), 268–277.
Somers, A., Jean, J.-C., Sommer, C. A., Omari, A., Ford, C. C., Mills, J. A., Ying, L., Sommer, A. G., Jean, J. M., Smith, B. W., & Lafyatis, R. (2010). Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells, 28(10), 1728–1740.
Ramos-Mejia, V., Fraga, M. F., & Menendez, P. (2012). IPSCs from cancer cells: Challenges and opportunities. Trends in Molecular Medicine, 18(5), 245–247.
Kadari, A., Lu, M., Li, M., Sekaran, T., Thummer, R. P., Guyette, N., Chu, V., & Edenhofer, F. (2014). Excision of viral reprogramming cassettes by Cre protein transduction enables rapid, robust and efficient derivation of transgene-free human induced pluripotent stem cells. Stem Cell Research & Therapy, 5(2), 47.
Baldo, A., van den Akker, E., Bergmans, H. E., Lim, F., & Pauwels, K. (2013). General considerations on the biosafety of virus-derived vectors used in gene therapy and vaccination. Current Gene Therapy, 13(6), 385–394.
Kaji, K., Norrby, K., Paca, A., Mileikovsky, M., Mohseni, P., & Woltjen, K. (2009). Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature, 458(7239), 771–775.
Sommer, C. A., & Mostoslavsky, G. (2013). The evolving field of induced pluripotency: Recent progress and future challenges. Journal of Cellular Physiology, 228(2), 267–275.
O’Malley, J., Woltjen, K., & Kaji, K. (2009). New strategies to generate induced pluripotent stem cells. Current Opinion in Biotechnology, 20(5), 516–521.
Dey, C., Narayan, G., Krishna Kumar, H., Borgohain, M. P., Lenka, N., & Thummer, R. P. (2017). Cell-penetrating peptides as a tool to deliver biologically active recombinant proteins to generate transgene-free induced pluripotent stem cells. Studies on Stem Cell Research and Therapy, 3(1), 6–15.
Seo, B. J., Hong, Y. J., & Do, J. T. (2017). Cellular reprogramming using protein and cell-penetrating peptides. International Journal of Molecular Medicine, 18(3), 552.
Ebrahimi, B. (2015). Reprogramming barriers and enhancers: Strategies to enhance the efficiency and kinetics of induced pluripotency. Cell Regeneration, 4(1), 4–10.
Haridhasapavalan, K. K., Raina, K., Dey, C., Adhikari, P., & Thummer, R. P. (2020). An insight into reprogramming barriers to iPSC generation. Stem Cells Reviews and Reports, 16(1), 56–81.
Borgohain, M. P., Narayan, G., Kumar, H. K., Dey, C., & Thummer, R. P. (2018). Maximizing expression and yield of human recombinant proteins from bacterial cell factories for biomedical applications. In P. Kumar, J. K. Patra, & P. Chandra (Eds.), Advances in microbial biotechnology (pp. 447–486). Apple Academic Press.
Bhatwa, A., Wang, W., Hassan, Y. I., Abraham, N., Li, X.-Z., & Zhou, T. (2021). Challenges associated with the formation of recombinant protein inclusion bodies in Escherichia coli and strategies to address them for industrial applications. Frontiers in Bioengineering and Biotechnology, 9, 65.
Haridhasapavalan, K. K., Sundaravadivelu, P. K., & Thummer, R. P. (2020). Codon optimization, cloning, expression, purification, and secondary structure determination of human ETS2 transcription factor. Molecular Biotechnology, 62(10), 485–494.
Thool, M., Dey, C., Bhattacharyya, S., Sudhagar, S., & Thummer, R. P. (2021). Generation of a recombinant stem cell-specific human SOX2 protein from Escherichia coli under native conditions. Molecular Biotechnology, 63(4), 327–338.
Narayan, G., Sundaravadivelu, P. K., Agrawal, A., Gogoi, R., Nagotu, S., & Thummer, R. P. (2021). Soluble expression, purification, and secondary structure determination of human PDX1 transcription factor. Protein Expression and Purification, 180, 105807.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Haridhasapavalan, K. K., Sundaravadivelu, P. K., Bhattacharyya, S., Harsh, S. R., Raina, K., & Thummer, R. P. (2021). Generation of cell-permeant recombinant human transcription factor GATA4 from E. coli. Bioprocess and Biosystems Engineering, 44(6), 1131–1146.
Micsonai, A., Wien, F., Bulyáki, É., Kun, J., Moussong, É., Lee, Y.-H., Goto, Y., Réfrégiers, M., & Kardos, J. (2018). BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Research, 46(W1), 315–322.
Kane, J. F. (1995). Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Current Opinion in Biotechnology, 6(5), 494–500.
Dey, C., Thool, M., Bhattacharyya, S., Sudhagar, S., & Thummer, R. P. (2021). Generation of biologically active recombinant human OCT4 protein from E. coli. 3 Biotech, 11(5), 207.
Joseph, B., Pichaimuthu, S., & Rao, M. S. (2015). An overview of the parameters for recombinant protein expression in Escherichia coli. Journal of Cell Science & Therapy, 6(5), 221.
Kaur, J., Kumar, A., & Kaur, J. (2018). Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. International Journal of Biology and Macromolecules, 106, 803–822.
Dong, G., Zhao, X., Guo, J., Ma, L., Zhou, H., Liu, Q., Zhao, X., Wang, C., & Wu, K. (2021). Functional expression and purification of recombinant full-length human ATG7 protein with HIV-1 Tat peptide in Escherichia coli. Protein Expression and Purification, 182, 105844.
Damough, S., Sabzalinezhad, M., Talebkhan, Y., Nematollahi, L., Bayat, E., Torkashvand, F., Adeli, A., Jahandar, H., Barkhordari, F., & Mahboudi, F. (2021). Optimization of culture conditions for high-level expression of soluble and active tumor necrosis factor-α in E. coli. Protein Expression and Purification, 179, 105805.
Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: Advances and challenges. Frontiers in Microbiology, 5, 172.
Berrow, N. S., Büssow, K., Coutard, B., Diprose, J., Ekberg, M., Folkers, G. E., Lévy, N., Lieu, V., Owens, R. J., Peleg, Y., & Pinaglia, C. (2006). Recombinant protein expression and solubility screening in Escherichia coli: A comparative study. Acta Crystallographica Section D: Biological Crystallography, 62(10), 1218–1226.
Tegel, H., Tourle, S., Ottosson, J., & Persson, A. (2010). Increased levels of recombinant human proteins with the Escherichia coli strain Rosetta (DE3). Protein Expression and Purification, 69(2), 159–167.
Maertens, B., Spriestersbach, A., von Groll, U., Roth, U., Kubicek, J., Gerrits, M., Graf, M., Liss, M., Daubert, D., Wagner, R., & Schäfer, F. (2010). Gene optimization mechanisms: A multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Science, 19(7), 1312–1326.
Ryan, B. J., & Henehan, G. T. (2013). Overview of approaches to preventing and avoiding proteolysis during expression and purification of proteins. Current Protocols in Protein Science, 71(1), 5–25.
Kelly, S. M., Jess, T. J., & Price, N. C. (2005). How to study proteins by circular dichroism? Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 1751(2), 119–139.
Greenfield, N. J. (2006). Using circular dichroism spectra to estimate protein secondary structure. Nature Protocol, 1(6), 2876–2890.
Grada, A., Otero-Vinas, M., Prieto-Castrillo, F., Obagi, Z., & Falanga, V. (2017). Research techniques made simple: Analysis of collective cell migration using the wound healing assay. Journal of Investigative Dermatology, 137(2), 11–16.
Ghasemi, Y., Ghoshoon, M. B., Taheri, M., Negahdaripour, M., & Nouri, F. (2020). Cloning, expression and purification of human PDGF-BB gene in Escherichia coli: New approach in PDGF-BB protein production. Gene Reports, 19, 100653.
Zamani, M., Berenjian, A., Hemmati, S., Nezafat, N., Ghoshoon, M. B., Dabbagh, F., Mohkam, M., & Ghasemi, Y. (2015). Cloning, expression, and purification of a synthetic human growth hormone in Escherichia coli using response surface methodology. Molecular Biotechnology, 57(3), 241–250.
Galluccio, M., Amelio, L., Scalise, M., Pochini, L., Boles, E., & Indiveri, C. (2012). Over-expression in E. coli and purification of the human OCTN2 transport protein. Molecular Biotechnology, 50(1), 1–7.
Bhat, E. A., Sajjad, N., Sabir, J. S. M., Kamli, M. R., Hakeem, K. R., Rather, I. A., & Bahieldin, A. (2020). Molecular cloning, expression, overproduction and characterization of human TRAIP Leucine zipper protein. Saudi Journal of Biological Sciences, 27(6), 1562–1565.
Tripathi, N. K., Sathyaseelan, K., Jana, A. M., & Rao, P. V. L. (2009). High yield production of heterologous proteins with Escherichia coli. Defence Science Journal, 59(2), 137–146.
Vincentelli, R., Cimino, A., Geerlof, A., Kubo, A., Satou, Y., & Cambillau, C. (2011). High-throughput protein expression screening and purification in Escherichia coli. Methods, 55(1), 65–72.
Pannunzio Carmignotto, G., & Rodrigues Azzoni, A. (2019). On the expression of recombinant Cas9 protein in E. coli BL21(DE3) and BL21(DE3) Rosetta strains. Journal of Biotechnology, 306, 62–70.
Søgaard, K. M., & Nørholm, M. H. H. (2016). Side effects of extra tRNA supplied in a typical bacterial protein production scenario. Protein Science, 25(11), 2102–2108.
Bosnali, M., & Edenhofer, F. (2008). Generation of transducible versions of transcription factors Oct4 and Sox2. Biological Chemistry, 389(7), 851–861.
Braun, P., Hu, Y., Shen, B., Halleck, A., Koundinya, M., Harlow, E., & LaBaer, J. (2002). Proteome-scale purification of human proteins from bacteria. Proceedings of the National Academy of Sciences of the United States of America, 99(5), 2654–2659.
Kleman, G. L., & Strohl, W. R. (1994). Acetate metabolism by Escherichia coli in high-cell-density fermentation. Applied and Environmental Microbiology, 60(11), 3952–3958.
de Mey, M., de Maeseneire, S., Soetaert, W., & Vandamme, E. (2007). Minimizing acetate formation in E. coli fermentations. Journal of Industrial Microbiology and Biotechnology, 34(11), 689–700.
Åkesson, M., Hagander, P., Axelsson, J. P., & Bioeng, B. (2001). Avoiding acetate accumulation in Escherichia coli cultures using feedback control of glucose feeding. Biotechnology and Bioengineering, 73(3), 223–230.
Eiteman, M. A., & Altman, E. (2006). Overcoming acetate in Escherichia coli recombinant protein fermentations. Trends in Biotechnology, 24(11), 530–536.
Martínez-Gómez, K., Flores, N., Castañeda, H. M., Martínez-Batallar, G., Hernández-Chávez, G., Ramírez, O. T., Gosset, G., Encarnación, S., & Bolivar, F. (2012). New insights into Escherichia coli metabolism: Carbon scavenging, acetate metabolism and carbon recycling responses during growth on glycerol. Microbial Cell factories, 11(1), 1–21.
Baneyx, F., & Mujacic, M. (2004). Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology, 22(11), 1399–1408.
Sørensen, H. P., & Mortensen, K. K. (2005). Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microbial Cell Factories, 4(1), 1–8.
San-Miguel, T., Pérez-Bermúdez, P., & Gavidia, I. (2013). Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus, 2(1), 89.
Jadeja, D., Dogra, N., Arya, S., Singh, G., Singh, G., & Kaur, J. (2016). Characterization of LipN (Rv2970c) of Mycobacterium Tuberculosis H37Rv and its probable role in xenobiotic degradation. Journal of Cellular Biochemistry, 117(2), 390–401.
Balbás, P. (2001). Understanding the art of producing protein and nonprotein molecules in Escherichia coli. Molecular Biotechnology, 19(3), 251–267.
Hu, J. H., Wang, F., & Liu, C. Z. (2015). Development of an efficient process intensification strategy for enhancing Pfu DNA polymerase production in recombinant Escherichia coli. Bioprocess and Biosystems Engineering, 38(4), 651–659.
Olaofe, O. A., Burton, S. G., Cowan, D. A., & Harrison, S. T. L. (2010). Improving the production of a thermostable amidase through optimising IPTG induction in a highly dense culture of recombinant Escherichia coli. Biochemical Engineering Journal, 52(1), 19–24.
Thier, M., Münst, B., & Edenhofer, F. (2010). Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. International Journal of Developmental Biology, 54(11–12), 1713–1721.
Thier, M., Münst, B., Mielke, S., & Edenhofer, F. (2012). Cellular reprogramming employing recombinant Sox2 protein. Stem Cells International, 2012, 549846.
Peitz, M., Münst, B., Thummer, R. P., Helfen, M., & Edenhofer, F. (2014). Cell-permeant recombinant Nanog protein promotes pluripotency by inhibiting endodermal specification. Stem Cell Research, 12(3), 680–689.
Münst, B., Thier, M. C., Winnemöller, D., Helfen, M., Thummer, R. P., & Edenhofer, F. (2016). Nanog induces suppression of senescence through downregulation of p27KIP1 expression. Journal of Cell Science, 129(5), 912–920.
Vadnais, C., Shooshtarizadeh, P., Rajadurai, C. V., Lesurf, R., Hulea, L., Davoudi, S., Cadieux, C., Hallett, M., Park, M., & Nepveu, A. (2014). Autocrine activation of the Wnt/b-catenin pathway by CUX1 and GLIS1 in breast cancers. Biology Open, 3(10), 937–946.
Haridhasapavalan, K. K., Ranjan, S. H., Bhattacharyya, S., & Thummer, R. P. (2021). Soluble expression, purification, and secondary structure determination of human MESP1 transcription factor. Applied Microbiology and Biotechnology, 105(6), 2363–2376.
Francis, D. M., & Page, R. (2010). Strategies to optimize protein expression in E. coli. Current Protocol in Protein Science, 61(1), 5–24.
Acknowledgements
We thank all the members of the Laboratory for Stem Cell Engineering and Regenerative Medicine (SCERM) for their critical reading and excellent support. This work was supported by North Eastern Region—Biotechnology Program Management Cell (NERBPMC), Department of Biotechnology, Government of India (BT/PR16655/NER/95/132/2015), and also by IIT Guwahati Institutional Top-Up on Start-Up Grant.
Author information
Authors and Affiliations
Contributions
CD was responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript; VV was responsible for collection and/or assembly of data, data analysis and interpretation, and final editing and approval of the manuscript; and RPT was responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, and financial support. All the authors gave consent for publication.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (TIF 1735 kb)
Figure S1 Schematic illustration of the tagging pattern in human GLIS1 gene. Representation of GLIS1 fusion genes tagged at both N- and C-terminal end. His polyhistidine tag (8X); TAT transactivator of transcription; NLS nuclear localization signal.
Supplementary file2 (TIF 1731 kb)
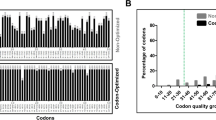
Figure S2 In silico analysis of GLIS1 gene before and after codon optimization using GRCA tool. The bar graph represents the percentage distribution of codon quality groups before (red) and after (blue) codon optimization. Codons above the threshold (green dotted line) are most likely not to interfere with the gene expression in E. coli.
Supplementary file3 (TIF 14945 kb)
Figure S3 In silico analysis of GLIS1 gene before and after codon optimization using GCUA tool. (A) Bar graph representation of the relative adaptiveness value (in %) of individual codons before (left) and after (right) codon optimization. All the 621 codons are represented for both the non-optimized and codon-optimized GLIS1 gene. Relative adaptiveness value of codon ≤ 30% is highlighted in magenta.
Supplementary file4 (TIF 4130 kb)
Figure S4 Restriction analysis of GLIS1 fusion genetic constructs. His polyhistidine tag (8X); TAT transactivator of transcription; NLS Nuclear localization signal.
Supplementary file5 (TIF 4144 kb)
Figure S5 Screening for lower inducer concentration for the expression of GLIS1-NTH fusion protein (n = 2).
Supplementary file6 (TIF 1773 kb)
Figure S6 Effect of the exogenously delivered recombinant GLIS1-NTH fusion protein on the rate of migration of BJ human fibroblast cells. Cells were seeded in 24-well culture dishes, and the respective wells were treated with protein or vehicle control for 24 h. Graphical representation of the rate of migration of protein vs. vehicle control-treated cells (p ≤ 0.05) (n = 2).
Rights and permissions
About this article

Cite this article
Dey, C., Venkatesan, V. & Thummer, R.P. Identification of Optimal Expression Parameters and Purification of a Codon-Optimized Human GLIS1 Transcription Factor from Escherichia coli. Mol Biotechnol 64, 42–56 (2022). https://doi.org/10.1007/s12033-021-00390-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-021-00390-z