
Abstract
The stem cell-specific SOX2 transcription factor is critical for early embryonic development and the maintenance of embryonic and neural stem cell identity. It is also crucial for the generation of induced pluripotent and neural stem cells, thus providing immense prospect in patient-specific therapies. Here, we report soluble expression and purification of human SOX2 protein under native conditions from a bacterial system. To generate this macromolecule, we codon-optimized the protein-coding sequence and fused it to a nuclear localization signal, a protein transduction domain, and a His-tag. This was then cloned into a protein expression vector and was expressed in Escherichia coli. Subsequently, we have screened and identified the optimal expression conditions to obtain recombinant fusion protein in a soluble form and studied its expression concerning the position of fusion tags at either terminal. Furthermore, we purified two versions of recombinant SOX2 fusion proteins to homogeneity under native conditions and demonstrated that they maintained their secondary structure. This molecular tool can substitute genetic and viral forms of SOX2 to facilitate the derivation of integration-free induced pluripotent and neural stem cells. Furthermore, it can be used in elucidating its role in stem cells, various cellular processes and diseases, and for structural and biochemical studies.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Generation of induced Pluripotent Stem Cells (iPSCs) by overexpression of reprogramming factors into somatic cells has transformed the field of regenerative medicine [1, 2]. iPSCs have tremendous potential for their applicability in understanding human developmental biology, a platform for drug discovery and toxicity screening, development of new human disease models, and an ideal source for autologous cell-based therapy [3]. The approaches used for the generation of iPSCs are divided into integration-based and integration-free techniques [4, 5]. Integration-based approaches employ γ-retro- and lenti-viral vectors that integrate into the genome for the derivation of iPSCs. However, these techniques adversely affect the developmental potential of the generated iPSCs and result in the formation of tumors [6], thus nullifying the clinical potential of the cells. The integration-free techniques involve Sendai viruses, episomal plasmids, mRNAs, microRNAs, small molecules, and recombinant proteins [4, 5, 7, 8]. These approaches overcome the risk of any genomic alteration and increase the potential of iPSCs for various biomedical applications [4, 5]. Among the integration-free techniques, the recombinant protein-based approach is considered as the safest to date [5, 7,8,9,10]. This technology involves direct transduction of biologically active proteins tagged with Protein Transduction Domains (PTDs) or Cell-Penetrating Peptides (CPPs) [5, 9]. It provides greater control over time and dosage of application and the flexibility to perform experimental variations in reprogramming factor combinations for investigating their stage-specific roles in the reprogramming process [5, 9].
To date, the major limitation of this technique is low reprogramming efficiency and slow kinetics of iPSC generation [5, 9]. This can be realized by tagging the reprogramming proteins with PTDs/CPPs to facilitate the cellular entry and Nuclear Localization Signal (NLS) for its directed translocation into the nucleus [9, 10]. Also, an ample amount of reprogramming proteins is required during the reprogramming process. This can be achieved by choosing an expression host that produces a high yield of bioactive eukaryotic proteins such as Escherichia coli (E. coli). The E. coli expression strains such as BL21(DE3) prevent proteolysis and provide stability to obtain full-length eukaryotic proteins [11, 12]. However, codon bias, inefficient expression, protein aggregation, and misfolded or partially folded protein post-purification are the major obstacles to overcome in the case of eukaryotic proteins. Codon optimization, appropriate expression vector and host strain, identification of optimal expression conditions, and purification under native conditions can address these issues [11, 12], eventually to produce bioactive proteins.
Using this protein transduction technology, many groups have generated human and mouse iPSCs from terminally differentiated somatic cells by delivering reprogramming transcription factors [13,14,15,16,17,18]. However, the major drawbacks in most of these studies were stability, the endosomal entrapment due to misfolded proteins, and perinuclear localization due to surface accumulation during nuclear entry, thereby compromising the overall reprogramming efficiency and kinetics. Alleviating these problems and generating transducible forms of reprogramming proteins is one of the primary requirements for successfully implementing protein transduction technology in iPSC generation. In this study, we aimed to produce a transducible version of recombinant human Sex determining region Y-box 2 (SOX2) protein from E. coli, which can be used for reprogramming and various biological applications.
SOX2 is a member of the SOXB1 transcription factor family and is the earliest factor of the subfamily to be expressed in mouse [19,20,21]. It plays a vital role in the embryonic development, where it is expressed in inner cell mass (the source of embryonic stem cells), epiblast, germ cells, and the multipotent cells of extraembryonic ectoderm [22]. Zygotic deletion of the gene causes early embryonic lethality due to failure in forming pluripotent epiblast cells [20, 22]. SOX2 is also expressed in adult stem and progenitor cells [20], and its major role is observed in the maintenance of neural stem cells and its subsequent differentiation into lineage-specific cell types [21, 23]. Downregulation of SOX2 showed direct implication on the self-renewal activity in both mouse and human ESCs [19, 21, 24]. Both showed a loss in maintaining pluripotency and subsequently differentiated into trophectodermal lineage. The role of SOX2 was also identified in the generation and maintenance of iPSCs in conjunction with other core reprogramming factors [1, 2]. It played a critical role in regulating the reprogramming network and assisted in the epigenetic reversal of the somatic cells into iPSCs [21]. Furthermore, the dosage of SOX2 is also reported to be crucial for efficient reprogramming [25].
Interestingly, the role of SOX2 is also implicated in multiple cancers such as human squamous cell carcinoma, osteosarcoma, glioblastoma, and melanoma [20]. SOX2+ cells are marked as a bonafide factor in identifying a potential tumor-causing cell and a SOX2-induced drug-resistant cell, where these drug-resistant cells are critical to be identified, especially after cancer therapies, including radiography and chemotherapy [26]. Mechanistically, SOX2 promotes cell proliferation and survival, metastasis through invasion, drug resistance, and cancer stemness, therefore making it a potential anti-cancer target [27, 28]. All these studies implicate the crucial function of SOX2 in various cellular processes and the panoply of diseases. Hence, the generation of recombinant human SOX2 will not only provide an opportunity to understand its stoichiometric and structural relevance with respect to the generation of integration-free human iPSCs and neural stem cells, but also help investigate its function in iPSCs, neural stem cells, and cancer cells.
In the current study, we have generated recombinant human SOX2 fusion proteins from E. coli followed by the determination of their secondary structure. This study highlights the influence in expression due to the position of tags and tackles the major experimental constraints related to heterologous protein expression and purification.
Materials and Methods
Construction of Human SOX2 Fusion Gene Constructs
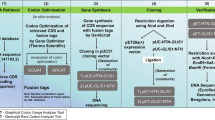
The protein-coding 951 bp full-length sequence of the SOX2 gene (NM_003106.3) was obtained from the National Center for Biotechnology Information RefSeq database. This protein-coding sequence was then codon-optimized using GeneOptimizer (Thermo Fisher Scientific) and analyzed using two independent online tools (GenScript Rare Codon Analysis and Graphical Codon Usage Analyser 2.0) as described recently [29]. Subsequently, this codon-optimized gene sequence was fused either before the start or stop codon with codon-optimized fusion tags [polyhistidine-tag (octahistidine; (H)), HIV-Trans-Activator of Transcription (TAT; (T)), and NLS; (N))] to generate HTN-SOX2 and SOX2-NTH, respectively. These customized gene inserts (HTN-SOX2 and SOX2-NTH) were procured from GenScript Biotech Corporation, China) and were cloned into the pET28a(+) expression vector (Novagen, Merck Millipore). The resulting pET28a(+) vectors harboring SOX2 fusion genes (pET28a-HTN-SOX2 or pET28a-SOX2-NTH) were analyzed using restriction digestion analysis and DNA sequencing.
Screening Gene Constructs and Expression Parameters to Achieve Soluble Expression of Recombinant Human SOX2 Protein
Gene constructs pET28a-HTN-SOX2 (hereafter, HTN-SOX2) and pET28a-SOX2-NTH (hereafter, SOX2-NTH) were transformed into E. coli BL21(DE3) strain. The transformants harboring HTN-SOX2 or SOX2-NTH gene constructs were grown as described earlier [29]. To identify the optimal induction concentration, secondary cultures were grown until the OD600 reached ~ 0.5 and then induced with different concentrations (0.05, 0.1, 0.25, and 0.5 mM) of Isopropyl β-d-1-thiogalactopyranoside (IPTG) (Sisco Research Laboratories) for 2 h at 37 °C with constant shaking. For optimizing the cell density at the time of induction, secondary cultures were induced with optimal IPTG concentration at different OD600 (~ 0.5, ~ 1.0, and ~ 1.5) for 2 h at 37 °C. The optimal post-induction incubation time was identified by inducing the cultures at the optimal cell density with the optimal inducer concentration at 37 °C with constant shaking, and samples were collected at an interval of 2, 4 and 8 h for analysis. To identify the optimal induction temperature, cultures with optimal cell density (OD600 ~ 0.5) were induced with an optimal IPTG concentration (0.25 mM) and then incubated at 37 °C (2 h) and 18 °C (36 h) with constant shaking. Post-induction, the cells were centrifuged and resuspended in cold lysis buffer [50 mM phosphate buffer (Sisco Research Laboratories), 150 mM sodium chloride (Sisco Research Laboratories), and 20 mM imidazole (Merck Millipore); pH 7.8] followed by ultrasonication using Vibra-Cell™ VCX-130 Ultrasonic Liquid Processor (Sonics and Materials Inc.) at refrigerated conditions to obtain the crude lysate. This crude lysate was separated further by centrifugation to obtain soluble supernatant and insoluble pellet fractions. The sonicated and centrifuged samples were further analyzed by Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and western blotting. Uninduced cultures were used as a negative control in all the experiments.
Immobilized Metal Ion Affinity Chromatography
For the purification of SOX2 fusion proteins, all the buffers were adjusted to pH 7.8 at room temperature and pre-chilled on ice. To purify the recombinant human SOX2 fusion proteins, we carried out immobilized metal ion affinity chromatography under native conditions. SOX2 was induced with the identified optimal expression parameters in 1.2 L culture volumes. Cells were then harvested and re-suspended in cold lysis buffer (20 ml) and ultrasonicated on ice for cell lysis and then centrifuged to obtain supernatant/soluble cell fractions. Further, the soluble cell fraction was diluted with an equal volume buffer (20 ml) of equilibration (50 mM phosphate buffer, 150 mM sodium chloride, and 20 mM imidazole) and loaded onto the purification column (charged with nickel-nitrilotriacetic acid; Bio-Rad) followed by incubation with constant shaking for ~ 2 h at 4 °C. After binding of the SOX2 fusion protein to the nickel, the unbound/bacterial proteins were discharged, and the column was washed with 20 column volumes of wash buffer 1 (50 mM phosphate buffer, 150 mM sodium chloride, 50 mM imidazole) with incubation at 4 °C for five times with incubation time of 15 min. Similarly, the column was washed sequentially with wash buffer 2 (50 mM phosphate buffer, 150 mM sodium chloride, and 100 mM imidazole) and wash buffer 3 (50 mM phosphate buffer, 150 mM sodium chloride, and 150 mM imidazole). The bound SOX2 fusion proteins were eluted with elution buffer (50 mM phosphate buffer, 150 mM sodium chloride, and 500 mM imidazole). Based on experimental design, purification samples collected at different steps were analyzed using SDS-PAGE and western blotting. Further, the purified proteins were desalted and/or buffer exchanged (as per the experimental design) using PD10 columns as per the manufacturer’s instructions (GE healthcare) against glycerol buffer [20% glycerol in 50 mM phosphate buffer (pH 7.8)] and then stored at − 80 °C until further use.
SDS-PAGE and Western Blotting
The total protein concentrations were measured using Bradford assay [30] (Bio-Rad) using bovine serum albumin (Bio-Rad) as a standard and measured with a multi-plate reader (Multiskan GO, Thermo Scientific). SDS-PAGE, coomassie staining, and western blotting were performed as described earlier [29]. The following primary [anti-His (1:5000; BioBharati, BB-AB0010) and anti-SOX2 (1:2000, Santacruz Biotechnology; sc-365823)] and secondary antibodies [1:5000; anti-rabbit IgG antibody (Invitrogen, 31460) and anti-mouse IgG-HRP Conjugated (1:5000; Invitrogen; 31430)] were used in the western blotting analysis.
Far Ultraviolet Circular Dichroism Spectroscopy
To identify the retention of secondary structure conformation of purified HTN-SOX2 and SOX2-NTH fusion proteins, far ultraviolet (UV) Circular Dichroism (CD) spectroscopy was carried out as described earlier using the same parameters [29]. Briefly, the spectra were recorded in the range of 260–190 nm of wavelength with the desalted purified protein [final concentration of 0.92 μM for HTN-SOX2 and 1.76 μM for SOX2-NTH in 50 mM phosphate buffer (pH 7.8)] using JASCO J-1500 Circular Dichroism Spectrophotometer. The final spectrum was analyzed after subtracting the background noise using an online tool, Beta Structure Selection (BeStSel) (http://bestsel.elte.hu/index.php) [31].
Results
Plasmid Construction and Gene Cloning of Codon-Optimized Human SOX2 Gene Sequence in a Protein Expression Vector
First, we performed codon optimization of the SOX2 gene sequence to remove codon bias and all the undesirable elements such as low or high GC content, codon bias, mRNA instability elements and secondary structures, cis-regulatory elements, common restriction sites, internal ribosome entry sites, intragenic poly(A) sites to obtain an increased expression of SOX2 in a heterologous system (in this study, E. coli). To accomplish this, the protein-coding sequence of the human SOX2 gene was codon-optimized using GeneOptimizer (Thermo Fisher Scientific). The sequence alignment of the non-optimized and codon-optimized SOX2 coding sequence showing the altered nucleotide substitutions are shown in Fig. 1. The quality of codon optimization of the sequence was evaluated using two independent online tools, GenScript Rare Codon Analysis (GRCA; Supplementary Fig. S1) and Graphical Codon Usage Analyser 2.0 (GCUA 2.0; Supplementary Fig. S2). Using the GRCA tool, it was found that 9% of the codons in the non-optimized sequence were having a codon usage frequency of ≤ 30%, which could hamper the expression of human SOX2 in E. coli [Supplementary Fig. S1 (in grey), Supplementary Table S1]. Using the GCUA 2.0 tool, seven codons have been found to have a relative adaptiveness value of ≤ 30% in the non-optimized sequence that might affect the expression of human SOX2 in E. coli [Supplementary Fig. S2 (left, shown by arrows)]. These codons as well as other codons that might impact the expression were replaced with the most suitable synonymous codons using the GeneOptimizer tool to enhance gene expression [Supplementary Figs. S1 (in black), S2 (right)]. Additionally, codon optimization resulted in an increased codon adaptation index value to 0.91 for the codon-optimized sequence from 0.70 of its non-optimized sequence (Supplementary Table S1). This analysis confirmed that the codon-optimized SOX2 sequence was devoid of rare codons that could affect its expression, favoring its heterologous expression in E. coli.

Comparison of non-optimized and codon-optimized SOX2 protein-coding sequence. The nucleotides highlighted in both were altered to achieve efficient expression in E. coli
This validated codon-optimized SOX2 coding sequence was fused to a set of tags, namely the Octa-His (H) tag to enable affinity chromatography-based purification, a PTD called TAT (T) for intracellular, and NLS (N) for intranuclear delivery. All the three tags were either placed before the start codon to generate HTN-SOX2 or before the stop codon to generate SOX2-NTH (Fig. 2a). The customized gene inserts (HTN-SOX2 and SOX2-NTH) obtained in pUC57 plasmid were excised using restriction endonucleases, NcoI and XhoI, and cloned into the pET28a(+) protein expression vector between NcoI and XhoI restriction sites. This gene was placed under the transcriptional control of a tightly regulated strong T7 promoter. The obtained plasmids pET28a-HTN-SOX2 (hereafter, HTN-SOX2) and pET28a-SOX2-NTH (hereafter, SOX2-NTH) were confirmed using restriction digestion analysis (Fig. 2b). The empty vector [pET28a(+)] was also taken as a control during this experiment to confirm the absence of a codon-optimized SOX2 coding sequence (data not shown). The fidelity of the cloned gene sequence was confirmed with DNA sequencing using standard T7 promoter (5′-TAATACGACTCACTATAGGG-3′) and terminator (5′-GCTAGTTATTGCTCAGCGG-3′) primers (data not shown).

Schematic representation of the SOX2 gene with fusion tags and confirmation of its cloning into the pET28a(+) expression vector. a Schematic illustrations of SOX2 fusion gene inserts [HTN-SOX2 (left) and SOX2-NTH (right); not drawn to scale]. Codon-optimized SOX2 protein-coding sequence was fused to His-tag for affinity chromatography, TAT to enable cell penetration, and NLS to facilitate nuclear translocation in mammalian cells. His Histidine (8×), TAT transactivator of transcription, NLS nuclear localization signal/sequence. b The gene inserts shown in a were cloned into a protein expression vector, pET28a(+). The resulting plasmids were then confirmed by restriction digestion using various restriction enzymes, as depicted
Identification of Gene Constructs and Optimal Expression Conditions to Achieve Maximal and Soluble Expression of Recombinant Human SOX2 Protein
Various studies have demonstrated that the identification of optimal expression parameters and specifically induction temperature is vital to attain the high and soluble expression of biologically active recombinant proteins from E. coli [32,33,34,35,36]. From these observations, various parameters such as inducer concentration (IPTG), optical density (OD), and post-induction incubation time were screened and identified for the maximal expression of SOX2 in E. coli (Table 1). The identified optimal inducer concentration, induction cell density and post-induction incubation time were 0.25 mM, ~ 0.5, and 2 h, respectively. These results indicate that higher amount of IPTG, late growth phase, or prolonged post-induction incubation time had no significant effect on the expression of SOX2 fusion proteins. Identification of these parameters was crucial to obtain maximal expression of SOX2 fusion proteins. Using these expression parameters, maximal expression of HTN-SOX2 and SOX2-NTH was observed in E. coli (Fig. 3a, b; L fraction at 37 °C). Further, to prevent protein aggregation and achieve maximal soluble expression of SOX2 fusion proteins in E. coli, the expression and solubility profiles of two gene constructs, HTN-SOX2 and SOX2-NTH at two different temperatures (37 vs. 18 °C) was investigated. The results indicated that the expression and solubility profiles of HTN-SOX2 was higher compared to SOX2-NTH when compared at the same temperature (37 or 18 °C; Fig. 3). Interestingly, more than 90% of the SOX2 protein was observed in the soluble fraction in the case of HTN-SOX2, whereas this was not observed in the case of SOX2-NTH. An uninduced sample was taken as a control to show no leaky expression of the SOX2 fusion proteins (Fig. 3). Surprisingly, few truncated fragments of SOX2 protein were observed in the case of SOX2-NTH, as shown in western blotting (Fig. 3b, bottom). This was irrespective of the various parameters and two different temperatures analyzed (Fig. 3b, bottom). A similar observation was also seen in earlier studies for a C-terminally tagged mouse SOX2 protein purified from E. coli [37, 38]. The addition of protease inhibitors did not decrease the formation of truncated fragments in SOX2-NTH (data not shown). These truncated products may be due to proteolytic cleavage at specific sites in some protein molecules during expression [35] or due to the presence of intragenic sequences mimicking E. coli ribosomal entry sites [39]. Although faint truncated fragments of SOX2 protein were observed in the case of SOX2-NTH, most of it was still retained as a full-length protein (Fig. 3). Interestingly, soluble expression was observed with both the gene constructs and at temperatures 37 and 18 °C; therefore, we chose these constructs and induction at both the temperatures for further experiments.

Soluble expression analysis of human SOX2 fusion proteins. BL21(DE3) cells were transformed with pET28a-HTN-SOX2 or pET28a-SOX2-NTH and grown until they reached OD600 ~ 0.5, followed by induction with 0.25 mM IPTG and incubated at 37 °C for 2 h and 18 °C for 36 h. Next, the cells were lysed using a lysis buffer and ultrasonication. The crude lysate (L) thus obtained was centrifuged to separate soluble supernatant fraction (S) and insoluble pellet fraction (P). Protein concentrations were measured and then normalized to 20 µg/well for L fraction, and an equal amount for P and S fractions corresponding to the respective L fraction were used for analysis. These samples were separated on 12% SDS-PAGE gels and stained with Coomassie Brilliant Blue G-250 (top), or western blotting was performed using anti-Histidine (α-His) antibody (bottom) (n = 2). *Truncations of SOX2 fusion proteins. M protein marker (kDa), UI uninduced, L crude lysate, P insoluble pellet fraction, S soluble supernatant fraction, Ab antibody
Purification of Recombinant Human SOX2 Fusion Proteins Under Native Conditions
We next sought to purify HTN-SOX2 and SOX2-NTH induced at both 37 and 18 °C using immobilized metal ion affinity chromatography under native conditions in a facile manner. This is a versatile technique employed to purify polyhistidine affinity-tagged proteins and also help accomplish a high yield of proteins with nearly 95% purity [40]. Although HTN-SOX2 and SOX2-NTH induced at 18 °C was pure, the yield of the recombinant proteins for HTN-SOX2 (0.67 mg/L) and SOX2-NTH (0.91 mg/L) was low compared to HTN-SOX2 (1.35 mg/L) and SOX2-NTH (1.52 mg/L) induced at 37 °C. Therefore, HTN-SOX2 and SOX2-NTH induced at 18 °C were excluded from further analysis. The purity of HTN-SOX2 and SOX2-NTH fusion proteins induced at 37 °C was confirmed by SDS-PAGE analysis [Fig. 4a (top), b, c (top), d], and the fusion proteins were detected by western blotting using an anti-His antibody (Fig. 4a, middle, c, middle). Further, the identity of purified SOX2 fusion proteins was confirmed using an anti-SOX2 antibody (Fig. 4a, bottom, c, bottom). The faint truncated protein fragments of SOX2 fusion proteins were also detected by both anti-His and anti-SOX2 antibody (Fig. 4a, c), indicating that these protein fragments were not of bacterial origin. Notably, most of the SOX2 fusion protein molecules were still intact (Fig. 4a, c). A band corresponding to full-length human SOX2 fusion protein was observed at ~ 40 kDa (Fig. 4). Thus, we demonstrate the homogeneous purification of recombinant SOX2 fusion proteins under native conditions from E. coli.

Generation of recombinant human SOX2 fusion proteins under native conditions using immobilized metal ion affinity chromatography. BL21(DE3) cells harboring HTN-SOX2 and SOX2-NTH were induced with 0.25 mM IPTG at OD600 ~ 0.5 and incubated at 37 °C for 2 h. Subsequently, the cells were harvested, lysed by ultrasonication, and centrifuged to obtain soluble supernatant fraction. From the supernatant fraction, the expressed fusion proteins were purified using Ni–NTA affinity chromatography under native conditions. HTN-SOX2 (a) and SOX2-NTH (c) protein samples collected during different stages of purification were separated on 12% SDS-PAGE gels with normalized loading. They were either stained with Coomassie Brilliant Blue G-250 (top), or western blotting was performed with anti-Histidine (α-His) antibody (bottom) and the anti-SOX2 antibody (n = 4). b, d Elution fractions (E1–4) resolved on 12% SDS-PAGE gels and stained with Coomassie Brilliant Blue G-250 of HTN-SOX2 and SOX2-NTH, respectively. The arrow (←) indicates HTN-SOX2 and SOX2-NTH protein, whereas the asterisk (*) indicates truncations of the SOX2 fusion protein. M protein marker (kDa), L crude lysate, S soluble/supernatant fraction, FT flow-through fraction, W1 wash buffer 1 fraction, W2 Wash buffer 2 fraction, W3 wash buffer 3 fraction, E eluted fraction, Ab antibody
Determination of the Secondary Structure of Purified Recombinant SOX2 Fusion Proteins
To the best of our knowledge, neither the crystal structure of full-length human SOX2 protein nor its secondary structure content has been reported to date. Therefore, we studied its secondary structure content using far UV CD spectroscopy. CD is a widely performed technique for the estimation of the secondary structure content and folding characteristics/conformation of purified proteins [41]. The characteristic CD spectrum indicates different secondary conformations, namely α-helix, β-sheet, turn, and random coil [41, 42]. The CD spectrum representing α-helix displays the negative peaks at 222 and 208 nm and a positive peak of about 193 nm [41]. Likewise, the distinct antiparallel β-pleated sheets (β-sheets) have a negative peak at 218 nm and a positive peak at 195 nm, while the disordered proteins containing random coil have a positive peak above 210 nm and a negative peak near 195 nm [41]. To estimate the secondary structure content of purified HTN-SOX2 and SOX2-NTH fusion proteins (induced at 37 °C), far UV CD spectroscopic analysis was performed. First, desalting and buffer exchange of the fusion proteins was carried out to remove salt and imidazole as this might interfere with the analysis. Subsequently, the fusion proteins were subjected to far UV CD spectroscopic analysis. The obtained far UV CD data were further quantified and analyzed using a recently developed online tool, Beta Structure Selection (BeStSel) [31]. It is a free web server developed to analyze the CD spectra recorded by CD spectrophotometer for prompt and reliable prediction of the secondary structure content of proteins [31]. The CD spectra (plotted using BeStSel result) of recombinant HTN-SOX2 protein shows that its secondary structure comprised 10% α-helices, 28% β-sheets, 16% turns, and 46% random coils (Fig. 5a, b). The CD spectrum and secondary structure content values for SOX2-NTH were also very similar: 11% α-helices, 28% β-sheets, 15% turns, and 46% random coils (Fig. 5a, b). Notably, these results established that the purified fusion proteins majorly comprised of random coils and β-sheets and a good proportion of turns and α-helices. This data established that the recombinant SOX2 fusion proteins had maintained their secondary structure, and they show great promise of being bioactive.

Determination of the secondary structure of human SOX2 fusion proteins. The secondary structure was determined using far UV CD spectroscopy for the purified recombinant HTN-SOX2 and SOX2-NTH fusion proteins in 50 mM phosphate buffer at pH 7.8. The CD data obtained were then evaluated using BeStSel online tool. From the results, CD spectra have been plotted with wavelength (nm) against Delta Epsilon (M−1cm−1) for purified HTN-SOX2 (dotted line) and SOX2-NTH (bold line) recombinant proteins as shown in a. The bar graph in b shows the percentage of secondary structures [α-helix, β-sheet, turn, and other (random coil)] for purified HTN-SOX2 and SOX2-NTH recombinant proteins (n = 4)
Discussion
In this study, we report the generation of highly pure recombinant human SOX2 fusion proteins, one of the critical transcription factors in embryonic development, stem cell identity, and iPSC and induced neural stem cell production. The generation of a transducible version of SOX2 protein is critical for overcoming the limitations associated with viral- and plasmid-based delivery systems [37, 43]. Viral-based gene delivery systems integrate into the genome, whereas plasmid-based systems are less efficient and screening of iPSC clones is more cumbersome; therefore, these approaches are not the most suitable for regenerative medicine [4, 5, 7, 8].
First, codon optimization of the protein-coding sequence of the human SOX2 gene was performed. Numerous studies have reported that codon optimization is critical for achieving enhanced expression of human genes in bacterial systems [29, 39, 44], which is a powerful tool to improve protein expression by altering the coding sequence of a gene of interest to make codon usage match the accessible tRNA pool within the desired host [44]. Various undesirable factors/elements such as codon bias, low or high GC content, mRNA instability elements and secondary structures, cis-regulatory elements, internal ribosome entry sites, intragenic poly(A) sites, and common restriction sites are eliminated while performing codon optimization [39]. Moreover, the codon optimization approach had a more positive impact than the tRNA overexpression strategy on heterologous (human) gene expression in E. coli [39, 44]. Based on these studies, as well as our in silico analysis, we have codon-optimized the coding sequence of the human SOX2 gene to achieve its enhanced heterologous expression in E. coli. Similar to our recent in silico analysis of the human ETS2 and PDX1 gene sequence [29, 45] we observed an increase in codon adaptation index value due to codon optimization, which further confirmed that the codon-optimized SOX2 sequence is devoid of rare codons that would interfere in its heterologous expression. Thus, codon optimization improved gene expression and translational efficiency giving a high expression of human SOX2 protein in E. coli.
The codon-optimized SOX2 gene was tagged with a set of tags at either terminal to generate two different SOX2 fusion gene constructs. This strategy enabled the determination of gene constructs with fusion tags that had no adverse effect on protein expression and purification. Earlier studies have reported that the position of fusion tags at either end of the terminal can influence expression, solubility, stability, folding, and functionality of recombinant protein undergoing heterologous expression in bacteria [29, 37, 46]. The expression and solubility profile analysis in this study clearly indicates that the placement of fusion tags at different terminals does influence the expression and solubility of the SOX2 protein. Notably, although variable, we could obtain soluble expression with both the genetic constructs at different temperatures, unlike in our previous study with ETS2 protein [29].
In the current study, E. coli was chosen as an expression host as it is the most preferred choice for heterologous protein production. This is mainly due to its fast growth, ease of handling, inexpensive media, well-characterized genetics, high expression level, and availability of versatile host strains and vector systems [39]. Moreover, this host is the most widely used for the production of recombinant human proteins for which post-translational modifications are not critical for their biological activity [47,48,49,50]. Specifically, the protease-deficient BL21(DE3) strain of E. coli offers the benefit of inducible protein expression and improved stability of expressed proteins. However, heterologous expression of human proteins in E. coli generally cannot retain its native conformation due to misfolding or failure in interacting with folding modifiers, thus resulting in insoluble aggregates, which are referred to as inclusion bodies [51]. These human proteins are biologically inactive and require extra denaturation-renaturation steps to restore biological activity [52, 53]. Therefore, an ideal approach is to obtain the soluble expression of the protein of interest in E. coli that retains its native folding. In this study, we have screened and identified optimal expression conditions to obtain soluble expression of SOX2 fusion proteins, which allowed us to purify it under native conditions. The recombinant fusion proteins are highly pure, and both these proteins have retained their secondary structure post-purification.
Various studies have reported the purification of human SOX2 protein from E. coli [38, 54,55,56,57]. However, all these studies have reported purification from inclusion bodies, therefore making the solubilization of the purified protein dependent on the use of harsh detergents, making refolding tedious and time-consuming, and commonly ends with a low yield of biologically active protein in its native conformation [58]. Although the purified SOX2 protein was biologically active in these studies [38, 54,55,56,57], this might have a profound effect on the structural integrity, ultimately compromising its biological activity. Moreover, the refolding procedure of purified proteins from inclusion bodies enhances protein aggregation and therefore results in poor recovery of biologically active protein [59]. Thus, purification under native conditions is a requisite. To the best of our knowledge, this is the first study to report the purification of human SOX2 protein from E. coli under native conditions in a facile manner. Although fusion with 30Kc19 peptide was reported to promote the solubility of SOX2 protein [60], this study has shown a more pronounced solubility and purity of SOX2 as compared to the previous report, without any bacterial protein contamination. The presence of bacterial protein contaminants in the final purified fractions could have deleterious effects on the transduced mammalian cells [61]. Moreover, the secondary structure post-purification was retained and comprised random coils and β-sheets and also constituted of turns and α-helices. To the best of our knowledge, this is the first study to report the secondary structure content of human SOX2 protein. Although the expression and solubility profiles varied based on the placement of fusion tags in the two genetic constructs, it did not alter the secondary structure of purified SOX2 protein. Prospectively, this purified SOX2 fusion proteins can be delivered into mammalian cells for various biological applications.
Moreover, the fusion of His-tag enabled affinity chromatography-mediated purification of SOX2 protein, and TAT and NLS will facilitate subcellular and subnuclear delivery, respectively. Previous studies have also used such strategies to enable intracellular and intranuclear delivery of mouse versions of stem cell-specific recombinant proteins SOX2, OCT4 and NANOG in mammalian cells [37, 43, 62,63,64]. These studies have also corroborated the fact that the fusion of both TAT and NLS can facilitate efficient translocation of mouse stem cell-specific transcription factors to the nucleus [5, 9, 37, 43, 62,63,64], and lack of NLS has shown inefficient translocation of SOX2 protein into the nucleus as observed in the microscopy images [38, 54]. Notably, these studies have also demonstrated that the presence of these fusion tags after the delivery of the protein of interest into the cells (via TAT) and nucleus (via NLS) did not affect biological activity of the protein of interest.
Transducible versions of mouse stem cell-specific transcription factors were able to substitute their viral and genetic counterparts [37, 43, 62,63,64] and generate integration-free and virus-free iPSCs by employing reprogramming proteins without any genetic manipulation [13,14,15,16,17,18]. This study successfully generated a purified SOX2 protein and is presumably the first to report its secondary structure and potential to transduce into mammalian cells to exert its biological activity. Generation of this biological tool will also be useful in the generation of integration-free induced neural stem cells [65], induced pluripotent mesenchymal stem cells [57], induced dopaminergic neural progenitor-like cells [55], and oligodendrocyte-like cells [56] from human fibroblasts, along with opening plethora of opportunities to investigating its structural, biochemical, cellular, and molecular functions in diverse cellular processes and diseases.
References
Takahashi, K., & Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126, 663–676.
Yu, J., Vodyanik, M. A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science, 318, 1917–1920.
Rowe, R. G., & Daley, G. Q. (2019). Induced pluripotent stem cells in disease modelling and drug discovery. Nature Reviews Genetics, 20, 377–388.
Haridhasapavalan, K. K., Borgohain, M. P., Dey, C., Saha, B., Narayan, G., Kumar, S., & Thummer, R. P. (2019). An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene, 686, 146–159.
Borgohain, M. P., Haridhasapavalan, K. K., Dey, C., Adhikari, P., & Thummer, R. P. (2019). An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Reviews and Reports, 15, 286–313.
Stadtfeld, M., & Hochedlinger, K. (2010). Induced pluripotency: History, mechanisms, and applications. Genes and Development, 24, 2239–2263.
Sommer, C. A., & Mostoslavsky, G. (2013). The evolving field of induced pluripotency: Recent progress and future challenges. Journal of Cellular Physiology, 228, 267–275.
O’Malley, J., Woltjen, K., & Kaji, K. (2009). New strategies to generate induced pluripotent stem cells. Current Opinion in Biotechnology, 20, 516–521.
Dey, C., Narayan, G., Krishna Kumar, H., Borgohain, M. P., Lenka, N., & Thummer, R. P. (2017). Cell-penetrating peptides as a tool to deliver biologically active recombinant proteins to generate transgene-free induced pluripotent stem cells. Studies on Stem Cell Research and Therapy, 3, 6–15.
Seo, B. J., Hong, Y. J., & Do, J. T. (2017). Cellular reprogramming using protein and cell-penetrating peptides. International Journal of Molecular Medicine, 18, 552.
Kaur, J., Kumar, A., & Kaur, J. (2018). Strategies for optimization of heterologous protein expression in E. coli: Roadblocks and reinforcements. Internaional Journal of Biology and Macromolecules, 106, 803–822.
Borgohain, M. P., Narayan, G., Kumar, H. K., Dey, C., & Thummer, R. P. (2018). Maximizing expression and yield of human recombinant proteins from bacterial cell factories for biomedical applications. In P. Kumar, J. K. Patra, & P. Chandra (Eds.), Advances in microbial biotechnology (pp. 447–486). Burlington: Apple Academic Press.
Cho, H.-J., Lee, C.-S., Kwon, Y.-W., Paek, J. S., Lee, S.-H., Hur, J., et al. (2010). Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood, 116, 386–395.
Zhou, H., Wu, S., Joo, J. Y., Zhu, S., Han, D. W., Lin, T., et al. (2009). Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell, 4, 381–384.
Zhang, H., Ma, Y., Gu, J., Liao, B., Li, J., Wong, J., & Jin, Y. (2012). Reprogramming of somatic cells via TAT-mediated protein transduction of recombinant factors. Biomaterials, 33, 5047–5055.
Nemes, C., Varga, E., Polgar, Z., Klincumhom, N., Pirity, M. K., & Dinnyes, A. (2014). Generation of mouse induced pluripotent stem cells by protein transduction. Tissue Engineering Part C: Methods, 20, 383–392.
Lee, J., Sayed, N., Hunter, A., Au, K. F., Wong, W. H., Mocarski, E. S., et al. (2012). Activation of innate immunity is required for efficient nuclear reprogramming. Cell, 151, 547–558.
Khan, M., Narayanan, K., Lu, H., Choo, Y., Du, C., Wiradharma, N., et al. (2013). Delivery of reprogramming factors into fibroblasts for generation of non-genetic induced pluripotent stem cells using a cationic bolaamphiphile as a non-viral vector. Biomaterials, 34, 5336–5343.
Masui, S., Nakatake, Y., Toyooka, Y., Shimosato, D., Yagi, R., Takahashi, K., et al. (2007). Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature Cell Biology, 9, 625–635.
Sarkar, A., & Hochedlinger, K. (2013). The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell, 12, 15–30.
Zhang, S., & Cui, W. (2014). Sox2, a key factor in the regulation of pluripotency and neural differentiation. World Journal of Stem Cells, 6, 305–311.
Avilion, A. A., Nicolis, S. K., Pevny, L. H., Perez, L., Vivian, N., & Lovell-Badge, R. (2003). Multipotent cell lineages in early mouse development depend on SOX2 function. Genes and Development, 17, 126–140.
Pevny, L. H., & Nicolis, S. K. (2010). Sox2 roles in neural stem cells. International Journal of Biochemistry and Cell Biology, 42, 421–424.
Adachi, K., Suemori, H., Yasuda, S., Nakatsuji, N., & Kawase, E. (2010). Role of SOX2 in maintaining pluripotency of human embryonic stem cells. Genes to Cells, 15, 455–470.
Haridhasapavalan, K. K., Raina, K., Dey, C., Adhikari, P., & Thummer, R. P. (2020). An insight into reprogramming barriers to iPSC generation. Stem Cells Reviews and Reports, 16, 56–81.
Chuang, H.-M., Huang, M.-H., Chen, Y.-S., & Harn, H.-J. (2020). SOX2 for stem cell therapy and medical use: Pros or cons? Cell Transplantation, 29, 0963689720907565.
Wuebben, E. L., & Rizzino, A. (2017). The dark side of SOX2: cancer-a comprehensive overview. Oncotarget, 8, 44917–44943.
Zhang, S., Xiong, X., & Sun, Y. (2020). Functional characterization of SOX2 as an anticancer target. Signal Transduction and Targeted Therapy, 5, 1–17.
Haridhasapavalan, K. K., Sundaravadivelu, P. K., & Thummer, R. P. (2020). Codon optimization, cloning, expression, purification, and secondary structure determination of human ETS2 transcription factor. Molecular Biotechnology, 62, 485–494.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Micsonai, A., Wien, F., Bulyáki, É., Kun, J., Moussong, É., Lee, Y.-H., et al. (2018). BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Research, 46, W315–W322.
Vasina, J. A., & Baneyx, F. (1997). Expression of aggregation-prone recombinant proteins at low temperatures: A comparative study of the Escherichia coli cspAandtacPromoter Systems. Protein Expression and Purification, 9, 211–218.
Sørensen, H. P., & Mortensen, K. K. (2005). Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microbial Cell Factories, 4, 1.
San-Miguel, T., Pérez-Bermúdez, P., & Gavidia, I. (2013). Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus, 2, 89.
Ryan, B. J., & Henehan, G. T. (2013). Overview of approaches to preventing and avoiding proteolysis during expression and purification of proteins. Current Protocols in Protein Science, 71, 5–25.
Huang, C.-J., Lin, H., & Yang, X. (2012). Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. Journal of Industrial Microbiology and Biotechnology, 39, 383–399.
Bosnali, M., & Edenhofer, F. (2008). Generation of transducible versions of transcription factors Oct4 and Sox2. Biological Chemistry, 389, 851–861.
Pan, C., Lu, B., Chen, H., & Bishop, C. E. (2010). Reprogramming human fibroblasts using HIV-1 TAT recombinant proteins OCT4, SOX2, KLF4 and c-MYC. Molecular Biology Reports, 37, 2117–2124.
Maertens, B., Spriestersbach, A., von Groll, U., Roth, U., Kubicek, J., Gerrits, M., et al. (2010). Gene optimization mechanisms: A multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Science, 19, 1312–1326.
Bornhorst, J. A., & Falke, J. J. (2000). Purification of proteins using polyhistidine affinity tags. In J. Thorner, S. D. Emr, & J. N. Abelson (Eds.), Methods in enzymology (Vol. 326, pp. 245–254). Amsterdam: Elsevier.
Greenfield, N. J. (2006). Using circular dichroism spectra to estimate protein secondary structure. Nature Protocol, 1, 2876–2890.
Kelly, S. M., Jess, T. J., & Price, N. C. (2005). How to study proteins by circular dichroism. Biochimica et Biophysica Acta (BBA): Proteins and Proteomics, 1751, 119–139.
Thier, M., Münst, B., Mielke, S., & Edenhofer, F. (2012). Cellular reprogramming employing recombinant sox2 protein. Stem Cells International, 2012, 549846.
Burgess-Brown, N. A., Sharma, S., Sobott, F., Loenarz, C., Oppermann, U., & Gileadi, O. (2008). Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expression and Purification, 59, 94–102.
Narayan, G., Sundaravadivelu, P. K., Agrawal, A., Gogoi, R., Nagotu, S., & Thummer, R. P. (2020). Soluble expression, purification, and secondary structure determination of human PDX1 transcription factor. Protein Expression and Purification, 180, 105807.
Braun, P., Hu, Y., Shen, B., Halleck, A., Koundinya, M., Harlow, E., & LaBaer, J. (2002). Proteome-scale purification of human proteins from bacteria. Proceedings of the National Academy of Sciences of the United States of America, 99, 2654–2659.
Ghasemi, Y., Ghoshoon, M. B., Taheri, M., Negahdaripour, M., & Nouri, F. (2020). Cloning, expression and purification of human PDGF-BB gene in Escherichia coli: New approach in PDGF-BB protein production. Gene Reports, 19, 100653.
Galluccio, M., Amelio, L., Scalise, M., Pochini, L., Boles, E., & Indiveri, C. (2012). Over-expression in E. coli and purification of the human OCTN2 transport protein. Molecular Biotechnology, 50, 1–7.
Bhat, E. A., Sajjad, N., Sabir, J. S. M., Kamli, M. R., Hakeem, K. R., Rather, I. A., & Bahieldin, A. (2020). Molecular cloning, expression, overproduction and characterization of human TRAIP Leucine zipper protein. Saudi Journal of Biological Sciences, 27, 1562–1565.
Zamani, M., Berenjian, A., Hemmati, S., Nezafat, N., Ghoshoon, M. B., Dabbagh, F., et al. (2015). Cloning, expression, and purification of a synthetic human growth hormone in Escherichia coli using response surface methodology. Molecular Biotechnology, 57, 241–250.
Baneyx, F., & Mujacic, M. (2004). Recombinant protein folding and misfolding in Escherichia coli. Nature Biotechnology, 22, 1399–1408.
Rosano, G. L., & Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: Advances and challenges. Frontiers in Microbiology, 5, 172.
Vincentelli, R., & Romier, C. (2013). Expression in Escherichia coli: becoming faster and more complex. Current Opinion in Biotechnology, 23, 326–334.
Hu, P. F., Guan, W. J., Li, X. C., & Ma, Y. H. (2012). Construction of recombinant proteins for reprogramming of endangered Luxi cattle fibroblast cells. Molecular Biology Reports, 39, 7175–7182.
Mirakhori, F., Zeynali, B., Rassouli, H., Salekdeh, G. H., & Baharvand, H. (2015). Direct conversion of human fibroblasts into dopaminergic neural progenitor-like cells using TAT-mediated protein transduction of recombinant factors. Biochemical and Biophysical Research Communications, 459, 655–661.
Pouya, A., Rassouli, H., Rezaei-Larijani, M., Salekdeh, G. H., & Baharvand, H. (2020). SOX2 protein transduction directly converts human fibroblasts into oligodendrocyte-like cells. Biochemical and Biophysical Research Communications, 525, 1–7.
Chen, F., Zhang, G., Yu, L., Feng, Y., Li, X., Zhang, Z., et al. (2016). High-efficiency generation of induced pluripotent mesenchymal stem cells from human dermal fibroblasts using recombinant proteins. Stem Cell Research and Therapy, 7, 99.
Tsumoto, K., Ejima, D., Kumagai, I., & Arakawa, T. (2003). Practical considerations in refolding proteins from inclusion bodies. Protein Expression and Purification, 28, 1–8.
Burgess, R. R. (2009). Refolding solubilized inclusion body proteins. In R. R. Burges & M. P. Deutscher (Eds.), Methods in enzymology (Vol. 463, pp. 259–282). Amsterdam: Elsevier.
Ryu, J., Park, H. H., Park, J. H., Lee, H. J., Rhee, W. J., & Park, T. H. (2016). Soluble expression and stability enhancement of transcription factors using 30Kc19 cell-penetrating protein. Applied Microbiology and Biotechnology, 100, 3523–3532.
Araki, Y., Hamafuji, T., Noguchi, C., & Shimizu, N. (2012). Efficient recombinant production in mammalian cells using a novel IR/MAR gene amplification method. PLoS ONE, 7, e41787.
Münst, B., Thier, M. C., Winnemöller, D., Helfen, M., Thummer, R. P., & Edenhofer, F. (2016). Nanog induces suppression of senescence through downregulation of p27KIP1 expression. Journal of Cell Science, 129, 912–920.
Peitz, M., Münst, B., Thummer, R. P., Helfen, M., & Edenhofer, F. (2014). Cell-permeant recombinant Nanog protein promotes pluripotency by inhibiting endodermal specification. Stem Cell Research, 12, 680–689.
Thier, M., Münst, B., & Edenhofer, F. (2010). Exploring refined conditions for reprograming cells by recombinant Oct4 protein. International Journal of Developmental Biology, 54, 1713.
Ring, K. L., Tong, L. M., Balestra, M. E., Javier, R., Andrews-Zwilling, Y., Li, G., et al. (2012). Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell, 11, 100–109.
Acknowledgments
We thank all the members of the Laboratory for Stem Cell Engineering and Regenerative Medicine (SCERM) for their critical reading and excellent support. The authors gratefully acknowledge the support of DBT Program Support (Prof. S.S. Ghosh), Department of Biosciences and Bioengineering, IIT Guwahati, for their assistance in Circular Dichroism experiments. This work was supported by North Eastern Region—Biotechnology Programme Management Cell (NERBPMC), Department of Biotechnology, Government of India (BT/PR16655/NER/95/132/2015), and also by IIT Guwahati Institutional Top-Up on Start-Up Grant.
Author information
Authors and Affiliations
Contributions
MT and CD were responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript; SB was responsible for collection and/or assembly of data, data analysis and interpretation, and final editing and approval of the manuscript; SS was responsible for conception and design, data analysis and interpretation, final approval of manuscript and financial support; and RPT was responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript and financial support.
Corresponding author
Ethics declarations
Conflict of Interest
All authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
All the authors gave consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12033_2021_305_MOESM1_ESM.tif
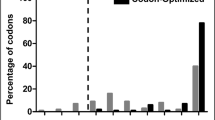
Supplementary file1 (TIF 14273 KB) Fig. S1 Evaluation of human SOX2 protein-coding sequence before (non-optimized) and after (codon-optimized) codon optimization using the GRCA tool. The graph shows the percentage distribution of codons that belong to a specific quality group. Codons with the most frequently used codon for a given amino acid in E. coli is assigned a value of 100. The codons which are distributed in codon quality groups having value ≤ 30 are used less frequently by that organism (threshold is shown as a dotted line) and are more likely to impede the expression. The presence of grey bars (i.e. non-optimized) with values ≤ 30 in the graph signified that it had codons that could affect its expression in E. coli. The absence of black bars (i.e. codon-optimized) with values ≤ 30 in the graph signified that it lacked codons that could affect its expression in E. coli.
12033_2021_305_MOESM2_ESM.tif
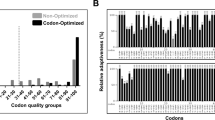
Supplementary file2 (TIF 2228 KB) Fig. S2 Evaluation of human SOX2 protein-coding sequence before (non-optimized) and after (codon-optimized) codon optimization using the GCUA 2.0 tool. The graph shows the codons of non-optimized and codon-optimized SOX2 coding sequence against the relative adaptiveness value. The relative adaptiveness value is based on the number of codons available concerning the organism for that particular amino acid. Therefore, for a given codon, relative adaptiveness value is determined by computing the multiplication of 100 with its usage frequency to the frequently used codon usage frequency. Codons with a relative adaptiveness value ≤ 30% (shown by arrows in black) are very likely to impede the expression in E. coli. The absence of grey bars in the codon-optimized graph shows that it was devoid of inhibitory codons.
Rights and permissions
About this article

Cite this article
Thool, M., Dey, C., Bhattacharyya, S. et al. Generation of a Recombinant Stem Cell-Specific Human SOX2 Protein from Escherichia coli Under Native Conditions. Mol Biotechnol 63, 327–338 (2021). https://doi.org/10.1007/s12033-021-00305-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-021-00305-y