
Abstract
Transcription factor GATA4 is expressed during early embryogenesis and is vital for proper development. In addition, it is a crucial reprogramming factor for deriving functional cardiomyocytes and was recently identified as a tumor suppressor protein in various cancers. To generate a safe and effective molecular tool that can potentially be used in a cell reprogramming process and as an anti-cancer agent, we have identified optimal expression parameters to obtain soluble expression of human GATA4 in E. coli and purified the same to homogeneity under native conditions using immobilized metal ion affinity chromatography. The identity of GATA4 protein was confirmed using western blotting and mass spectrometry. Using circular dichroism spectroscopy, it was demonstrated that the purified recombinant protein has maintained its secondary structure, primarily comprising of random coils and α-helices. Subsequently, this purified recombinant protein was applied to human cells and was found that it was non-toxic and able to enter the cells as well as translocate to the nucleus. Prospectively, this cell- and nuclear-permeant molecular tool is suitable for cell reprogramming experiments and can be a safe and effective therapeutic agent for cancer therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
GATA binding protein 4 (GATA4) belongs to the family of GATA transcription factors that has a pair of highly conserved zinc-finger domains, which recognizes the core WGATAR (W: A/T; R: A/G) motif present in the promoters and cis-regulatory elements of numerous target genes [1]. During mouse embryogenesis, GATA4 is expressed in the primitive endoderm (hypoblast), extra-embryonic endoderm, and in cells associated with cardiac and gonadal development [1, 2]. In addition, it is also crucial for the induction of definitive endoderm, and therefore essential for the formation of endodermal lineages [3]. The deletion of the Gata4 gene in mice resulted in early embryonic lethality with severe developmental defects [4,5,6]. These mice failed to form a proper heart tube and showed defects in ventral morphogenesis [4, 5], indicating that it is a critical factor during the early stage of embryonic development. It is also a crucial factor for the survival and proliferation of both embryo and adult cardiomyocytes and vital for proper cardiac development [6], as its loss perturbed cardiomyocyte formation and resulted in acardia in mice [7]. Besides, mutations in the human GATA4 gene showed cardiac septal defects and dilated cardiomyopathy [8]. Recently, the role of GATA4 has been implicated in the development of the heart, neural crest and craniofacial skeleton (bone and teeth) in mice [9]. All these studies implicate GATA4 as a crucial factor for proper endoderm, gonadal, neural crest, and cardiac development.
In addition, various studies have identified GATA4 as a core-reprogramming factor in the cocktail of transcription factors to generate functional cardiomyocytes from mouse and human fibroblasts using different gene delivery approaches [10, 11]. These studies have demonstrated that GATA4 is an indispensable cardiac reprogramming factor for reprogramming non-myocyte cells to derive functional cardiomyocytes. Transplantation of functional cardiomyocytes to replenish the lost or replace the non-functional cardiomyocytes in diseased heart patients could potentially support normal physiological functions and facilitate heart regeneration [12]. Notably, GATA4 was identified as a tumor suppressor protein in different types of cancers [13,14,15,16]. This novel function of GATA4 has improved its prospects to be used as a potential anti-cancer agent for the treatment of various cancers. In the current study, we aimed to produce pure GATA4 recombinant protein, which can further be used for cell reprogramming, as an anti-cancer agent, and for various biological applications.
The production of recombinant proteins has revolutionized the biotechnology field. So far, numerous biologically active recombinant proteins have reached the market in a short period for various applications, such as therapeutics, diagnostics, research, and to comprehend fundamental biological questions [17]. Recombinant proteins are considered to be one of the safest approaches for cell reprogramming and cancer therapy, where the protein of interest is delivered into the target cell to perform the desired function [18,19,20,21,22,23]. In addition, recombinant proteins allow precise control over time and dosage of application of the protein of interest into the target cell, which will help researchers to identify its biological role [20]. Importantly, the recombinant protein-based approach does not modify or alter the genome of the target cells, which is ideal for generating genetically stable cells for regenerative medicine [18, 20, 22]. In general, these human therapeutic proteins are produced in the recombinant form either from prokaryotic or eukaryotic expression systems [20]. Commonly, the bacterial system is chosen to produce human recombinant proteins in large quantities with simple purification techniques in a cost-effective manner [17, 24]. However, generating a highly pure and stable recombinant protein remains challenging due to codon bias, gene product toxicity, mRNA stability, low protein expression, protein degradation by host cell proteases, and in vitro solubility and stability [17, 25]. In this study, we aim to address these bottlenecks associated with heterologous expression in the production of recombinant human GATA4 protein, which can be transduced into mammalian cells for various biological applications.
Recently, we have reported the heterologous expression and purification of recombinant human ETS2 and PDX1 proteins from Escherichia coli (E. coli) [26, 27]. In the present study, we report codon optimization, cloning, expression, and purification of human GATA4 from E. coli, followed by the determination of its secondary structure. This is the first study to report screening and identification of optimal expression parameters in E. coli to express and purify a highly pure human GATA4 under native conditions and giving an insight into its specific secondary structure content. Further, we demonstrate the ability of the purified protein to enter the cell and translocate to the nucleus of human cells.
Materials and methods
Gene constructs
The pET28a(+)-HTN-GATA4 and pET28a(+)-GATA4-NTH gene constructs: the full length, 1326 bp coding sequence of human GATA4, was obtained from the NCBI reference sequence (RefSeq) database (NM_002052). This sequence was codon-optimized for the heterologous expression in E. coli using ThermoFisher Scientific GeneOptimizer online tool (https://www.thermofisher.com/in/en/home/life-science/cloning/gene-synthesis/geneart-gene-synthesis/geneoptimizer.html). The codon-optimized sequence was validated using Graphical Codon Usage Analyser 2.0 (http://gcua.schoedl.de) and Genscript Rare Codon Analysis (https://www.genscript.com/tools/rare-codon-analysis) online tools. Subsequently, the codon-optimized sequence was tagged with codon-optimized nucleotide sequences of polyhistidine-tag [His; (H); octahistidine], HIV-Trans-Activator of Transcription [TAT (T); a short peptide sequence and also called a cell-penetrating peptide], and nuclear localization signal/sequence [NLS (N)]. All of these tags were flanked by restriction sites for easy removal of any individual component that might affect the protein functionality. This customized HTN-GATA4 and GATA4-NTH inserts were then gene synthesized with NcoI (at 5′ end) and XhoI (at 3′ end) restriction sites and obtained from GenScript Biotech Corporation, Nanjing, China. Both the gene inserts were then cloned into the protein expression vector pET28a(+) (Novagen, Merck Millipore) using restriction endonucleases NcoI and XhoI (Fermentas). The resulting plasmids, pET28a(+)-HTN-GATA4 and pET28a(+)-GATA4-NTH, were verified by restriction digestion analysis and then by DNA sequencing (Eurofins Genomics India Pvt. Ltd. Bengaluru, Karnataka, India).
Identification of optimal expression conditions for heterologous expression of GATA4 fusion proteins in E. coli
To identify various parameters such as inducer concentration, optical density (OD), induction time, and induction temperature, E. coli BL21(DE3) host cells were transformed with pET28a(+)-GATA4-NTH plasmid and grown overnight in Luria–Bertani broth (HiMedia) supplemented with 50 μg/ml of kanamycin (HiMedia) and used as an inoculum for the protein expression analysis. For optimizing inducer concentration, secondary cultures were incubated at 37 °C with continuous shaking at 180 rpm (Orbital incubator shaker, IKON instruments, India) until the OD600 reached ~ 0.5. Cultures were then induced with different inducer concentrations (0.05, 0.1, 0.25, and 0.5 mM) of Isopropyl β-d-1-thiogalactopyranoside (IPTG) (HiMedia) and incubated for the next 2 h at 37 °C. In order to identify the optimal OD of E. coli BL21(DE3) host cells at the time of induction, secondary cultures were induced at different OD600 (~ 0.5, ~ 1.0, ~ 1.5) with optimal IPTG concentration (0.25 mM) for 2 h at 37 °C with continuous shaking at 180 rpm. The post-induction incubation time was optimized by inducing the cultures with the optimal inducer concentration (0.25 mM) at the optimal cell density (~ 0.5 OD) for 8 h at 37 °C with continuous shaking at 180 rpm, and samples were collected every two hours for analysis. For identifying the optimal induction temperature, the secondary cultures with optimal cell density (~ 0.5 OD) were induced at different temperatures (37 °C for 2 h, 30 °C for 12 h, 25 °C for 18 h, and 18 °C for 24 h with continuous shaking at 180 rpm) with an optimal concentration (0.25 mM) of IPTG. In all the above experiments to identify optimal expression parameters, un-induced cultures were used as a control. After the induction, cells were harvested by centrifugation and resuspended in pre-chilled lysis buffer (pH 8.0) containing 20 mM phosphate buffer (PB) (HiMedia), 300 mM sodium chloride (HiMedia), 20% glycerol (Merck Millipore), and 20 mM imidazole (Merck Millipore), and then lysed by ultrasonication using Vibracell™ VCX-130 cell disruptor (Sonics and Materials Inc., Newtown, CT, USA) on ice. Samples were then resolved and analyzed using 12% Sodium Dodecyl Sulfate‒Polyacrylamide Gel Electrophoresis (SDS-PAGE) and western blotting to observe the effect of inducer concentration, OD, induction time, and induction temperature on the expression of the fusion protein.
Purification of recombinant GATA4 fusion protein
Immobilized metal ion affinity chromatography (IMAC) was performed to obtain purified recombinant GATA4 fusion proteins (HTN-GATA4 and GATA4-NTH). Briefly, HTN-GATA4 and GATA4-NTH expression were induced in large culture volumes (1.2 L) with the identified optimal expression conditions (temperature: 37 °C; cell density: OD600 = ~ 0.5; IPTG concentration: 0.25 mM; induction time: 2 h). The harvested cell pellets were resuspended in pre-chilled lysis buffer and further lysed by ultrasonication using Vibracell™ VCX-130 cell disruptor (Sonics and Materials Inc., Newtown, CT, USA) on ice. The cell suspension was clarified by centrifugation at 11,000 rpm, 4 °C for 30 min.
Purification under native conditions
The clarified soluble fraction was diluted in a 1:1 ratio with equilibration buffer (20 mM PB, 300 mM NaCl, 20% glycerol, and 20 mM imidazole) prior to purification. Subsequently, the nickel-nitrilotriacetic acid (Ni–NTA) charged purification column (Bio-Rad) was equilibrated with equilibration buffer, and then the diluted soluble fraction was loaded onto the column and incubated with continuous shaking at 4 °C for 8–14 h. After the incubation, the flow-through was collected, reloaded onto the column, and re-incubated for 15–20 min. The unbound proteins were drained out, and the column was washed with 20 column volumes of wash buffer 1 (20 mM PB, 300 mM NaCl, 20% glycerol, and 50 mM imidazole) with incubation at 4 °C for 5–20 min. This step was repeated thrice with wash buffer 1. Similarly, the column was washed with wash buffer 2 (20 mM PB, 300 mM NaCl, 20% glycerol, 100 mM imidazole) and wash buffer 3 (20 mM PB, 300 mM NaCl, 20% glycerol, 150 mM imidazole) sequentially. After the wash buffers drained out completely, the bound proteins were eluted with elution buffer (20 mM PB, 300 mM NaCl, 20% glycerol, 500 mM imidazole; 6 fractions in total). The eluted proteins were collected and stored at -80 °C. Samples were collected at different stages based on the analysis requirement. The purification samples were then resolved and analyzed using 12% SDS-PAGE and western blotting. All the purification buffers were adjusted to pH 8.0 at room temperature and prechilled on ice.
Purification under mild denaturation conditions
The clarified soluble fraction was diluted accordingly with equilibration buffer (80 mM PB, 500 mM NaCl, 20% glycerol, and 40 mM imidazole) and denaturation buffer containing 80 mM PB, 500 mM NaCl, 20% glycerol, 40 mM imidazole, and 8 M urea, to the final concentration of 0 or 2 or 4 M urea and incubated under shaking condition overnight at 4 °C prior to loading on to the purification column. The Ni–NTA charged column was equilibrated with equilibration buffer, and then the diluted or denatured soluble fraction was loaded onto the column and incubated with continuous shaking at 4 °C for 8 to 14 h. Remaining procedures were carried out similar to native purification; however, the buffer compositions were different from the native purification: lysis buffer (80 mM PB, 500 mM NaCl, 20% glycerol, and 40 mM imidazole), wash buffer 1 (80 mM PB, 500 mM NaCl, 20% glycerol, 50 mM imidazole, and 0 or 2 or 4 M urea), wash buffer 2 (80 mM PB, 500 mM NaCl, 20% glycerol, 100 mM imidazole, and 0 or 2 or 4 M urea), wash buffer 3 (80 mM PB, 500 mM NaCl, 20% glycerol, 150 mM imidazole, and 0 or 2 or 4 M urea), and elution buffer (80 mM PB, 500 mM NaCl, 20% glycerol, 500 mM imidazole, and 0 or 2 or 4 M urea). The purification samples were then resolved and analyzed using 12% SDS-PAGE and western blotting. All the purification buffers were adjusted to pH 8.0 at room temperature.
The purified proteins were further dialyzed using snakeskin dialysis tubing (HiMedia) or buffer exchanged using PD10 columns (GE Healthcare) against 20 mM PB (pH 8.0) or sterile phosphate buffer saline (PBS) with 20% glycerol (glycerol buffer) depending on the experimental requirement. Purified proteins were concentrated using Amicon® Ultra-15 10 K centrifugal filter device (Merck Millipore Limited, Co. Cork, Ireland) and stored at -80 °C until further use.
SDS-PAGE and western blotting
Protein concentrations were determined using Bradford assay (Bradford reagent (Bio-Rad)) using bovine serum albumin (Bio-Rad) as a standard and measured with a multi-plate reader (Multiskan GO, Thermo Scientific).
For Coomassie staining, the protein samples were resolved on 12% SDS-PAGE gel and stained with staining solution containing 50% (v/v) methanol (Merck Millipore), 10% (v/v) acetic acid (Merck Millipore), and 0.4% (w/v) Coomassie Brilliant Blue G-250 (Merck Millipore) in deionized water. The stained gel was then destained with 50% (v/v) methanol, and 10% (v/v) acetic acid in deionized water and the image was recorded in the molecular imager (ChemiDoc™ XRS +) equipped with Image Lab™ software (Bio-Rad, California, USA).
For western blotting, the proteins were resolved on 12% SDS-PAGE gel and the resolved samples were transferred onto the nitrocellulose membrane (Bio-Rad) in Pierce Power Blotter XL System (Thermo Scientific™, Bremen, Germany). After confirming the transfer with Ponceau S staining (HiMedia), the membrane with transferred proteins was washed (destained) and then blocked with 5% (w/v) fat-free milk in Tris-buffered saline (TBS) (HiMedia) with 0.1% (v/v) Tween-20 (TBST) (Invitrogen) for 2 h at room temperature to reduce non-specific binding, followed by a wash with TBST for 10 min. The membrane was then incubated with primary antibody overnight at 4 °C and washed thrice with TBST for 5 min, followed by horseradish peroxidase-conjugated secondary antibody incubation for 1 h at room temperature. After washing thrice with TBST, immunoblot was developed in the presence of a chemiluminescence substrate (Bio-Rad), and the image was recorded in the molecular imager (ChemiDoc™ XRS + ; Bio-Rad) equipped with Image Lab™ software. All primary [anti-His (1:5000, BB-AB0010, BioBharati LifeScience Pvt. Ltd., India); anti-GATA4 (1:1000, sc-25310, Santa Cruz Biotechnology Inc., USA)] and secondary antibodies [anti-rabbit IgG-HRP Conjugated (1:5000, BB-SAB01A, BioBharati LifeScience Pvt. Ltd., India); anti-mouse IgG-HRP Conjugated (1:5000; 31430; Invitrogen, USA)] were diluted with 5% (w/v) bovine serum albumin (HiMedia) in TBST and 5% (w/v) fat-free milk in TBST, respectively.
Mass spectrometry (MS)
In-gel digestion
The purified recombinant GATA4 fusion protein (GATA4-NTH) was run on SDS-PAGE gel and stained with staining solution containing 50% (v/v) methanol, 10% (v/v) acetic acid, 0.4% (w/v) Coomassie Brilliant Blue G-250 in deionized water. The desired band was excised and destained with 40 mM ammonium bicarbonate (HiMedia) in a 40% (v/v) acetonitrile solution (Merck Millipore). The destained gel was then treated with reduction solution (5 mM dithiothreitol (Sigma-Aldrich) in 40 mM ammonium bicarbonate) for 30 min at 60 °C, followed by alkylation solution (20 mM iodoacetamide (Sigma-Aldrich) in 40 mM ammonium bicarbonate) for 10 min at room temperature (dark). The excised gel slice was dehydrated by adding 100% (v/v) acetonitrile and then digested with trypsin (Promega). After overnight digestion, the peptides were extracted using extraction buffer (5% formic acid (Merck Millipore) and 40% acetonitrile (Merck Millipore)) and were vacuum-dried using SpeedVac and stored at − 80 °C.
LC–MS/MS analysis
The dried peptides were reconstituted in 0.1% formic acid (Merck Millipore) and analyzed using Q Exactive™ (Thermo Scientific™) mass spectrometer coupled with the Proxeon Easy nLC system (Thermo Scientific™). For peptide enrichment, protein fragments were passed on to an Acclaim™ PepMap™ trap column (Michrom Biosciences Inc.). Peptides were separated on an analytical column employing a linear gradient of 7–30% acetonitrile for 80 min. MS and MS/MS scan acquisitions were executed in the quadrupole Orbitrap mass analyzer.
Data analysis
The MS data were searched against NCBI HsRefSeq81 (human protein database; version 81) and analyzed using the Mascot search engine (Matrix Science, London, UK; version 2.2.0) and SequestHT program and Proteome Discoverer software (Thermo Fisher Scientific; version 1.4.0.288).
Circular dichroism spectroscopy
The secondary structure of the purified GATA4 fusion proteins (GATA4-NTH and HTN-GATA4) were determined using far ultraviolet (UV) circular dichroism (CD) spectra from J-815/J-1500 spectropolarimeter (Jasco, Japan) equipped with a thermoelectric cooling-based temperature control unit. Far UV CD spectra of the protein were recorded as an average of ten accumulations from wavelength 260–190 nm in a 0.1 cm path length quartz cuvette at a scan rate of 100 nm/min with a data integration time of 1 s. From the sample spectrum, background noise due to the PB was subtracted, and the final spectrum was analyzed and quantified using in silico Beta Structure Selection (BeStSel) online tool (http://bestsel.elte.hu/index.php) to estimate the secondary structure of the purified protein. Detailed information about the BeStSel algorithm is described elsewhere [28, 29].
Cell culture
Human foreskin fibroblasts (HFF), BJ (ATCC® CRL-2522™), were cultured in fibroblast growth medium containing Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen), 1% penicillin/streptomycin (P/S) (Invitrogen), 1X non-essential amino acids (Invitrogen), and 1X Glutamax (Invitrogen) at 37 °C with 5% CO2 in a humidified atmosphere. Cells were passaged at 70–80% confluence in the 1:4 ratio, with 0.25% trypsin–EDTA (Invitrogen).
GATA4 fusion protein transduction
The HFF cells were adjusted to 1 × 105 cells/well and seeded in 6-well culture plates. Cells were grown till 40–50% confluency at 37 °C with 5% CO2 in a humidified atmosphere. The medium was then replaced with filter sterile protein transduction medium (DMEM, 2% FBS, 1% Penicillin and Streptomycin (P/S)), and optimal concentrations of purified recombinant protein (GATA4-NTH) or glycerol buffer as a control) and re-incubated for 12 or 24 h. After the incubation, cells were washed with PBS and used for further analysis.
Cell viability assay
The 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay was performed and optimized for the HFF cell line. In 96-well culture plate, 4 × 104 HFFs/well were seeded and incubated overnight at 37 °C with 5% CO2 in a humidified atmosphere. Once cells adhered to the plate, cells were washed with sterile PBS and treated with filter sterile protein transduction medium (DMEM, 2% FBS, 1% P/S, and varying concentrations of purified recombinant protein (GATA4-NTH) or glycerol buffer (control)) and re-incubated for 24 h. For the comparable experimental conditions, glycerol buffer was added accordingly to the cell culture media containing low concentrations of purified GATA4 protein. After the incubation, cells were washed with PBS and incubated in the dark with fresh DMEM with 0.05% (w/v) of MTT (Sigma-Aldrich) for 2 h. Dimethyl sulfoxide (HiMedia) was added to dissolve the crystals and incubated for 10 min with gentle shaking, and then absorbance at 570 nm was measured using a multi-plate reader (Multiskan GO, Thermo Scientific).
Immunocytochemistry and microscopy
Cells were washed with a heparin solution (HiMedia) followed by PBS and fixed with 2% paraformaldehyde (Merck Millipore). Fixed cells were then permeabilized by treating with 0.1% Triton™ X-100 (Sigma-Aldrich) in PBS for 15 min. After permeabilization, cells were blocked with a blocking solution (0.5% bovine serum albumin, 0.15% glycine in PBS) for an hour at room temperature. The primary antibody [anti-Vimentin (1:1000, MA5-14564, Invitrogen, USA); anti-GATA4 (1:300, sc-25310, Santa Cruz Biotechnology Inc., USA)] was then added and incubated overnight at 4 °C in a moist chamber. Cells were washed three times with PBS and then incubated with corresponding secondary antibody [anti-rabbit IgG, Alexa Fluor 488 (1:2000, A-11034, Invitrogen, USA); anti-mouse IgG, Alexa Fluor 594 (1:1000, A-11032, Invitrogen, USA)] for 1 h in a moist chamber at room temperature. After incubation, cells were washed three times with PBS and then stained with 4′,6-diamidino-2-phenylindole (DAPI) (1:15000; Sigma-Aldrich) for 10 min. Excess DAPI staining was removed with PBS and then visualized under an inverted fluorescent microscope (ZOE Fluorescent Cell Imager, Bio-Rad, California, USA).
Statistical analysis
A statistical test (One-way ANOVA analysis followed by Dunnett's multiple comparison test) was carried out for the results obtained for MTT assay using Graphpad Prism 5 software. The results are expressed as the mean ± standard deviations. p values < 0.05 were considered to be statistically significant.
Results and discussion
Codon optimization of human GATA4 gene sequence
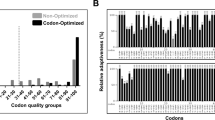
Codon optimization is one of the numerous factors that contribute to the success of heterologous protein expression. It is a process that involves synonymous codon substitution to obtain enhanced protein expression in a heterologous system by improving the gene expression and translational efficiency of a gene of interest [30]. Various studies have reported enhanced protein expression of human genes in E. coli as well as in other model systems after codon optimization [30, 31]. Therefore, to achieve a heterologous expression of GATA4 in a bacterial system (E. coli in this study), the full-length coding sequence of the human GATA4 gene was codon-optimized for the expression in E. coli using GeneOptimizer (ThermoFisher Scientific) online tool. The most preferred synonymous codons substituted codons in the non-optimized sequence without altering the protein sequence. The codon-optimized and non-optimized sequences were then analyzed using two independent online tools, Graphical Codon Usage Analyser 2.0 (Fig. 1a; Suppl. Figure 1) and GenScript Rare Codon Analysis (Fig. 1b; Suppl. Table 1). This was performed to confirm that the codon-optimized sequence lacked codons that can impede protein expression and was very likely to give a high expression in E. coli compared to the non-optimized sequence.

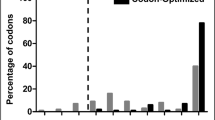
Validation and comparison of codon-optimized over non-optimized GATA4 gene sequence for the heterologous expression in E. coli using Graphical codon usage analyzer and GenScript rare-codon-analysis tools. a Codon optimization analysis using Graphical codon usage analyser. The bar graphs shown here are only for the first 50 codons of non-optimized and codon-optimized GATA4 gene sequence as a representation. This analysis depicts codon quality as a measure of relative adaptiveness value. The relative adaptiveness value of each codon is based on the ratio of the usage of each codon to that of the most abundant codon for a particular amino acid multiplied by 100. All the codons were analyzed by the relative adaptiveness value; therefore, the value above 30% means that those codons should be suitable for expression in E. coli. b Codon optimization analysis using GenScript Rare Codon Analysis Tool. The bar graph shows the percentage distribution of codons in computed codon quality groups for the non-optimized (gray) and codon-optimized (black) GATA4 gene sequences. Codons with the highest usage frequency for a given amino acid in the desired expression organism are assigned a value of 100. Codons with values ≤ 30 are likely to hamper the expression in the desired expression organism (in this study; expression organism is E. coli)
The codon quality of the coding sequences can be analyzed and represented in two ways: either based on relative adaptiveness value or codon usage frequency. Using the Graphical Codon Usage Analyser tool, 15 codons with a relative adaptiveness value of ≤ 30% were identified to affect the expression in E. coli in the non-optimized sequence (Suppl. Figure 1 (left)). These 15 codons, as well as other codons (e.g., fifth codon TGG coding for leucine was substituted to CTG changing the relative adaptiveness value from 34 to 100%; Fig. 1a) that could affect expression, was substituted with the most preferred synonymous codons using GeneOptimizer tool to improve the gene expression in E. coli (Fig. 1a; Suppl. Figure 1). The relative adaptiveness value calculated for each codon is the ratio of the frequency of the used codon to that of the most abundant codon for the same amino acid multiplied by 100. Using GenScript Rare Codon Analysis 2.0 tool, 9% of the codons with codon usage frequency of ≤ 30% were identified to affect the expression in E. coli in the non-optimized sequence (Fig. 1b (in gray); Suppl. Table 1). These codons, as well as other codons that could affect expression, were also substituted with the most preferred synonymous codons using the GeneOptimizer tool to improve the gene expression in E. coli (Fig. 1b (black)). Therefore, codon optimization resulted in an increase in the codon adaptation index value from 0.68 of the non-optimized sequence to 0.89 of the codon-optimized sequence (Suppl. Table 1). Codon adaptation index is the relative adaptiveness of the codon usage of a protein-coding gene towards the codon usage of highly expressed (reference set) genes [26]. A codon adaptation index between 0.8 and 1.0 is considered as ideal for gene expression in the desired organism. The lower the codon adaptation index, the higher the chance that the gene of interest will be expressed poorly. In our analysis, the codon adaptation index of the codon-optimized GATA4 gene sequence was 0.89 in contrast to 0.68 for the non-optimized sequence (Suppl. Table 1), which signified that codon optimization would favor the expression of our gene of interest in our desired expression organism (E. coli). Therefore, this analysis indicated that the codon-optimized GATA4 gene sequence would give a high expression in E. coli.
Cloning of codon-optimized GATA4 gene with fusion tags in a protein expression vector
The codon-optimized human GATA4 gene sequence was fused with tags (fusion tags were also codon-optimized for the expression in E. coli), either before the start codon (to generate HTN-GATA4) or at the end of the coding sequence (to generate GATA4-NTH) as shown in Fig. 2a and Suppl. Figure 2A. The purpose to incorporate the fusion tags at either end is for the reason that these tags can influence expression level, stability and solubility of human proteins expressed in E. coli [26, 27, 32, 33]. Three fusion tags were used: poly-His (H) tag for affinity chromatography, TAT (T) to enable cell penetration, and NLS (N) to facilitate sub-nuclear localization (Suppl. Table 2). A similar approach was employed in earlier studies to enable efficient cell- and nuclear delivery of transcription factors in mammalian cells [32, 34, 35].

Schematic representation of the human GATA4 gene insert and confirmation of cloning the gene insert into the protein expression vector. a Schematic diagram of GATA4-NTH insert (not drawn to scale). The GATA4 gene was fused at 3′ end with nucleotide sequences of NLS (N) and TAT (T) to facilitate nuclear translocation and cell penetration in mammalian cells, respectively, followed by His tag for affinity chromatography-based purification. b Restriction digestion analysis was performed to confirm the successful cloning of GATA4-NTH insert into the protein expression vector. The synthetic gene insert was cloned in the protein expression vector, pET28a(+), using restriction endonucleases, NcoI, and XhoI. The resulting plasmid, pET28a(+)-GATA4-NTH (in short GATA4-NTH), was then confirmed by restriction digestion using various restriction enzymes, as depicted in a and b. NLS (N): nuclear localization signal/sequence; TAT (T): transactivator of transcription; His tag (H): polyhistidine (8X)
To express GATA4 in E. coli, HTN-GATA4 and GATA4-NTH inserts were cloned under the control of a strong inducible T7 promoter of the protein expression vector, pET28a(+). These gene inserts were cloned between NcoI and XhoI sites using restriction endonucleases, NcoI and XhoI, to remove the poly-His tag (present before the start codon) and thrombin cleavage site sequence present in the pET28a(+) vector. The resulting plasmids (pET28(a) + HTN-GATA4 (hereafter, HTN-GATA4) and pET28(a) + GATA4-NTH (hereafter, GATA4-NTH)) were primarily verified by restriction digestion analysis (Fig. 2b and Suppl. Figure 2B). The empty vector, pET28a(+) only, was also included in the analysis as a control to confirm the absence of gene of interest (data not shown). Further, these gene constructs, HTN-GATA4 and GATA4-NTH, were confirmed via DNA sequencing from both the ends using T7 promoter and T7 terminator primers. The sequencing results of the cloned inserts confirmed the fidelity of the gene sequence and fusion tags.
Identification of optimal conditions for the heterologous expression of recombinant GATA4 fusion protein
One of the most critical factors in the production of recombinant proteins is the selection of an appropriate host expression system. In this study, the widely used bacterium E. coli was used as an expression host. This organism has a high transformation efficiency, fast growth rate, well-understood genetics, and cost-effective protein production [17, 30]. This expression host is commonly used for human proteins for which post-translational modifications are not essential for their bioactivity [36,37,38]. Recently, it was reported that even in the absence of post-translational modifications, the GATA4 protein has retained its biological activity [39]. Specifically, the commonly used E. coli BL21(DE3) strain was used, which is devoid of lon and OmpT proteases, allowing a high-level of heterologous protein expression without any degradation.
Several studies have demonstrated the importance of identifying the optimal expression parameters for obtaining high yield of biologically active recombinant proteins in a soluble form [40,41,42,43,44,45]. Based on these studies, the effect of inducer concentrations (IPTG) on the expression of GATA4 in E. coli BL21(DE3) transformed with GATA4-NTH plasmid was investigated. SDS-PAGE and western blot analysis revealed that the expression level varied with different inducer concentrations (for the induction of T7 RNA polymerase-mediated expression of the protein), and it reached the maximum when induced with 0.25 mM of IPTG (Fig. 3a). The results confirmed the absence of leaky expression and demonstrated that the inducer concentration had a strong effect on protein expression. However, the growth of induced cultures decreased exponentially until 0.25 mM of IPTG and was stable up to 0.5 mM (Fig. 3b), which indicated that GATA4-NTH induction increased the doubling time of E. coli. Next, the optimal cell density at the time of induction was determined by inducing with an optimal concentration (0.25 mM) of IPTG at three different growth phases. Various studies have reported that the bacterial system overexpresses heterologous proteins when induced at the log phase (OD600 = 0.3–0.8) [45]. In contrast, Gavidia and colleagues reported that the induction at an early log phase (OD600 = 0.1), resulted in ~ three-fold higher soluble expression, unlike inducing at the log phase (OD600 = 0.6) [44]. Apart from these, few reports state that high-density induction enhances the solubility of heterologous mammalian proteins under controlled conditions [40, 41]. Hence, identifying the optimal cell density at the time of induction is crucial for the maximal expression of recombinant proteins. In this study, from the SDS-PAGE analysis (Fig. 3c), the maximal expression of the GATA4 fusion protein was observed when induced during the log phase, OD600 = ~ 0.5 in comparison to late log phase (OD600 = ~ 1 or ~ 1.5). Moreover, the same was confirmed using western blotting with anti-His antibody (Fig. 3c). The effect of pre-induction cell density on the final growth of the culture is shown in Fig. 3d.

Identification of optimal conditions for the maximal expression of the recombinant GATA4 fusion protein in E. coli. a Screening of minimal inducer concentration required for maximal expression of recombinant GATA4 protein*. E. coli BL21(DE3) cells transformed with GATA4-NTH were induced with different concentrations of IPTG at the early log phase (OD600 = ~ 0.5) for 2 h at 37 °C with continuous shaking at 180 rpm (n = 3). b Effect of IPTG concentration on the final growth of E. coli BL21(DE3)#. c Screening of optimal cell density at the time of induction for the maximal expression of recombinant GATA4 protein*. E. coli BL21(DE3) cells transformed with GATA4-NTH was induced with an optimal IPTG concentration (0.25 mM) at different cell densities for 2 h at 37 °C with continuous shaking at 180 rpm (n = 3). d Effect of pre-induction growth on the final cell density#. e Screening of post-induction incubation time for the maximal expression of recombinant GATA4 protein*. Transformed E. coli BL21(DE3) cells with GATA4-NTH was induced at optimal cell density (OD600 = ~ 0.5) with an optimal concentration of IPTG (0.25 mM) for 2/4/6/8 h at 37 °C with continuous shaking at 180 rpm (n = 2). f Effect of post-induction incubation on the bacterial growth#. g Soluble expression analysis of C-terminal tagged recombinant GATA4 protein*. E. coli BL21(DE3) cells transformed with GATA4-NTH was induced with an optimal IPTG concentration (0.25 mM) at an optimal cell density (OD600 = ~ 0.5) for 2 h at 37 °C with continuous shaking at 180 rpm. The total cell lysate fractions (L) were then centrifuged to obtain a pellet/insoluble (P) and a soluble (S) fractions (n = 3). h Quantification analysis on the solubility of the expressed recombinant GATA4 protein. The intensities from the western blot of the soluble expression analysis were measured using Image J 1.48 V software. From the measured values, the amount of GATA4 protein (%) was calculated, and a graph was plotted using GraphPad Prism 5 software. The bar graph represents the amount of GATA4 protein (%) with respect to different fractions (L, P, and S) of this fusion protein. i Screening of induction temperature to obtain maximal soluble expression of recombinant GATA4 protein*. The transformed E. coli BL21(DE3) cells with GATA4-NTH was induced at optimal cell density (OD600 = ~ 0.5) with an optimal concentration of IPTG (0.25 mM) at different temperatures (37 °C for 2 h, 30 °C for 12 h, 25 °C for 18 h and 18 °C for 24 h) with continuous shaking at 180 rpm. The total cell lysate fractions (L) were then centrifuged to obtain a pellet/insoluble (P) and a soluble (S) fractions (n = 2). j Summary of identified optimal expression condition for soluble expression of GATA4. *Harvested cells were lysed by ultrasonication, and total lysates were run on SDS-PAGE with normalized protein loading concentration of 20 μg/lane (For g and i equal volume corresponding to the respective L fractions was loaded for P and S fractions). The resolved SDS–polyacrylamide gel was stained with Coomassie Brilliant Blue G-250 (top) or transferred to nitrocellulose membrane and performed western blotting with Histidine antibody (bottom). #The final growth of the un-induced and induced cultures was recorded before harvesting the cells, and a graph was plotted using GraphPad Prism 5 software. M, Protein Marker (kDa); L, Total cell lysate; P, Pellet/insoluble cell fraction; S, Soluble cell fraction; Ab, Antibody
After the identification of optimal inducer concentration and cell density at the time of induction, the relationship between post-induction incubation time, cellular growth, and GATA4-NTH expression were studied. To identify the optimal post-induction incubation time, the culture at the log phase (OD600 = ~ 0.5) was induced with 0.25 mM IPTG at 37 °C for 8 h, and samples were collected every 2 h for analysis. As shown in Fig. 3E, the expression level reached the maximum within 2 h of induction and remained unchanged up to 8 h. This result signified that the recombinant GATA4-NTH protein was stable in E. coli. However, the growth rate of uninduced and induced cultures varied significantly (Fig. 3f), and that confirmed the burden on the growth of the bacterial system upon GATA4-NTH induction. Further, the solubility of the expressed recombinant GATA4 fusion protein was determined by analyzing the cell lysate fractions. Interestingly, the fusion protein was found in both the pellet and supernatant fractions of the cell lysate (Fig. 3g). Quantitative analysis of western blotting using ImageJ showed that ~ 60% of the fusion protein was in the insoluble pellet fraction and the remaining in the soluble fraction (Fig. 3h). However, other studies reported that the reduction in the temperature during induction enhanced the solubility of the protein of interest [42, 44]. Based on these studies and to improve solubility, the effect of temperature on the HTN-GATA4 and GATA4-NTH protein solubility was examined. In both cases, the reduction in the induction temperatures did not enhance the solubility; instead, a decrease in the expression of the GATA4 fusion proteins (Fig. 3i and Suppl. Figure 2C) was observed. Therefore, the optimal temperature to obtain the maximal soluble expression of the recombinant GATA4 fusion protein was found to be 37 °C. The overall identified optimal expression conditions of the GATA4 fusion protein are compiled in Fig. 3j. This is the first study to report identification of optimal expression conditions in E. coli to obtain high and soluble expression of the recombinant GATA4 protein to achieve native purification.
Purification of recombinant GATA4 fusion proteins
Recombinant proteins purified under native conditions often retain native-like conformations and are bioactive [25]. Therefore, to retain the secondary structure and biological activity of GATA4 fusion proteins (GATA4-NTH and HTN-GATA4), purification using IMAC under native conditions (from soluble fraction) was performed. IMAC is a widely used purification technique that depends on the interactions between the polyhistidine residues and charged transition metal ions such as Ni2+ in this study, immobilized on a matrix such as NTA. The recombinant GATA4-NTH and HTN-GATA4 proteins were expressed at the identified parameters (temperature: 37 °C; cell density: OD600 = ~ 0.5; IPTG concentration: 0.25 mM; induction time: 2 h) and purified using IMAC to homogeneity at 4 °C. The high purity of the recombinant GATA4 fusion proteins was confirmed by SDS-PAGE analysis (Fig. 4a and Suppl. Figure 2D), which was not reported in the earlier study [39]. In fact, no biochemical data showing purification of human GATA4 protein were reported [39]. Importantly, application of impure proteins on the target mammalian cells could have detrimental effects [46], which was not the case in this study. A band of ~ 55 kDa corresponding to GATA4 fusion proteins was observed when the purified fractions were run on 12% SDS-PAGE (Fig. 4a and Suppl. Figure 2D). Although the overall expression of HTN-GATA4 protein was higher than GATA4-NTH, the yield of HTN-GATA4 was very low compared to its counterpart (Fig. 4a and Suppl. Figure 2D). Based on the western blotting analysis (Fig. 4a and Suppl. Figure 2D; bottom), loss of GATA4 proteins was detected in the flow-through/unbound fractions of both GATA4-NTH and HTN-GATA4 proteins. The main reason behind this low yield of HTN-GATA4 was the loss of the majority of protein molecules in flow-through fractions during purification. These observations indicated that the poly-His tag could have got buried during protein folding in these protein molecules. For this reason, many protein molecules did not efficiently bind to the Ni–NTA resin and were eventually lost in the flow-through fraction. To prevent the loss, purification of GATA4-NTH under mild denaturing conditions was performed by exposing the poly-His tag with 2 or 4 M urea. The SDS-PAGE and western blot analysis confirmed that GATA4-NTH was purified successfully, and based on the intensity, an increase in protein yield in 4 M urea treated soluble fraction was observed compared to 0 or 2 M treated soluble fraction (Suppl. Figure 3A and 3B). These results confirm that the loss of the majority of the molecules of GATA4 fusion proteins in flow-through could be due to the unexposed poly-His tags. Also, the terminal at which the poly-His tag was fused with GATA4 plays a crucial role in protein expression (in terms of low or high) and purification (in terms of protein yield). In this study, we report for the first time the biochemical data showing the purification of human GATA4 protein to homogeneity under native conditions.

Purification and characterization of the recombinant GATA4 fusion protein. a E. coli BL21(DE3) transformed with recombinant plasmid GATA4-NTH was induced under optimal expression conditions (temperature: 37 °C; cell density: OD600 = ~ 0.5; IPTG concentration: 0.25 mM; induction time: 2 h) with continuous shaking at 180 rpm. Harvested cells were lysed by ultrasonication, and the expressed protein was purified under native conditions from the soluble fraction using Ni–NTA affinity chromatography. The purification samples were run on SDS-PAGE with normalized loading volume. The resolved polyacrylamide gel was stained with Coomassie Brilliant Blue G-250 (top) or transferred to nitrocellulose membrane and performed western blotting with Histidine antibody (middle) or GATA4 antibody (bottom). M, protein marker (kDa); L, total cell lysate; S, soluble cell fraction; F, flow-through fraction; W1, wash buffer 1; W2, wash buffer 2; W3, wash buffer 3; E, elution; Ab, antibody. b and c Characterization of the purified recombinant GATA4-NTH protein. The desired GATA4 band was excised from the Coomassie-stained polyacrylamide gel and then destained, processed followed by trypsin digestion. The resulting peptide fragments were then analyzed using mass spectrometry, and the identified 12 unique peptide sequences were highlighted in the full-length amino acid sequence of the GATA4-NTH protein. From the identified peptide sequences, annotated spectra have been plotted for one of the unique peptide, HPNLDMFDDFSEGR
Characterization of recombinant protein GATA4
The purified recombinant GATA4-NTH protein was primarily characterized using Liquid chromatography-tandem mass spectrometry (LC–MS/MS). LC–MS/MS is a widely used powerful analytical technique to identify and quantify peptides or the proteome of an organism [47]. In the present study, the Orbitrap LC–MS/MS technique was utilized. This powerful technique is used to confirm the identity of the purified recombinant proteins [47]. Therefore, the purified GATA4-NTH protein was trypsin digested to produce small peptides, and the resulting peptides were then separated, fragmented, ionized, and analyzed using Orbitrap mass spectrometry. The MS/MS study of digested peptides generated a match with human GATA4 protein, and from that twelve unique peptide sequences (including two pairs of overlapping sequences) were identified (Fig. 4b) to be a part of human GATA4 (Accession no.: NP_002043.2) using Mascot, SequestHT algorithm and Proteome Discoverer software. Identified twelve unique peptide sequences are shown in Fig. 4b. The annotated spectra of one of these twelve unique peptides (HPNLDMFDDFSEGR) are shown in Fig. 4c. Furthermore, this purified fusion protein was detected and confirmed using western blotting with the GATA4 antibody (Fig. 4a). Thus, with the mass spectrometry and western blotting (with GATA4 antibody) analysis, the identity of recombinant GATA4 protein was confirmed.
The secondary structure content of human GATA4 protein using experimental techniques is not reported to date. Therefore, its secondary structure was investigated using far UV CD spectroscopy. This spectroscopic technique is used to estimate the secondary structure content of proteins purified from cells/tissues [48], particularly for proteins whose secondary structure content is not known, such as GATA4. The characteristic shape and magnitude of the far UV CD spectrum represent different secondary structures, namely α-helix, β-sheet, turn, and random coil [48, 49]. α-helix has negative peaks at 222 nm and 208 nm and a positive peak at 193 nm, while the β-sheet has a negative peak at 218 nm and a positive peak at 195 nm [48]. Likewise, the random coil shows a positive peak at 210 nm and a negative peak of about 195 nm [48]. Firstly, the purified recombinant GATA4 fusion proteins (under native conditions) were desalted or buffer exchanged (to avoid background noise due to NaCl), and then used for the far UV CD spectroscopy. The far UV CD data obtained were analyzed and quantified using BeStSel online tool [28, 29]. BeStSel is a recently developed tool to estimate the secondary structure content and fold recognition from a CD spectra [28, 29]. From the CD spectrum that was plotted using BeStSel results (Fig. 5a and Suppl. Figure 2E), it is evident that the recombinant GATA4 fusion proteins have maintained their secondary structure. Notably, the characteristic shape and magnitude of the CD spectra of both fusion proteins (HTN-GATA4 and GATA4-NTH) were similar (Fig. 5a and Suppl. Figure 2E), signifying that the secondary structure of the GATA4 protein was independent of the terminal at which the tags were fused. Further, the estimated secondary structure of the purified GATA4-NTH protein revealed that GATA4 protein predominantly has random coils and α-helices in its secondary structure (Fig. 5b). The estimated secondary structure of the purified GATA4-NTH protein indicates that it constitutes of 34% random coils, 32% α-helices, 16% β-sheets and 18% turns (Fig. 5b). Similarly, the purified recombinant GATA4-NTH protein under mild denaturing conditions was dialyzed against 20 mM PB (to refold) and/or buffer exchanged, and then analyzed using far UV CD spectroscopy. From the CD spectra, it is clear that the secondary structure was not regained after several attempts of refolding approaches (Suppl. Figure 3C). To summarize these results, the recombinant GATA4 fusion proteins purified under native conditions, without undergoing any denaturation treatment, retained the secondary structure after purification.

Determination of the secondary structure of the purified recombinant GATA4 fusion protein. Far UV CD spectroscopy was performed to determine the secondary structure content of the purified recombinant GATA4-NTH protein in 20 mM PB (pH 8.0 at room temperature). The far UV CD data obtained were analyzed using in silico BeStSel online tool. From the BeStSel results, a CD spectra have been plotted with Delta Epsilon (M−1 cm−1; Y-axis) against wavelength (nM; X-axis) and a bar graph b shows the percentage of secondary structures (α-helices, β-sheets, turns, and others (mostly random coils)) for the purified recombinant GATA4-NTH protein (n = 3)
Stability and transduction ability of recombinant GATA4 fusion protein
To achieve successful protein transduction, the recombinant proteins have to be soluble and stable at in vitro cell culture conditions [20, 32]. Therefore, in vitro cell culture solubility and stability were analyzed for the purified recombinant GATA4-NTH protein. Although both the GATA4 fusion proteins were purified to homogeneity under native conditions and retained their secondary structure, GATA4-NTH protein was used for further experiments due to its high yield in the purification procedure compared to HTN-GATA4. Therefore, the glycerol stock of purified GATA4-NTH protein was diluted with cell culture media and then incubated at standard mammalian cell culture conditions (37 °C in a humidified atmosphere containing 5% CO2 in a CO2 incubator). After 24 h of incubation, the media was centrifuged to separate aggregated or insoluble proteins, and the soluble fractions were analyzed using western blotting with GATA4 antibody. From the result (Fig. 6a), it is clear that GATA4-NTH recombinant protein was detected at 100 and 200 nM concentrations using western blotting. This signified that the GATA4 protein was soluble and stable at standard cell culture conditions for up to 24 h. Importantly, the recombinant protein-based cellular reprogramming generally requires multiple rounds of transduction with one round every 24 h [20, 32]. Our purified recombinant GATA4-NTH protein passed the minimum criteria for reprogramming in terms of solubility and stability at cell culture conditions.

Cell culture stability, transduction ability, and effect of the purified GATA4 fusion protein on human cells. a Cell culture stability analysis of GATA4-NTH recombinant protein. The purified GATA4-NTH protein was diluted to the final concentration of 50/100/200 nM with cell culture medium containing DMEM supplemented with 2% FBS, 1% P/S, 1X NEAA, and 1X Glutamax. These GATA4-NTH transduction media were incubated under standard cell culture conditions for 24 h in a 6-well culture dish. The incubated transduction media were then analyzed using western blotting with the GATA4 antibody. b Effect of GATA4-NTH recombinant protein on HFFs. In 96-well culture plate, 4 × 104 BJ cells/well were grown overnight under standard cell culture conditions and treated with purified GATA4 recombinant protein or PBS for 24 h. After the treatment, cells were incubated with MTT for 2 h, and the crystals were dissolved with dimethyl sulfoxide. The absorbance at 570 nm was then measured using a multi-plate reader, and the cell viability (%) was calculated. A bar graph was plotted with the calculated values, and statistical tests (one-way ANOVA) were performed using GraphPad Prism 5 software (n = 3). c Characterization of BJ fibroblasts using the Vimentin antibody. Nuclear staining was carried out with a DAPI solution. Scale bar: 100 μm. d Protein transduction ability of purified GATA4-NTH recombinant protein in HFFs (using GATA4 antibody). BJ fibroblast cell line (1 × 105 cells/well) was grown in a 6-well culture-dish to 50% confluency, and purified GATA4-NTH protein (200 nM) or an equal volume of PBS was added to the cells and incubated for 24 h under standard cell culture conditions. After the incubation, cells were washed with PBS, fixed, permeabilized, and incubated with primary antibody followed by respective secondary antibody incubation. Nuclear staining was carried out with a DAPI solution. Then the cellular uptake of the fusion protein was analyzed using fluorescence microscopy. For nuclear localization analysis, the images obtained in different modes were merged. White and gray arrows indicate the nuclear-transduced cells (NTCs) and non-nuclear-transduced cells (non-NTCs), respectively. Scale bar: 100 μm (n = 2). e Quantitative analysis of GATA4 nuclear translocation in human cells. The total number of cells and GATA4 nuclear-transduced cells were counted using Image J 1.48 V software, and the percentage of nuclear-transduced cells (NTCs) and non-nuclear-transduced cells (non-NTCs) were calculated. Using GraphPad Prism 5 software, a bar graph was plotted with the calculated values, which represents the quantitative analysis of nuclear transduction ability of the purified GATA4-NTH recombinant protein in HFFs
Further, the sub-cellular and sub-nuclear delivery of the purified GATA4-NTH recombinant protein into HFFs (BJ cells) was investigated. This cell line was selected for the analysis due to its large size, which is more convenient in visualizing the subcellular and nuclear localization of this fusion protein. Additionally, this cell line is also widely used for cell reprogramming experiments [20]. At first, the effect of purified GATA4-NTH protein on the viability of HFFs was studied. As shown in Fig. 6b, the varying concentrations of GATA4-NTH protein did not significantly affect the viability of HFFs even after 24 h of exposure. This result confirmed that the purified GATA4-NTH protein was not toxic to cells and safe for cell culture analysis. Further, the characterization of human BJ fibroblasts with immunocytochemistry was performed, and the result showed that these cells express vimentin, a fibroblast-specific marker (Fig. 6c). Also, it was confirmed that these vimentin+ cells did not express GATA4 protein (Fig. 6d; top). In order to study the transduction ability of GATA4-NTH protein into human cells, HFFs were incubated with this fusion protein (GATA4-NTH; 200 nM) or PBS (as control) for 12 h and washed with heparin to remove extracellularly bound protein. Then the transduced cells were visualized and analyzed using fluorescence microscopy. Microscopy results confirmed that the PBS (vehicle control) did not stimulate the expression of GATA4 or did not lead to any false positive signal during analysis (Fig. 6d; top). The TAT-mediated protein transduction resulted in efficient transduction of GATA4 fusion protein into HFFs (Fig. 6d; bottom). Interestingly, almost all the cells showed successful uptake of purified GATA4-NTH protein into their cytoplasm. Of these, ~ 70% of the cells showed nuclear localization of GATA4 fusion protein (Fig. 6e). Being a transcription factor [1], translocation of GATA4 to the nucleus is critical for its biological activity. Although GATA4 transcription factor has its own NLS, the fusion of an extra NLS at the C-terminal end should further enhance the nuclear translocation [20, 32, 34, 35]. However, a detailed investigation is required to confirm that the efficient nuclear localization of this GATA4-NTH protein is due to the fusion of additional NLS at the C-terminal end. Thus, our strategy using TAT and NLS fused to the GATA4 protein resulted in efficient sub-cellular and sub-nuclear delivery into the HFFs. The fusion of these two tags (TAT and NLS) with the recombinant protein can efficiently deliver protein of interest to the target site to perform its biological function. The earlier study required the usage of protein transduction reagent for efficient delivery of GATA4 protein into cells [39], whereas no such reagent was required in our study. In this study, the authors showed that GATA4 was biologically active and was one of the core reprogramming factors to derive functional cardiac progenitor cells [39]. In our study, we have expressed and purified full-length human GATA4 protein that is stable at cell culture conditions and has cell and nuclear translocation ability, unlike the commercial ones which are generally available in truncated versions and are devoid of cell and nuclear translocation ability. Other studies have also reported similar results using TAT-mediated cell penetration and NLS-mediated nuclear translocation for other transcription factors to generate desired cells for biological applications [20, 32, 34, 35]. These studies have also reported that the presence of these fusion tags did not affect the biological activity of the protein of interest. Additionally, we have demonstrated that this purified protein has retained its secondary structure, which indicates that this recombinant protein has great promise of being biologically active. This molecular tool can be used to generate integration-free cells and for other biological applications, avoiding the serious concerns associated with plasmid or viral integration into the host cell chromosomes [20, 50].
Conclusion
In this study, codon optimization, cloning, expression and purification of full-length human GATA4 transcription factor from E. coli was performed to obtain a highly pure recombinant protein. This is the first study reporting the identification of optimal expression parameters in E. coli and showing the generation of a highly pure recombinant protein. The protein purification protocol employed is economical, simple and reproducible. In addition, this protocol involves purification from soluble cell lysate fraction to obtain a native protein, and precludes purification from inclusion bodies; the latter is expensive, cumbersome, time consuming and requires the refolding of the denatured protein. One aspect where the purification procedure still requires further attention is to improve the yield of the protein as a good amount of protein is lost in the flow-through fraction. Notably, we generated a stable recombinant human GATA4 protein having cell penetration and nuclear translocation ability, which did not require the usage of a protein transduction reagent. Importantly, the purified recombinant protein has retained its secondary structure. Also, we report for the first time that the human GATA4 protein predominantly comprises of random coils and α-helices. In addition, fusion of tags at any end of the protein does not alter its secondary structure. Soon, we aim to demonstrate that this recombinant cell-permeant GATA4 protein is biologically active using this protein for cell reprogramming assays substituting for a viral and genetic form of GATA4 to generate functional human cardiomyocytes. Furthermore, we aim to demonstrate its ability to function as a tumor suppressor and inhibit cell proliferation in different types of cancer. In addition, this recombinant protein can be a useful biological tool for further structural analysis, identifying potential novel interaction partners, and investigating its molecular function in different cell types.
References
Arceci RJ, King A, Simon MC, Orkin SH, Wilson DB (1993) Mouse GATA-4: a retinoic acid-inducible GATA-binding transcription factor expressed in endodermally derived tissues and heart. Mol Cell Biol 13:2235–2246
Heikinheimo M, Scandrett JM, Wilson DB (1994) Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Develop Biol 164:361–373
Viger RS, Guittot SM, Anttonen M, Wilson DB, Heikinheimo M (2008) Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol 22:781–798
Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, Soudais C, Leiden JM (1997) GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 11:1048–1060
Molkentin JD, Lin Q, Duncan SA, Olson EN (1997) Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11:1061–1072
Rojas A, Kong SW, Agarwal P, Gilliss B, Pu WT, Black BL (2008) GATA4 is a direct transcriptional activator of cyclin D2 and Cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol 28:5420–5431
Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA (2008) Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Develop Biol 317:614–619
Li J, Liu W-D, Yang Z-L, Yuan F, Xu L, Li R-G, Yang Y-Q (2014) Prevalence and spectrum of GATA4 mutations associated with sporadic dilated cardiomyopathy. Gene 548:174–181
Guo S, Zhang Y, Zhou T, Wang D, Weng Y, Chen Q, Ma J, Li Y-p, Wang L (2018) GATA4 as a novel regulator involved in the development of the neural crest and craniofacial skeleton via Barx1. Cell Death Differ 25:1996–2009
Ieda M, Fu J-D, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142:375–386
Nam Y-J, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R (2013) Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci 110:5588–5593
van den Bos E, van der Giessen W, Duncker D (2008) Cell transplantation for cardiac regeneration: where do we stand? Netherlands Heart J 16:88–95
Gong Y, Zhang L, Zhang A, Chen X, Gao P, Zeng Q (2018) GATA4 inhibits cell differentiation and proliferation in pancreatic cancer. PLoS ONE 2018:13
Gao L, Hu Y, Tian Y, Fan Z, Wang K, Li H, Zhou Q, Zeng G, Hu X, Yu L (2019) Lung cancer deficient in the tumor suppressor GATA4 is sensitive to TGFBR1 inhibition. Nature Commun 10:1–15
Han X, Tang J, Chen T, Ren G (2019) Restoration of GATA4 expression impedes breast cancer progression by transcriptional repression of ReLA and inhibition of NF-κB signaling. J Cell Biochem 120:917–927
Xiang Q, Zhou D, He X, Fan J, Tang J, Qiu Z, Zhang Y, Qiu J, Xu Y, Lai G (2019) The zinc finger protein GATA4 induces mesenchymal-to-epithelial transition and cellular senescence through the nuclear factor-κB pathway in hepatocellular carcinoma. J Gastroenterol Hepatol 34:2196–2205
Borgohain MP, Narayan G, Kumar HK, Dey C, Thummer RP (2018) Maximizing expression and yield of human recombinant proteins from bacterial cell factories for biomedical applications. In: Kumar P, Patra JK, Chandra P (eds) Advances in microbial biotechnology (pp. 447–486). Apple Academic Press
Sommer CA, Mostoslavsky G (2013) The evolving field of induced pluripotency: recent progress and future challenges. J Cell Physiol 228:267–275
Serna N, Sánchez-García L, Unzueta U, Díaz R, Vázquez E, Mangues R, Villaverde A (2018) Protein-based therapeutic killing for cancer therapies. Trends Biotechnol 36:318–335
Borgohain MP, Haridhasapavalan KK, Dey C, Adhikari P, Thummer RP (2019) An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Rev Rep 15:286–313
Nezafat N, Sadraeian M, Rahbar MR, Khoshnoud MJ, Mohkam M, Gholami A, Banihashemi M, Ghasemi Y (2015) Production of a novel multi-epitope peptide vaccine for cancer immunotherapy in TC-1 tumor-bearing mice. Biologicals 43:11–17
O’Malley J, Woltjen K, Kaji K (2009) New strategies to generate induced pluripotent stem cells. Curr Opin Biotechnol 20:516–521
Kintzing JR, Interrante MVF, Cochran JR (2016) Emerging strategies for developing next-generation protein therapeutics for cancer treatment. Trends Pharmacol Sci 37:993–1008
Khow O, Suntrarachun S (2012) Strategies for production of active eukaryotic proteins in bacterial expression system. Asian Pacific J Trop Biomed 2:159–162
Wingfield PT (2015) Overview of the purification of recombinant proteins. Curr Protocols Protein Sci 80:611–6135
Haridhasapavalan KK, Sundaravadivelu PK, Thummer RP (2020) Codon optimization, cloning, expression, purification and secondary structure determination of human ETS2 transcription factor. Mol Biotechnol 2020:1–10
Narayan G, Sundaravadivelu PK, Agrawal A, Gogoi R, Nagotu S, Thummer RP (2021) Soluble expression, purification, and secondary structure determination of human PDX1 transcription factor. Protein Express Purif 2021:105807
Micsonai A, Wien F, Kernya L, Lee Y-H, Goto Y, Réfrégiers M, Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Natl Acad Sci 112:E3095–E3103
Micsonai A, Wien F, Bulyáki É, Kun J, Moussong É, Lee Y-H, Goto Y, Réfrégiers M, Kardos J (2018) BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res 46:W315–W322
Burgess-Brown NA, Sharma S, Sobott F, Loenarz C, Oppermann U, Gileadi O (2008) Codon optimization can improve expression of human genes in Escherichia coli: A multi-gene study. Protein Expr Purif 59:94–102
Maertens B, Spriestersbach A, von Groll U, Roth U, Kubicek J, Gerrits M, Graf M, Liss M, Daubert D, Wagner R (2010) Gene optimization mechanisms: a multi-gene study reveals a high success rate of full-length human proteins expressed in Escherichia coli. Protein Sci 19:1312–1326
Bosnali M, Edenhofer F (2008) Generation of transducible versions of transcription factors Oct4 and Sox2. Biol Chem 389:851–861
Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J (2002) Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci 99:2654–2659
Münst B, Thier MC, Winnemöller D, Helfen M, Thummer RP, Edenhofer F (2016) Nanog induces suppression of senescence through downregulation of p27KIP1 expression. J Cell Sci 129:912–920
Peitz M, Münst B, Thummer RP, Helfen M, Edenhofer F (2014) Cell-permeant recombinant Nanog protein promotes pluripotency by inhibiting endodermal specification. Stem Cell Res 12:680–689
Bhat EA, Sajjad N, Sabir JS, Kamli MR, Hakeem KR, Rather IA, Bahieldin A (2020) Molecular cloning, expression, overproduction and characterization of human TRAIP Leucine zipper protein. Saudi J Biol Sci 27:1562–1565
Stefan A, Calonghi N, Schipani F, Dal Piaz F, Sartor G, Hochkoeppler A (2018) Purification of active recombinant human histone deacetylase 1 (HDAC1) overexpressed in Escherichia coli. Biotech Lett 40:1355–1363
Lili W, Chaozhan W, Xindu G (2006) Expression, renaturation and simultaneous purification of recombinant human stem cell factor in Escherichia coli. Biotech Lett 28:993–997
Li X-H, Li Q, Jiang L, Deng C, Liu Z, Fu Y, Zhang M, Tan H, Feng Y, Shan Z (2015) Generation of functional human cardiac progenitor cells by high-efficiency protein transduction. Stem Cells Transl Med 4:1415–1424
Galloway CA, Sowden MP, Smith HC (2003) Increasing the yield of soluble recombinant protein expressed in E. coli by induction during late log phase. Biotechniques 34:524–530
Ou J, Wang L, Ding X, Du J, Zhang Y, Chen H, Xu A (2004) Stationary phase protein overproduction is a fundamental capability of Escherichia coli. Biochem Biophys Res Commun 314:174–180
Sørensen HP, Mortensen KK (2005) Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact 4:1
Rabhi-Essafi I, Sadok A, Khalaf N, Fathallah DM (2007) A strategy for high-level expression of soluble and functional human interferon α as a GST-fusion protein in E. coli. Protein Eng Des Sel 20:201–209
San-Miguel T, Pérez-Bermúdez P, Gavidia I (2013) Production of soluble eukaryotic recombinant proteins in E. coli is favoured in early log-phase cultures induced at low temperature. Springerplus 2:89
García-Fraga B, Da Silva AF, López-Seijas J, Sieiro C (2015) Optimized expression conditions for enhancing production of two recombinant chitinolytic enzymes from different prokaryote domains. Bioprocess Biosyst Eng 38:2477–2486
Araki Y, Hamafuji T, Noguchi C, Shimizu N (2012) Efficient recombinant production in mammalian cells using a novel IR/MAR gene amplification method. PLoS ONE 7:e41787
Karpievitch YV, Polpitiya AD, Anderson GA, Smith RD, Dabney AR (2010) Liquid chromatography mass spectrometry-based proteomics: biological and technological aspects. Ann Appl Stat 4:1797
Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1:2876
Kelly SM, Jess TJ, Price NC (2005) How to study proteins by circular dichroism. Biochim Biophys Acta Proteins Proteomics 1751:119–139
Haridhasapavalan KK, Borgohain MP, Dey C, Saha B, Narayan G, Kumar S, Thummer RP (2019) An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 686:146–159
Acknowledgements
We thank all the members of the Laboratory for Stem Cell Engineering and Regenerative Medicine (SCERM) for their critical reading and excellent support. The authors gratefully acknowledge the support of DBT Program Support (Prof. S.S. Ghosh), Department of Biosciences and Bioengineering, IIT Guwahati for their assistance in Circular Dichroism experiments. This work was supported by a research grant from Science and Engineering Research Board (SERB), Department of Science and Technology, Government of India (Early Career Research Award; ECR/2015/000193) and IIT Guwahati Institutional Start-Up Grant.
Author information
Authors and Affiliations
Contributions
KKH was responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing and final approval of the manuscript; PKS, SB, SHR, KR were responsible for collection and/or assembly of data, data analysis and interpretation and final editing and approval of the manuscript; RPT was responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript and financial support. All the authors gave consent for publication.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.

449_2021_2516_MOESM3_ESM.jpg
Fig. S1 Codon optimization analysis using Graphical codon usage analyzer. The bar graphs shown here are the codons (Codon number 51 onwards) of non-optimized and codon-optimized GATA4 gene sequences against relative adaptiveness values (details as explained in Fig. 1a legend). (JPG 4111 KB)
449_2021_2516_MOESM4_ESM.tif
Fig. S2 Expression analysis, purification and characterization of the recombinant GATA4 fusion protein. a Schematic diagram of human HTN-GATA4 insert (not drawn to scale). The GATA4 gene was fused with nucleotide sequences of His tag (H) for affinity chromatography-based purification followed by TAT (T) and NLS (N) and to facilitate cell penetration and nuclear translocation in mammalian cells, respectively. b Restriction digestion analysis was performed to confirm the successful cloning of human HTN-GATA4 insert into the protein expression vector. The synthetic gene insert was cloned in the protein expression vector, pET28a(+), using restriction endonucleases, NcoI, and XhoI. The resulting plasmid, pET28a(+)-HTN-GATA4 (in short HTN-GATA4) was then confirmed by restriction digestion using various restriction enzymes, as depicted in (a) and (b). c Soluble expression analysis of HTN-GATA4 protein, induced at two different temperatures. E. coli BL21(DE3) transformed with recombinant plasmid HTN-GATA4 was induced at the early log phase (OD600 = ~0.5) with 0.25 mM of IPTG at two different temperatures (37 °C for 2 hours, and 18 °C for 24 hours). Harvested cells were lysed by ultrasonication to obtain total cell lysate (L) fraction. These fractions were then centrifuged to obtain a pellet/insoluble (P) and a soluble (S) fractions. These fractions (L, P, and S) were run on SDS-PAGE with normalized protein loading concentration of 20 μg for L fractions. Equal volume corresponding to the respective L fraction was loaded for P and S fractions*. d Purification of recombinant HTN-GATA4 protein. E. coli BL21(DE3) transformed with recombinant plasmid HTN-GATA4 was induced at the early log phase (OD600 = ~0.5) with 0.25 mM of IPTG at 37 °C for 2 hours with continuous shaking at 180 rpm. Harvested cells were lysed by ultrasonication, and the expressed protein was purified under native conditions using Ni-NTA affinity chromatography. The purification samples were run on SDS-PAGE with normalized loading volume*. e Secondary structure determination for the purified recombinant HTN-GATA4 protein. The secondary structure was determined using far UV CD spectroscopy for the purified recombinant HTN-GATA4 protein in 20 mM PB (pH 8.0 at room temperature). The CD data obtained were then analyzed, and CD spectra have been plotted with Delta Epsilon (M-1 cm-1; Y-axis) against wavelength (nM; X-axis).*The resolved polyacrylamide gel was stained with Coomassie Brilliant Blue G-250 (top) or transferred to nitrocellulose membrane and performed western blotting with His antibody (bottom). NLS: nuclear localization signal/sequence; TAT: transactivator of transcription; His tag: polyhistidine (8X); M: Protein Marker (kDa); L: Total cell lysate; P: Pellet/insoluble cell fraction; S: Soluble cell fraction; F: Flow-through fraction; W1: Wash buffer 1; W2: Wash buffer 2; W3: Wash buffer 3; E: Elution; Ab: Antibody. (TIF 5633 KB)
449_2021_2516_MOESM5_ESM.tif
Fig. S3 Purification of human GATA4 fusion protein under mild denaturing conditions. E. coli BL21(DE3) transformed with recombinant plasmid GATA4-NTH was induced under optimal expression conditions (temperature: 37 °C; cell density: OD600 = ~0.5; IPTG concentration: 0.25 mM; induction time: 2 hours). Harvested cells were lysed by ultrasonication and clarified by centrifugation. Then, the clarified soluble cell fraction was treated with different concentrations of urea (0/2/4 M) at 4 °C for 12 hours with continuous shaking followed by purification using Ni-NTA affinity chromatography. a Identification of the minimal denaturant concentration required for the maximum purification yield. b Purification of GATA4-NTH under mild denaturing conditions using an optimal concentration of urea (4 M). The purification samples were run on SDS-PAGE with normalized loading volume. The resolved polyacrylamide gel was stained with Coomassie Brilliant Blue G-250 (top) or transferred to nitrocellulose membrane and performed western blotting with Histidine antibody (bottom). *Truncated GATA4-NTH protein. c Secondary structure determination for the purified recombinant GATA4-NTH protein under mild denaturing conditions. The secondary structure was determined using far UV CD spectroscopy for the purified and dialyzed recombinant GATA4-NTH protein in 20 mM PB (pH 8.0 at room temperature). The CD data obtained were then analyzed, and CD spectra have been plotted with wavelength (nM; X-axis) against Delta Epsilon (M-1 cm-1; Y-axis). M: Protein Marker (kDa); L: Total cell lysate; S: Soluble cell fraction; F: Flow-through fraction; W1: Wash buffer 1; W2: Wash buffer 2; W3: Wash buffer 3; E: Elution; Ab: Antibody (TIF 4103 KB)
Rights and permissions
About this article

{kind=link}
Cite this article
Haridhasapavalan, K.K., Sundaravadivelu, P.K., Bhattacharyya, S. et al. Generation of cell-permeant recombinant human transcription factor GATA4 from E. coli. Bioprocess Biosyst Eng 44, 1131–1146 (2021). https://doi.org/10.1007/s00449-021-02516-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00449-021-02516-8