
Abstract
Sphingolipids (SPs) comprise a highly diverse class of lipids that serve biological roles both as structural components of cell membranes and as mediators of cell signaling. Pharmacologic and genetic manipulation of SPs and their signaling systems have underscored their importance in most biological processes, including central nervous system development and function. Likewise, perturbations of SP accumulation or signaling have been associated with a number of disease states, such as neural tube defects, neuroinflammation, stroke, and dementia. SPs can be endogenously synthesized de novo, and their metabolism is a well-regulated process, so their value as nutraceuticals has not been scrutinized. However, there is evidence that sphingolipid-rich diets can affect lipid homeostasis, and several mycotoxins are SP analogs that are known to cause profound derangement of SP metabolism or signaling. Furthermore, plants and invertebrates have SP species that are not present in mammals. Several of these have been shown to induce biological responses in mammalian cells. These findings suggest that dietary intake of SPs or SP analogs may have significant effects on human health or disease outcome. This manuscript provides an overview of SP metabolism and signaling, their perturbations in neurological diseases, as well as potential impacts of modulating this system in the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction to Sphingolipid Structure and Metabolism
Structural characteristics. SPs comprise one of the eight major, structurally distinct classes of lipids (Fahy et al. 2005). They are ubiquitous in all eukaryotic cells and in some prokaryotes such as anaerobic bacteria (Fyrst and Saba 2010; Olsen and Jantzen 2001). They are highly abundant in mammalian plasma membranes and make up approximately 20 % of the lipids in blood plasma (Shui et al. 2011). The defining characteristic of all SPs is the presence of a sphingoid backbone or “long-chain base” (LCB), which consists of a nitrogenous amine headgroup and an acyl chain (Fig. 1a). While there is some variability in the chain length (see below), the majority of sphingoid backbones in mammalian SPs have 18 carbons (Quehenberger et al. 2010). These are typically saturated or monounsaturated, but quantitatively minor species of LCBs have been identified that contain two double bonds at stereotyped positions (sphingadienes, see Renkonen and Hirvisalo 1969; Panganamala et al. 1969). The simplest members of the family (such as the canonical SP, sphingosine) consist only of an LCB, but modifications to this primary structure provide the material for highly diverse structural heterogeneity. Indeed, due to potential combinations of the known variations in the headgroup, the N-linked acyl chain, hydroxylation, and the length and degree of saturation of the LCB, theoretically, there are tens of thousands of unique SPs. However, of these possible combinations, 620 have been systematically classified as confirmed species or presumptive intermediates (Quehenberger et al. 2010; Merrill et al. 2005; Fahy et al. 2009).

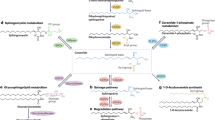
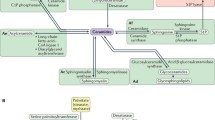
Sphingolipid structure and metabolism. a Minimum structural requirement of typical SPs. R1 = H, carbohydrate(s), sialic acid(s), sulfonic acid. R2 = saturated or mono unsaturated acyl chain. R3 = H, or acyl chain. b Sphingolipid metabolic pathway. De novo synthesis is the major source of SPs in mammals, which begins with the condensation of serine and a fatty acyl-CoA, eventually leading to the formation of ceramide, a key metabolic intermediate which may be converted to sphingomyelins or complex ceramides. These “higher-order” SPs may be readily used to regenerate ceramide via the sphingomyelinase pathway or the salvage pathway, respectively. Irreversible degradation of all SPs is catalyzed by the activity of S1P lyase, resulting in the generation of a fatty aldehyde and phosphoethanolamine, which may serve as a substrate for the generation of phospholipids via the Kennedy pathway
General Overview of SP Metabolism
SPs can be obtained through the diet and incorporated into endogenous lipid metabolic pathways, but the majority of functionally active SPs are generated by de novo synthesis (Pyne and Pyne 2000). De novo synthesis begins with the condensation of a saturated fatty acyl-CoA and an amino acid, usually serine, via the action of serine palmitoyltransferase (SPT) as the first committed step in the generation of SPs (Fig. 1b). This results in saturated LCB intermediates (“sphinganines”) that are rapidly N-acetylated to form dihydroceramide, which is then acted upon by a sphingolipid Δ4-desaturase to introduce a double bond that generates ceramide. Much of the structural diversity of SPs results from head group modifications to ceramide. The addition of phosphorylcholine or phosphorylethanolamine results in the formation of sphingomyelin, the most abundant SP making up to 20 % of the plasma membrane. Complex ceramides, or “higher-order sphingolipids,” can be generated by the addition of one or more carbohydrate groups. A single monosaccharide head group, typically glucose or galactose, results in the generation of a cerebroside. Increasing complexity occurs through elongation of this head group by further addition of monosaccharides (glucose, galactose, mannose, or fucose) or sialic acid, or by sulfonation. Complex ceramides are abundant in many tissues, particularly the brain, and can serve as a reservoir for the rapid generation of signaling pools of ceramide through the “salvage pathway” (Kitatani et al. 2008).
SP Degradation
Ceramide is deacylated to form sphingosine, the first described SP (Thudichum et al. 1884). By convention, the term “sphingosine” typically refers to 18-carbon, Δ4-monounsaturated, 1,3-dihydroxy sphingosine (d-erythro-sphingosine), which is the most abundant LCB in mammals. It is interesting to note that there is no known pathway by which sphingosine can be formed without an N-acetylated intermediate. That is, non-acetylated LCBs cannot be directly desaturated (Kitatani et al. 2008). Once formed, sphingosine can then be phosphorylated to sphingosine 1-phosphate (S1P) via the action of two isoforms of sphingosine kinase: SPHK1 and SPHK2. S1P can then be irreversibly degraded by the action of S1P lyase (Saba et al. 1997), representing the only means by which SPs eliminated. While S1P was originally considered to be an inert intermediate in SP metabolism, it has become the most intensively studied SP after it was identified as a highly potent ligand for a family of cell surface receptors (Lee et al. 1998; Mutoh et al. 2012). Since SPHK1/SPHK2 activity controls S1P content in most tissues, this places these two enzymes as key regulators of many S1P signaling processes, as described below. S1P degradation via S1P lyase may contribute to the regulation of other phospholipid classes in that the breakdown of SPs results in the production of a fatty aldehyde and phosphoethanolamine, the latter of which is a substrate for phosphatidylethanolamine synthesis via the Kennedy pathway. Although SP metabolism is a relatively minor source of ethanolamine in mammals (Gibellini and Smith 2010), it has been shown that this may play a significant role in the regulation of the Kennedy pathway in some organisms (Zhang et al. 2007).
Phylogenic Variations in SP Structure
While SPs are ubiquitously found in eukaryotes, there are significant differences in the LCB structures that are utilized by different organisms. For example, insect SPs are typically shorter than their mammalian counterparts, with the most abundant LCBs containing 14 rather than 18 carbons (Fyrst et al. 2004). Yeast, on the other hand, contains LCBs with 16–20 carbons, but is typically saturated with backbones consisting of dihydrosphingosine or phytosphingosine (Cowart and Obeid 2007; Dickson 2008; Fig. 2). Increasingly divergent structures have been identified in plants, fungi, and marine invertebrates. LCBs with branched backbones (Pruett et al. 2008) or “bifunctional” SPs with multiple headgroups (Bensemhoun et al. 2008) have been isolated from phylogenetically distant organisms but appear to be rare or absent in mammals. Interestingly, however, some of these compounds have been shown to have potent biological effects on mammalian cells.

Structures of key sphingolipids and sphingolipid analogs
Effects of Bioactive Sphingolipids on Brain Development and Function
SPs are ubiquitous and, as such, regulate biological processes in many tissues. This review will emphasize the roles of SPs in the CNS since its biomedical relevance is underscored by the fact that the only FDA-approved drug specifically acting on the SP system was approved for a neurological indication (see “Myriocin/Fingolimod” section below). However, other systems will be briefly discussed when necessary for context.
Taken as a whole, SPs are known to play critical roles in most aspects of normal brain function, such as neurogenesis and maintenance of neural stem cells (Kornhuber et al. 2014; Wang et al. 2014); integral structural components of both white and gray matter (O’Brien and Sampson 1965); formation and maintenance of lipid rafts which in turn regulate neuronal signaling (Aureli et al. 2015); and signaling molecules themselves modulating a wide range of responses, including neurotransmission, inflammation, cellular stress, autophagy, and apoptosis (Ghasemi et al. 2016; Ong et al. 2015; Pyne and Pyne 2000). Not surprisingly, perturbations of SP synthesis or regulation have been implicated in several neurological diseases, and specific phospholipids as well as associated enzymes and receptors have been investigated as potential therapeutic targets or biomarkers (Gomez-Munoz et al. 2016; Kornhuber et al. 2014; Mielke and Haughey 2012; Mutoh et al. 2012; Ong et al. 2015). In the following sections, we provide an overview of the biological functions of selected SPs, as well as their established or putative involvement in neurological diseases.
Ceramides
Ceramides are particularly well-studied SPs, primarily known for their pro-apoptotic effects, but also for regulating a number of important cellular processes (Fyrst and Saba 2010). Because of the remarkable structural diversity of ceramides, a comprehensive discussion of their roles is beyond the scope of this manuscript. Additional information may be found in more focused reviews (Reynolds et al. 2004; Ben-David and Futerman 2010). Ceramide can be rapidly generated in the CNS by tapping into the large reservoir of sphingomyelin in the plasma membrane. This occurs through the action of sphingomyelinase and can induce a number of effects such as neurotransmitter release, inflammation, and apoptosis of a number of cell types in the brain (Ong et al. 2015). Studies have also provided evidence for a possible bimodal role of ceramide on the effect of ethanol on the CNS. Excessive ceramide accumulation is thought to be generally neurodegenerative (Jana et al. 2009), and it has been suggested that this may mediate the harmful effect of alcohol consumption (de la Monte et al. 2009). Indeed, acute administration of ethanol in rats causes an increase in brain ceramide in adult and fetal brain (Saito et al. 2010). However, chronic voluntary consumption by ethanol-preferring rats resulted in a decrease in brain ceramides (Godfrey et al. 2015), suggesting that moderate ethanol intake may promote healthful bioactive lipid profiles. To further illustrate the conditional nature of ceramide function, a recent genetic analysis of ceramide metabolism identified a complicated relationship between ceramide and autophagy (Hebbar et al. 2015). When autophagic flux is inhibited in the brain, ceramide clearance is reduced and ceramides accumulate, accompanied by neurodegeneration (Hebbar et al. 2015; Finley et al. 2003). Unexpectedly, genetic reduction of ceramide (by reducing de novo synthesis) exacerbates the neurodegenerative phenotype, while increasing ceramide production through the salvage pathway reduces neurodegeneration. Apparently, although disruption of autophagy elevates ceramides, they remain sequestered in autophagosomes and are unable to engage their normal signaling functions. This provides evidence for a functional distinction among different metabolic sources of ceramide (Fig. 1b) and for the importance of subcellular localization in SP function.
Given the functional and pathogenic potential of ceramide perturbations, much research effort has understandably been focused on sphingomyelinase whose action is a main source of brain ceramides (see above). Altered regulation of sphingomyelinase activity has now been implicated in several neurological diseases or systemic diseases with CNS involvement, the best characterized of which are the Niemann–Pick disease types A and B arising from a deficiency of the acid sphingomyelinase SMPD1 gene. The resulting symptoms from the accumulation of ceramides in multiple organs are often systemic and may include cerebellar atrophy and dementia (Hulette et al. 1992; Obenberger et al. 1999; Macauley et al. 2008). Besides Niemann–Pick and related sphingolipidoses, altered sphingomyelinase function is thought to be involved in the progression of a number of diseases including Alzheimer’s disease, pain, and stroke, and sphingomyelinase may also be a target for novel antidepressant therapies (Gulbins et al. 2015; Ong et al. 2015).
Ceramides can be phosphorylated by the action of ceramide kinase (CERK) to form another bioactive lipid, ceramide 1-phosphate (C1P). C1P has a number of documented effects on mammalian cells that are generally proliferative or pro-inflammatory (reviewed in: Hoeferlin et al. 2013; Gomez-Munoz et al. 2016). These effects are mediated by intracellular targets rather than cell surface receptors and involve the activation of phospholipase A2, the MAPK/ERK pathway, and phosphoinositol 3-kinase (Lamour and Chalfant 2005; Arana et al. 2010). The role of C1P in the brain is largely unknown; however, several lines of evidence suggest biological significance in the CNS. For example, CERK activity was first identified in brain synaptic vesicles (Bajjalieh et al. 1989), and C1P is particularly abundant in the brain tissue (Yamashita et al. 2016). Furthermore, it was shown that C1P can regulate calcium entry in pituitary cells, suggesting that it may modulate neuroendocrine secretion (Tornquist et al. 2004).
Sphingosine 1-Phosphate (S1P)
A paradigm shift in our understanding of the biological roles of sphingolipids occurred when it was discovered that some SP species could act as high-affinity, cognate ligands for G protein-coupled receptors (GPCRs; Lee et al. 1998). While a number of reports demonstrate that S1P may act as a second messenger on intracellular targets such as histone deacetylase and tumor necrosis factor-associated factor 2 (Kunkel et al. 2013), the majority of the biological effects of S1P are known to be mediated by cell surface receptors. Specifically, a family of 5 GPCRs has been identified as bona fide sphingosine 1-phosphate (S1P) receptors (Kihara et al. 2014), which has instigated a profound research effort into understanding the biological roles of S1P. These receptors (S1P1–S1P5) are expressed in distinct, but overlapping patterns in most mammalian cell types. As such, S1P can exert a wide spectrum of physiological responses depending on the cellular context, i.e., the complement of S1P receptors, and the presence of downstream effectors for receptor-specific signaling pathways. In the central nervous system, S1P receptors are expressed in neurons, glia, microglia, and endothelial cells and have been shown to participate in the regulation of multiple aspects of tissue development and cellular function (Herr and Chun 2007). The biological relevance of this signaling system has been established by in vitro and in vivo studies and by clinical evidence.
Numerous studies demonstrate that S1P induces responses in cultured astrocytes suggestive of an overall pro-inflammatory effect. Notably, S1P treatment stimulates growth and proliferation (Pebay et al. 2001) and induces the production of growth factors such as nerve growth factor (Furukawa et al. 2007), fibroblast growth factor-2 (Malchinkhuu et al. 2003; Sato et al. 1999), glial-derived neurotrophic factor (Yamagata et al. 2003), and arachidonic acid (Rao et al. 2003). This was shown to be functionally relevant in that S1P-primed astrocytes were able to promote the differentiation of neural progenitor cells (Spohr et al. 2012). These effects appear to be mediated by S1P1, S1P2, and S1P3 (Rao et al. 2003). S1P receptors are also expressed in oligodendrocytes and mediate cell growth pathways (Yu et al. 2004). S1P5 may be the most relevant mediator of S1P signaling for myelination since CNS expression of S1P5 appears to be enriched in oligodendrocytes (Jaillard et al. 2005), although its function is not well characterized. While S1P5 does induce process retraction and cell survival in oligodendrocytes, there is no obvious myelination defect in S1P5-null mice (Jaillard et al. 2005). In addition to S1P5, there is evidence for the involvement of S1P1 in regulation of myelination in that oligodendrocyte-specific conditional deletion of S1P1 results in delayed differentiation of oligodendrocyte precursors (Dukala and Soliven 2016).
A number of mouse models have provided clear evidence for the involvement of S1P receptor-mediated signaling in CNS function in vivo (Yang et al. 2002). For example, genetic deletion of both isoforms of sphingosine kinase results in a severe depletion of S1P and embryonic lethality (Mizugishi et al. 2005). This is due to vascular defects and disruption of neurogenesis, demonstrating that S1P is essential for CNS development. On the other hand, increased S1P was found to accompany white matter lesions in an adult rodent bilateral carotid stenosis model of chronic hypoperfusion, both of which were ameliorated with sphingosine kinase inhibitor (SKI-II) treatment (Yang et al. 2016), although the effects of SKI-II may also involve its activity as an inhibitor of dihydroceramide desaturase (McNaughton et al. 2016; Cingolani et al. 2014). Taken together, these results suggest differential effects of S1P and sphingosine kinase alterations in different neurodevelopmental stages. Furthermore, S1P2-specific knockout mice present with several rather pronounced neural phenotypes. Notably, young S1pr2 −/− mice display frequent seizures characterized by wild-running episodes followed by tonic–clonic episodes that are occasionally fatal (MacLennan et al. 2001; Akahoshi et al. 2011). This is associated with increased excitability of cortical neurons (MacLennan et al. 2001). Furthermore, surviving S1pr2 −/− mice uniformly display progressive hearing loss due to degeneration of hair cells and the afferent neurons of the spiral ganglia (Herr et al. 2007). This phenotype has recently been observed in humans who carry missense mutations of the S1P2 gene (Santos-Cortez et al. 2016). Recently, we demonstrated that selective activation of S1P2 is cytoprotective to neural cell lines by reduction of oxidative stress, suggesting that an S1P2 agonist may be neuroprotective and/or otoprotective (Herr et al. 2016). Cumulatively, this demonstrates that S1P signaling plays an important role in the development and function of the nervous system.
It is likely that the effects of S1P in the CNS, particularly the glia, are relevant to neurological diseases. For example, it was shown that intracranial injection of S1P results in the robust activation of reactive astrogliosis (Sorensen et al. 2003), indicating that S1P is a regulator of neuroinflammation. This was validated in a mouse model for multiple sclerosis, whereby astrocyte-specific deletion of S1P1 results in a significant reduction in inflammatory cytokine production due to autoimmune challenge (Choi et al. 2011). Furthermore, S1P signaling has been shown to regulate neuronal loss in stroke models. Notably, transient middle cerebral artery occlusion/reperfusion results in an increase in S1P, S1P3, and neuroinflammation, which can be attenuated by broad-spectrum antagonism of S1P receptors (Moon et al. 2015). In addition, S1P2 may be an important regulator of vascular permeability and blood–brain barrier integrity during ischemia–reperfusion (Kim et al. 2015). These processes are likely to be clinically relevant since numerous studies collectively demonstrate that the S1P receptor modulator, fingolimod, reduces stroke damage in animal models (Liu et al. 2013).
Non-canonical Sphingolipids with Known Effects on Mammalian Cells
Mammalian SPs are generally characterized as structures containing the canonical sphingosine backbone. Variations of this typical structure are present, but at relatively low concentrations. For example, LCBs can contain 16–20 carbons, but more than 90 % are 18-carbon species (Quehenberger et al. 2010). Similarly, there are highly constrained variations in degree of saturation and hydroxylation. Recently, a number of studies have provided evidence that these non-canonical SPs have distinct biological effects, which suggests that they may be well-regulated mediators of cellular processes. This section describes variants that are emerging as potentially important, bioactive lipids.
Variations in Chain Length
The sphingoid base of SPs can vary in a number of ways, but much of the diversity in sphingoid bases is given by their different chain lengths. In mammals, the most abundant and ubiquitous long-chain base variant has 18 carbons (C18; Merrill 2011), but other forms, with shorter or longer chains, have been found in lower amounts. SPs containing a C16 sphingoid base have been detected in plasma (Quehenberger et al. 2010), heart tissue (Russo et al. 2013), and bovine milk (Byrdwell and Perry 2007), while a C20 sphingoid base has been found at high levels in brain SPs (especially gangliosides; Ikeda and Taguchi 2010), and in low levels in plasma (Narayanaswamy et al. 2014) and mucosal tissues (Keranen 1976). The presence of odd-chain sphingosines in low amounts has also been confirmed, and C17 or C19 (possibly also including branched chains) has been detected in human plasma (Quehenberger et al. 2010) and skin (Pruett et al. 2008). Of all tissues analyzed so far, skin has demonstrated the highest diversity in SPs, apparently due to their fundamental role in maintaining epidermal permeability barrier (t’Kindt et al. 2012; Stahlberg et al. 2015). According to t’Kindt et al. (2012), the number of unique ceramide species present in human skin is around a thousand. The sphingoid base length distribution in skin SPs can vary between 16 and 26 carbons. Interestingly, phytosphingosine (with carbon numbers between 16 and 22), usually present in non-detectable amount in the majority of tissues, is the most abundant LCB in skin. A very unusual characteristic of skin ceramides is also the large content of C26 dihydrosphingosine, usually present with 18–20 carbon atoms in other tissues. It is still not clear whether the heterogeneous SP composition of the skin is only due to a different SP-related gene expression in epithelial cells, or whether it is also a result of a contribution of lipids derived from the microorganisms that colonize the skin surface (De Luca and Valacchi 2010).
In addition to the chain length diversity of the SP backbone, there is also remarkable variation in the chain length of the N-linked fatty acid (FA) of ceramides (Fig. 1b). The most structurally complex ceramides have been also observed in skin, containing extremely FAs, up to C36. Interestingly, skin ceramides have been reported that contain a unique modification where an ω-carbon group is hydroxylated and esterified to another fatty acid (typically linoleic acid), forming “ω-O-acyl ceramide” (Novotny et al. 2010). This type of ceramide crucially contributes to the permeability properties of the skin lipid layers, due to its increased hydrophobicity, and its synthesis is often deregulated in skin-related pathologies like ichthyosis (Aldahmesh et al. 2011) or psoriasis (Motta et al. 1994). Ceramides with very short fatty acids, C2-ceramide, have also been found in mammals (Van Overloop et al. 2007). They are formed by the transfer of an acetyl group from platelet-activating factor (PAF) to sphinganine and have been detected in murine brain and liver (Lee et al. 1996), although they are present at a very low level when compared to more common molecular species. This C2-ceramide can also be converted to ceramide 1-phosphate, but its function is still unclear (Van Overloop et al. 2007).
At this point of time, not much is known about the biological effect of this chain length variability despite their recent detailed structural analyses. The reason behind this growing interest is that different structures present different biophysical properties, and this can influence membrane stability (Slotte 2016), binding with cognate receptors (Hanson et al. 2012), transporter function (Christoffersen et al. 2011), or enzymatic activity (Levy and Futerman 2010). S1P serves as an example of a specific function linked to structural requirements. S1P1 is the best studied S1P receptor and preferentially binds C18 S1P (Hanson et al. 2012). The interactions between S1P and S1P1 are both polar, with the zwitterionic headgroup of S1P, and hydrophobic, with its acyl chain tail (Yuan et al. 2013). All five S1P receptors have conserved residues for the binding to the S1P headgroup, but they show an interesting diversity in the residues that line the hydrophobic cavity where the acyl chain of S1P is bound (Pulkoski-Gross et al. 2015). Ligands of S1P1 can behave as agonists or antagonists depending on the length and position of their acyl chain in the binding pocket of the receptor (Hanson et al. 2012), where increasing chain lengths can shift the effect from antagonism to agonism. Since more molecular variants of S1P have been identified in mammals (Narayanaswamy et al. 2014)—variable in both chain length and degree of saturation—new developments in this field will likely identify how such variants affect the selectivity and signaling of their cognate receptors. This is of fundamental importance as structural analogues of S1P are used to treat multiple sclerosis (Willis and Cohen 2013) and rheumatoid arthritis (Fleischmann 2012).
The heterogeneity of the LCB chain length depends on the activity of the SPT enzyme, which is responsible for the first step in de novo synthesis (Fig. 1b). The combination of different catalytic subunits (SPTLC2 or 3; Hornemann et al. 2007, 2009) and other putative small subunits (ssSPTa, ssSPTb; Han et al. 2009) can shift the acyl-CoA substrate preference of the enzyme. The myocardium has highest expression of SPTLC3 of any tissue evaluated (Hornemann et al. 2009), and as a consequence its content in SPs containing C16 sphingoid bases is unusually high. This observation brought to the discovery that in mice, a diet rich in myristate enhances the expression of SPTLC3, promoting the synthesis of d16 SLs in the myocardium. To study the physiological effect of C16 sphinganine on mammalian cells, cultured myocytes were treated with C16 or C18 sphinganine. The treatment with C16 resulted in PARP-mediated apoptosis; however, C18 sphinganine did not show the same effect and instead induced autophagy, clearly suggesting that different chain length-containing SPs can have different functions (Russo et al. 2013).
Sphingadienes
The predominant LCB species in mammalian cells are dihydrosphingosine (also known as sphinganine) and sphingosine (Merrill et al. 1988; Valsecchi et al. 1996). Sphingosine contains an unsaturated hydrocarbon chain with one double bond at the Δ4 position, which is absent in dihydrosphingosine (Fig. 2). Besides dihydrosphingosine and sphingosine, studies have also revealed the presence of other LCBs, which contribute to the formation of non-canonical sphingolipids (Fyrst et al. 2008; Renkonen and Hirvisalo 1969). These LCB species were shown to possess an additional double bond at various positions along the hydrocarbon chain (T. U. Abeytunga 2015) and are commonly referred to as sphingadienes or sphingadienines (SDs; Renkonen and Hirvisalo 1969; Fyrst et al. 2008). The exact biosynthesis pathway(s) of SDs remain(s) elusive, but it has been suggested to involve desaturase enzymatic activity on sphingosine to introduce the second double bond into the hydrocarbon chain (Fyrst et al. 2008). These can then be readily incorporated into more complex SPs such as glucosylceramides (Abeytunga et al. 2008). Regulation of SDs was found to be highly dependent on S1P lyase (SPL) activity as shown by an increased accumulation of SDs in the Drosophila SPL mutant (Sply; Fyrst et al. 2008). SDs are most commonly found in plants such as soybean (Lynch and Dunn 2004), as well as being present across various organisms where the second double bond is located at different positions along the hydrocarbon chain (Table 1).
SPs obtained from the diet will undergo enzymatic lipolyzation in the intestine to form free LCBs and ceramide (Imai et al. 1997; Ohlsson et al. 2010; Sugawara et al. 2003; Rozema et al. 2012b; Nyberg et al. 1997). This suggests that diets high in SD-rich food such as soy may result in significant exposure to SD isoforms that are not endogenously produced. While there is no consensus regarding the impact of this on human health, there are several lines of evidence that suggest that SDs exert potent effects on mammalian cells. For example, Δ4,8-C18-sphingadiene and 9-methyl-Δ4,8-C18-sphingadiene are the major constituents of plants cerebrosides, which were found to exhibit calcium ion ionophoretic properties and anti-ulcerogenic effects in addition to inhibiting replicative DNA polymerase activities (Okuyama and Yamazaki 1983; Shibuya et al. 1990; Mizushina et al. 1998). Moreover, dietary monoglucosylceramide from rice bran, which contains a Δ4,8-C18-sphingadiene backbone, was found to inhibit preneoplastic lesions in the colon of F344 rats (Inamine et al. 2005). Sphingoid bases from lactic yeast and maize were also shown to induce apoptosis in the Caco-2 human colon cancer cell line via caspase activation and reduction of intracellular β-catenin levels (Aida et al. 2004). In addition, sea cucumber sphingoid bases comprising sphingadienes were shown to dose-dependently inhibit the viability of three different human colon cancer cell lines (Sugawara et al. 2006). Soy glucosylceramide, which similarly contains a Δ4,8-C18-sphingadiene backbone, was found to inhibit colon tumorigenesis and reduce gene expression of transcription factors linked to cancer in two different mouse models (Symolon et al. 2004). The role of SDs as the active constituent of SPs in reducing colon tumorigenesis was further validated when both Δ4,6-C14-sphingadiene and Δ4,8-C18-sphingadiene reduced colon cancer cell viability in a time- and dose-dependent manner (Fyrst et al. 2009). SD-induced cell death was found to involve the PI3K/Akt pathway since translocation, activation, and cellular signaling of Akt in the colon cancer cells were repressed after SD treatment (Fyrst et al. 2009). Additionally, the chemopreventive effects of SDs were suggested to involve Wnt signaling inhibition via a protein phosphatase 2A/Akt/GSK3β-dependent pathway (Kumar et al. 2012). Administration of Δ4,6-C14-sphingadiene also reduced intestinal tumorigenesis in vivo (Fyrst et al. 2009). Interestingly, both Δ4,6-C14-sphingadiene and Δ4,8-C18-sphingadiene had more pronounced cytotoxic effects on the colon cancer cells as compared to soy glucosylceramide, suggesting that enzymatic metabolism of glucosylceramide in the gut to release SDs is essential for their cytotoxic and tumor repression activities to occur (Fyrst et al. 2009).
The cytotoxicity of SDs is not limited to colon cancer cells. Novel ceramide analogs with the Δ4,6-C18-SD backbone induced apoptotic cell death in the MCF-7 and MDA-MB-231 breast cancer cell lines via mitochondrial activation (Struckhoff et al. 2004). Besides inducing cytotoxicity in different cancer cell lines such as HL-60 and RAW 264.7, Δ4,8-C18-sphingadiene also decreased cell viability and proliferation in non-cancerous cell lines such as HUVEC and HEK-293 (Rozema et al. 2012b). Nonetheless, ceramide analogs with the SD backbone exhibited reduced cytotoxicity toward normal breast epithelial cells, while non-cancerous human colon mucosal epithelial cells also showed lower sensitivity to SD-induced cytotoxicity (Struckhoff et al. 2004; Fyrst et al. 2009).
The reason that this minor structural change results in significantly increased cytotoxicity is unclear; however, it may in part be explained by derangement of the sphingolipid rheostat. The oncogenic enzyme sphingosine kinase 1 (SPHK1), which is elevated in colon carcinogenesis, can phosphorylate sphingosine to form S1P. This shifts the rheostat toward phosphorylation, which is implicated in angiogenesis formation, Akt activation, and pro-tumorigenic inflammatory pathway signaling (Lee et al. 1999; Lee et al. 2010; Xia et al. 2000; Kawamori et al. 2006, 2009; Chumanevich et al. 2010; Kumar et al. 2012). The presence of the second double bond in sphingadienes was also suggested to contribute to their higher cytotoxicity in cancer cells as compared to ceramide or sphingosine (Struckhoff et al. 2004).
The suggested pharmacological actions of sphingadienes were not just limited to their cytotoxicity in cancer cell lines. Δ4,8-C18-sphingadiene was also shown to exert anti-inflammatory effects via inhibition of lipopolysaccharide (LPS) and tumor necrosis factor-α (TNF-α)-induced cytokine interleukin-8 (IL-8) and the inflammatory adhesion molecule E-selectin (Rozema et al. 2012a). Interestingly, lower concentrations of Δ4,8-C18-sphingadiene were required for its anti-inflammatory effects compared to the concentrations where cytotoxic effects were observed (Rozema et al. 2012a). In addition, sphingolipids containing a Δ4,6-C14-sphingadiene backbone were found to induce neurotrophic effects and increased neurite outgrowth in PC12 cells similar to the effects of the nerve growth factor (NGF; Kwon et al. 2003).
Results from these studies suggest the possibility of utilizing SDs not only in colon cancer treatment but also in neurodegenerative diseases, particularly when inflammation and neuronal loss are involved. Added to the fact that they can be readily obtained from the diet, SDs represent an attractive nutraceutical compound for possible treatment of cancer and brain neurodegeneration. Nonetheless, there is still much work to be done regarding the role of SDs in human physiology and pathology, their direct targets and signaling mechanisms, and the metabolic pathways involved in SDs generation.
Deoxysphingolipids
Deoxysphingolipids are atypical constituents of the sphingolipidome (Merrill et al. 2009) that were originally identified in fungal species and marine sponges. They have only recently been shown to be present at low concentrations in mammalian tissues (Duan and Merrill 2015). Deoxysphingolipids such as 1-deoxysphinganine differ from the typical sphingoid base (e.g., sphingosine) due to the absence of a hydroxyl group at C1 position (Bode et al. 2015), but they are structurally diverse and can range from simple to complex. 1-deoxy- and 1-deoxymethyl-sphingoid bases are structurally simple deoxysphingolipids that are generated by many organisms including mammals (Zitomer et al. 2009), while at the opposite end of the spectrum, the “two headed” structure of oceanapiside isolated from marine sponges represents one of the more unique and complex structures seen in SPs (Nicholas and Molinski 2000; Fig. 2).
Deoxysphingolipids were initially thought to be a class of mycotoxins that are particularly cytotoxic to plant and animal cells (Abbas et al. 1998). Recently, however, two groups made the surprising discovery that they are synthesized in mammalian cells in relatively low concentrations (Penno et al. 2010; Zitomer et al. 2009). SPT is mutated in hereditary sensory and autonomic neuropathy type 1 (HSAN1; Penno et al. 2010). These mutations have been shown to alter substrate specificity of SPT, from l-serine to l-alanine or glycine, which results in the synthesis of 1-deoxysphingolipid compounds such as 1-deoxysphinganine (Penno et al. 2010). In addition to synthesis of deoxysphingolipids by mutant SPT, the synthesis of 1-deoxysphingolipid from wild-type SPT was seen in cell lines incubated with fumonisin B1, which inhibits ceramide synthases and results in the accumulation of deoxysphingolipids (Zitomer et al. 2009; Venkataraman et al. 2002). CHO-LyB cells, which carry an inactive SPT subunit (SPT1), failed to synthesize 1-deoxysphingolipids from l-alanine but was genetically rescued with wild-type SPT, confirming that generation of deoxysphingolipids is the result of normal mammalian SP metabolism (Zitomer et al. 2009).
Ongoing work aims to understand the effects of these atypical lipids on human cells to develop a better understanding of their biological roles in health and disease. 1-deoxysphinganine has been studied for its cellular effects, as it is among the more abundant 1-deoxysphingolipids present in humans. While it can enhance proliferation in Swiss 3T3 fibroblasts (Schroeder et al. 1994), it has been shown to inhibit cell growth as in Vero kidney epithelial cells (Zitomer et al. 2009) and is cytotoxic to LNCaP and PC-3 prostate cancer cells at low micromolar concentrations (Sanchez et al. 2008). The growth inhibition and cytotoxicity of deoxysphingolipids have been attributed to de novo synthesis of ceramide, phosphorylation of P53, and activation of apoptosis (Sanchez et al. 2008; Salcedo et al. 2007).
HSAN1 patients show elevated plasma concentration of 1-deoxysphingolipids, which has been attributed to the neurodegeneration seen in HSAN1 (Penno et al. 2010). Similarly, the occurrence of diabetic neuropathy, a common and debilitating sequela of diabetes, correlates with elevation of 1-deoxysphingolipids in plasma samples of type II diabetes patients (Bertea et al. 2010). This was corroborated by an in vivo model in which supplementation of l-serine in a diabetic rat results in decreased plasma deoxysphingolipids and decreased neuropathic behavior (Othman et al. 2015). These findings support a possible connection between deoxysphingolipids and neuroinflammation/neurodegeneration. Recent evidence also indicates that 1-deoxysphingolipids play a role in TNF-mediated neurotoxicity of dopaminergic neurons (Martinez et al. 2012). Cumulatively, these data provide compelling evidence that deoxysphingolipids, although a quantitatively minor class of SLs, are important regulators of neuronal pathology. It has been proposed, however, that this cytotoxicity could be exploited therapeutically. For example, Enigmol, which is a synthetic 1-deoxysphingoid base, has shown to suppress tumors in colon and prostate cancer mouse models (Symolon et al. 2011; Garnier-Amblard et al. 2011).
Naturally Occurring Sphingolipid Modulators
It is likely that the dietary intake of structurally diverse SPs exerts significant biological effects on mammals. In addition, it is known that naturally occurring compounds, present in diet, have potent effects on SP metabolism and/or signaling. This can have a rather large impact on the composition of the sphingolipidome or may profoundly affect biological processes that are regulated by SP signal transduction. This section describes the most notable examples of SP analogs with known biological effects.
Myriocin/Fingolimod
Successful therapeutic relevance of sphingolipids has been realized through approval of a sphingosine analog known initially as FTY720 (Chiba et al. 1996) that has, through a tortuous process, become a drug known as Gilenya (generic name, fingolimod; Kappos et al. 2006; Brinkmann et al. 2010; Chun and Brinkmann 2011; Groves et al. 2013) that was FDA-approved in 2010 as a first oral treatment for relapsing forms of the autoimmune disease, multiple sclerosis (MS; Chun and Brinkmann 2011). The compound was a chemical derivative of a naturally occurring immunosuppressant molecule initially identified from the fungus Myriococcum albomyces and named “Myriocin” (St-Jacques 1973; Kluepfel et al. 1972). Years later, myriocin was identified from another fungus, the sac fungus Ascomycota (formerly known as Ascomycetes) and includes the entomopathogenic fungus Isaria sinclarii (Fig. 3) as “ISP-1” that is myriocin. Its antifungal properties were extended to mammalian systems where it was observed to have immunosuppressant properties in culture attributed to SPT inhibition (Miyake et al. 1995). These properties suggested utility in a variety of therapeutic applications like organ transplantation, and myriocin was studied for possible in vivo use; however, exposure in animals was associated with unacceptable toxicity (Napoli 2000), which lead to a concerted effort from academia and pharmaceutical collaborations in Japan involving chemical derivatization of myriocin to produce a more desirable compound, which resulted in the identification and synthesis of FTY720 (Fig. 2; the names reflecting the contribution by Professor Tetsuro Fujita at Kyoto University; and the companies Taito and Yoshitomi; Chiba et al. 1996; Fujita et al. 1994).

Reprinted with permission from An Illustrated Guide to New Zealand Soil Invertebrates, http://soilbugs.massey.ac.nz. © Massey University
Source of myriocin. Myriocin was originally isolated from the fungus Isaria sinclairii, which is commonly used in traditional Chinese medicine. Like other Cordyceps species it parasitizes insects, such as cicada larvae under the soil. The structures of FTY720 (derived from myriocin) and cyclosporine (widely used immunosuppressant drug also of fungal origin) are shown for comparison.
FTY720 was tested in a wide range of animal models, in which it was thought to be acting as a strong immunosuppressant (Suzuki et al. 1996; Chueh et al. 1997; Mitsusada et al. 1997; Ueda et al. 2000). Its mechanism, however, was distinct from Myriocin that inhibits SPT, whereas FTY720 did not (Napoli 2000). Nevertheless, basic studies on Myriocin indicated that its roles in sphingosine metabolism (Miyake et al. 1995) might be relevant to derivatives like FTY720; however, its precise actions were unclear. Another fungal agent, cyclosporine, was isolated from Ascomycota fungus Tolypocladium inflatum that is the same fungal order (Hypocreales) as Isaria sinclarii, the source of myriocin/ISP-1 from which FTY720 emerged. Discovered by Sandoz scientists as an important immunosuppressant (Heusler and Pletscher 2001), cyclosporine was observed in animals to synergize with FTY720 (Chiba et al. 1996; Enosawa et al. 1996; Hoshino et al. 1996; Kawaguchi et al. 1996; Masubuchi et al. 1996). However, studies at Novartis and in academia produced less clear activities for immunosuppression, culminating with a pivotal phase III clinical trial for renal transplantation rejection that combined reduced-dose cyclosporine—a proven immunosuppressant—with high-dose FTY720 that failed to reach its desired clinical endpoints (Tedesco-Silva et al. 2006). However, other preclinical work supported activity of FTY720 in an animal model of multiple sclerosis (MS; Webb et al. 2004; Brinkmann et al. 2002; Fujino et al. 2003), experimental autoimmune encephalomyelitis (EAE), which led to human clinical trials for MS and ultimately resulted in regulatory approval of the compound that is known by its generic name “fingolimod” and commercial name Gilenya (Imusera in Japan; Kappos et al. 2006; Chun and Hartung 2010; Cohen and Chun 2011; Chun and Brinkmann 2011). In the USA, Gilenya received FDA approval in 2010 for treatment of relapsing forms of MS as well as regulatory approval globally where it has been used in the treatment of over 125,000 patients worldwide by 2015 (www.gilenya.com).
During the period of myriocin modification that produced FTY720, a completely separate research effort identified the first of a novel GPCR family (Hecht et al. 1996; Fukushima et al. 2001; Kihara et al. 2014) that mediates the effects of lysophospholipids (LPs) that included lysophosphatidic acid (LPA) and S1P. As a family, LP receptors currently represent over 40 % of lipid-interacting GPCRs. The discovery by scientists at Novartis (Brinkmann et al. 2002) and Merck (Mandala et al. 2002) that FTY720 phosphorylation (FTY-P) produced an S1P receptor non-selective agonist brought these two fields together and has allowed mechanistic understanding of important physiological activities. FTY-P interacts with 4 of 5 S1P receptors with high affinity (all except S1P2). Notably, despite initial identification as an agonist, the prominent effects of FTY720 in disease models involve receptor loss through ubiquitin-mediated receptor degradation that produces functional antagonism (Welsch et al. 2003; Bohler et al. 2005; Oo et al. 2007), thereby explaining phenocopying of results between prolonged FTY720 exposure and mice deficient for S1P receptors (Matloubian et al. 2004; Pappu et al. 2007; Schwab and Cyster 2007; Choi et al. 2011). The primary effects of FTY720 in MS appear to involve disruption of normal lymphocyte egress from lymphoid organs produced by loss of S1P receptors (Matloubian et al. 2004). In addition, there are direct effects of FTY720 mediated by S1P signaling within the central nervous system (CNS) that involve astrocytes, whereby removal of S1P1 receptors from astrocytes reduces disease and eliminates FTY720 efficacy in EAE (Choi et al. 2011; Groves et al. 2013). The direct CNS effect was further supported by the observation that FTY720 can promote the regeneration of sciatic nerve following crush injury (Szepanowski et al. 2016).
Beyond the proven efficacy that FTY720 has in improving MS outcomes, it has also been suggested that it may be useful in other neurological indications, such as stroke (Moon et al. 2015; Liu et al. 2013) and Alzheimer’s disease (Aytan et al. 2016). In addition, there is evidence for the potential efficacy of FTY720 in treating certain cancers (Patmanathan et al. 2015), and it was recently shown that it induces apoptosis and attenuates proliferative signaling pathways in oral squamous cell carcinoma cells (Patmanathan et al. 2016). It is notable that the possible anticancer effects of FTY720 may in part be independent of its direct effects on S1P receptors. It has been reported to affect S1P metabolism by inhibiting SPHK and S1P lyase (Tonelli et al. 2010; Bandhuvula et al. 2005) and to act directly on other intracellular targets, such as protein phosphatase 2A (Matsuoka et al. 2003) and the 14-3-3 adaptor protein (Woodcock et al. 2010). The clinical relevance of these reports is unclear.
These effects and the success of FTY720 have spurred research into other therapeutics based upon lysophospholipid signaling (Kihara et al. 2015), and it will not be surprising to encounter new biology and therapeutics in view of the vast range of phenotypes observed in deletion mutants of S1P receptors (Ishii et al. 2001, 2002; Yang et al. 2002; Baudhuin et al. 2004; Nofer et al. 2004; Herr et al. 2007, 2013; Keller et al. 2007; Means et al. 2007; Tolle et al. 2008; Choi et al. 2011; Hopson et al. 2011; Means et al. 2008), some of which may be accessed by modified fungal metabolites.
Fumonisin B1
Fumonisin B1 is one of a family of mycotoxins produced by filamentous fungi from the genus Fusarium that commonly infect corn (maize). It is a naturally occurring SP analog that contains tricarballylic acid and methyl modifications to an LCB (Fig. 2). This compound is particularly notable in that it has been shown to inhibit the activity of ceramide synthase and cause the accumulation of dihydrosphingosine in vivo (E. Wang et al. 1991; Wang et al. 1999). Sub-toxic exposure of fumonisin B1 routinely occurs through normal dietary intake in many countries including the USA and is safely tolerated at levels below 2 µg/kg/day (Stockmann-Juvala and Savolainen 2008). However, high level of fumonisin B1 intake is characterized by an increased dihydrosphingosine/sphingosine ratio (Riley et al. 1993) and may lead to deleterious effect on the CNS. Notably, fumonisin B1 exposure and elevated dihydrosphingosine were associated with an outbreak of neural tube defects that occurred in a population with a corn-rich diet that lived along the US–Mexico border in 1990–1991 (Missmer et al. 2006). This was experimentally reproduced in mice, whereby exposure of embryos to as little as 2 µM Fumonisin B1 caused neural tube defects (Sadler et al. 2002), confirming that regulation of SPs is essential for normal development of the CNS. Interestingly, the occurrence of neural tube defects could be attenuated by co-administration of folic acid, suggesting that derangement of SP metabolism may disrupt the uptake or bioavailability of folate.
d,l-Threo-dihydrosphingosine and N,N-Dimethyl sphingosine
Studies involving the use of sphingosine analogs have demonstrated that a number of structural variants are recognized by SP metabolic enzymes, but may act as inhibitors. Two notable compounds are d,l-threo-dihydrosphingosine (DHS) and N,N-dimethyl sphingosine (DMS; Fig. 2), which can inhibit sphingosine kinase activity. DHS, an enantiomer of naturally occurring d-erythro-dihydrosphingosine, was shown to inhibit SPHK1 (Buehrer and Bell 1992), but acts as a substrate for SPHK2 (Pyne and Pyne 2002). Similarly, L-threo-dihydrosphingosine (safingol) is known to inhibit SPHK1, but also acts on protein kinases, such as protein kinase C (Canals and Hannun 2013). Phase I clinical trials have been completed for the use of safingol for solid tumors in combination with doxorubicin (Schwartz et al. 1997) or cisplatin (Dickson et al. 2011). Both studies provided evidence for safety, but did not evaluate efficacy. DMS was found to be endogenously present at low levels in splenocytes (Felding-Habermann et al. 1990) and epidermoid carcinoma cells (Igarashi et al. 1990) and can inhibit both isoforms of sphingosine kinase (Yatomi et al. 1996; Pyne and Pyne 2002). Both of these compounds have served as useful tools for elucidating the biological roles of sphingosine kinase and further demonstrate that dietary SPs and SP analogs not only act as substrates for lipid metabolism, but also may have significant effects on endogenous metabolic pathways.
Conclusions and Implications
In conclusion, SPs are abundant and well-studied lipids that are critical mediators of many functions of the CNS and other tissues. The metabolic pathways that regulate SPs and the signaling pathways that they influence are abundantly characterized and are biomedically relevant. However, the importance of dietary SPs and the effects of naturally occurring SP analogs on endogenous SP metabolism are not fully realized. Atypical SPs can be absorbed from the diet and incorporated into the mammalian sphingolipidome, and several analogs found in normal diet or herbal supplements (myriocin, fumonisin B1) have been shown to exert potent biological effects. It is a near certainty that novel, naturally occurring SPs will continue to be identified and that therapeutically useful effects will be discovered from these nutraceuticals.
References
Abbas, H. K., Duke, S. O., Merrill, A. H, Jr., Wang, E., & Shier, W. T. (1998). Phytotoxicity of australifungin, AAL-toxins and fumonisin B1 to Lemna pausicostata. Phytochemistry, 47(8), 1509–1514. doi:10.1016/S0031-9422(97)00781-4.
Abeytunga, T. U. (2015). Occurrence, structure elucidation, biosynthesis, functions and synthesis of sphingadienes. Mini-Reviews in Organic Chemistry, 12(3), 282–292.
Abeytunga, D. T., Glick, J. J., Gibson, N. J., Oland, L. A., Somogyi, A., Wysocki, V. H., et al. (2004). Presence of unsaturated sphingomyelins and changes in their composition during the life cycle of the moth Manduca sexta. Journal of Lipid Research, 45(7), 1221–1231. doi:10.1194/jlr.M300392-JLR200.
Abeytunga, D. T., Oland, L., Somogyi, A., & Polt, R. (2008). Structural studies on the neutral glycosphingolipids of Manduca sexta. Bioorganic Chemistry, 36(2), 70–76. doi:10.1016/j.bioorg.2007.10.002.
Aida, K., Kinoshita, M., Sugawara, T., Ono, J., Miyazawa, T., & Ohnishi, M. (2004). Apoptosis inducement by plant and fungus sphingoid bases in human colon cancer cells. Journal of Oleo Science, 53, 503–510.
Akahoshi, N., Ishizaki, Y., Yasuda, H., Murashima, Y. L., Shinba, T., Goto, K., et al. (2011). Frequent spontaneous seizures followed by spatial working memory/anxiety deficits in mice lacking sphingosine 1-phosphate receptor 2. Epilepsy & Behavior, 22(4), 659–665. doi:10.1016/j.yebeh.2011.09.002.
Aldahmesh, M. A., Mohamed, J. Y., Alkuraya, H. S., Verma, I. C., Puri, R. D., Alaiya, A. A., et al. (2011). Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. American Journal of Human Genetics, 89(6), 745–750. doi:10.1016/j.ajhg.2011.10.011.
Arana, L., Gangoiti, P., Ouro, A., Trueba, M., & Gomez-Munoz, A. (2010). Ceramide and ceramide 1-phosphate in health and disease. Lipids in Health and Disease, 9, 15. doi:10.1186/1476-511X-9-15.
Aureli, M., Grassi, S., Prioni, S., Sonnino, S., & Prinetti, A. (2015). Lipid membrane domains in the brain. Biochimica et Biophysica Acta, 1851(8), 1006–1016. doi:10.1016/j.bbalip.2015.02.001.
Aytan, N., Choi, J. K., Carreras, I., Brinkmann, V., Kowall, N. W., Jenkins, B. G., et al. (2016). Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Scientific Reports, 6, 24939. doi:10.1038/srep24939.
Bajjalieh, S. M., Martin, T. F., & Floor, E. (1989). Synaptic vesicle ceramide kinase. A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. Journal of Biological Chemistry, 264(24), 14354–14360.
Bandhuvula, P., Tam, Y. Y., Oskouian, B., & Saba, J. D. (2005). The immune modulator FTY720 inhibits sphingosine-1-phosphate lyase activity. Journal of Biological Chemistry, 280(40), 33697–33700. doi:10.1074/jbc.C500294200.
Baudhuin, L. M., Jiang, Y., Zaslavsky, A., Ishii, I., Chun, J., & Xu, Y. (2004). S1P3-mediated Akt activation and cross-talk with platelet-derived growth factor receptor (PDGFR). FASEB Journal, 18(2), 341–343.
Ben-David, O., & Futerman, A. H. (2010). The role of the ceramide acyl chain length in neurodegeneration: Involvement of ceramide synthases. Neuromolecular Medicine, 12(4), 341–350. doi:10.1007/s12017-010-8114-x.
Bensemhoun, J., Bombarda, I., Aknin, M., Faure, R., Vacelet, J., & Gaydou, E. M. (2008). Marine bifunctional sphingolipids from the sponge Oceanapia ramsayi. Molecules, 13(4), 772–778.
Bertea, M., Rutti, M. F., Othman, A., Marti-Jaun, J., Hersberger, M., von Eckardstein, A., et al. (2010). Deoxysphingoid bases as plasma markers in diabetes mellitus. Lipids in Health and Disease, 9, 84. doi:10.1186/1476-511X-9-84.
Bode, H., Bourquin, F., Suriyanarayanan, S., Wei, Y., Alecu, I., Othman, A., et al. (2015). HSAN1 mutations in serine palmitoyltransferase reveal a close structure-function-phenotype relationship. Human Molecular Genetics,. doi:10.1093/hmg/ddv611.
Bohler, T., Budde, K., Neumayer, H. H., & Waiser, J. (2005). Novel mediators of FTY720 in human lymphocytes. Transplantation, 79(4), 492–495.
Brinkmann, V., Billich, A., Baumruker, T., Heining, P., Schmouder, R., Francis, G., et al. (2010). Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. [Review]. Nature Reviews Drug Discovery, 9(11), 883–897. doi:10.1038/nrd3248.
Brinkmann, V., Davis, M. D., Heise, C. E., Albert, R., Cottens, S., Hof, R., et al. (2002). The immune modulator FTY720 targets sphingosine 1-phosphate receptors. Journal of Biological Chemistry, 277(24), 21453–21457.
Buehrer, B. M., & Bell, R. M. (1992). Inhibition of sphingosine kinase in vitro and in platelets. Implications for signal transduction pathways. Journal of Biological Chemistry, 267(5), 3154–3159.
Byrdwell, W. C., & Perry, R. H. (2007). Liquid chromatography with dual parallel mass spectrometry and 31P nuclear magnetic resonance spectroscopy for analysis of sphingomyelin and dihydrosphingomyelin. II. Bovine milk sphingolipids. Journal of Chromatography A, 1146(2), 164–185. doi:10.1016/j.chroma.2007.01.108.
Canals, D., & Hannun, Y. A. (2013). Novel chemotherapeutic drugs in sphingolipid cancer research. Handbook of Experimental Pharmacology, 215, 211–238. doi:10.1007/978-3-7091-1368-4_12.
Chiba, K., Hoshino, Y., Suzuki, C., Masubuchi, Y., Yanagawa, Y., Ohtsuki, M., et al. (1996). FTY720, a novel immunosuppressant possessing unique mechanisms. I. Prolongation of skin allograft survival and synergistic effect in combination with cyclosporine in rats. Transplantation Proceedings, 28(2), 1056–1059.
Choi, J. W., Gardell, S. E., Herr, D. R., Rivera, R., Lee, C. W., Noguchi, K., et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proceedings of the National Academy of Sciences of the United States of America, 108(2), 751–756. doi:10.1073/pnas.1014154108.
Christoffersen, C., Obinata, H., Kumaraswamy, S. B., Galvani, S., Ahnstrom, J., Sevvana, M., et al. (2011). Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proceedings of the National Academy of Sciences of the United States of America, 108(23), 9613–9618. doi:10.1073/pnas.1103187108.
Chueh, S. C., Tian, L., Wang, M., Wang, M. E., Stepkowski, S. M., & Kahan, B. D. (1997). Induction of tolerance toward rat cardiac allografts by treatment with allochimeric class I MHC antigen and FTY720. Transplantation, 64(10), 1407–1414.
Chumanevich, A. A., Poudyal, D., Cui, X., Davis, T., Wood, P. A., Smith, C. D., et al. (2010). Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis, 31(10), 1787–1793. doi:10.1093/carcin/bgq158.
Chun, J., & Brinkmann, V. (2011). A mechanistically novel, first oral therapy for multiple sclerosis: The development of fingolimod (FTY720, Gilenya). [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Discovery Medicine, 12(64), 213–228.
Chun, J., & Hartung, H. P. (2010). Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clinical Neuropharmacology, 33(2), 91–101. doi:10.1097/WNF.0b013e3181cbf825.
Cingolani, F., Casasampere, M., Sanllehi, P., Casas, J., Bujons, J., & Fabrias, G. (2014). Inhibition of dihydroceramide desaturase activity by the sphingosine kinase inhibitor SKI II. Journal of Lipid Research, 55(8), 1711–1720. doi:10.1194/jlr.M049759.
Cohen, J. A., & Chun, J. (2011). Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Annals of Neurology, 69(5), 759–777. doi:10.1002/ana.22426.
Cowart, L. A., & Obeid, L. M. (2007). Yeast sphingolipids: Recent developments in understanding biosynthesis, regulation, and function. Biochimica et Biophysica Acta, 1771(3), 421–431. doi:10.1016/j.bbalip.2006.08.005.
de la Monte, S. M., Longato, L., Tong, M., DeNucci, S., & Wands, J. R. (2009). The liver-brain axis of alcohol-mediated neurodegeneration: Role of toxic lipids. International Journal of Environmental Research and Public Health, 6(7), 2055–2075. doi:10.3390/ijerph6072055.
De Luca, C., & Valacchi, G. (2010). Surface lipids as multifunctional mediators of skin responses to environmental stimuli. Mediators of Inflammation, 2010, 321494. doi:10.1155/2010/321494.
Dickson, R. C. (2008). Thematic review series: Sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. Journal of Lipid Research, 49(5), 909–921. doi:10.1194/jlr.R800003-JLR200.
Dickson, M. A., Carvajal, R. D., Merrill, A. H, Jr., Gonen, M., Cane, L. M., & Schwartz, G. K. (2011). A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clinical Cancer Research, 17(8), 2484–2492. doi:10.1158/1078-0432.CCR-10-2323.
Duan, J., & Merrill, A. H, Jr. (2015). 1-Deoxysphingolipids encountered exogenously and made de novo: Dangerous mysteries inside an enigma. Journal of Biological Chemistry, 290(25), 15380–15389. doi:10.1074/jbc.R115.658823.
Dukala, D. E., & Soliven, B. (2016). S1P1 deletion in oligodendroglial lineage cells: Effect on differentiation and myelination. Glia, 64(4), 570–582. doi:10.1002/glia.22949.
Enosawa, S., Suzuki, S., Kakefuda, T., Li, X. K., & Amemiya, H. (1996). Induction of selective cell death targeting on mature T-lymphocytes in rats by a novel immunosuppressant, FTY720. Immunopharmacology, 34(2–3), 171–179.
Fahy, E., Subramaniam, S., Brown, H. A., Glass, C. K., Merrill, A. H, Jr., Murphy, R. C., et al. (2005). A comprehensive classification system for lipids. Journal of Lipid Research, 46(5), 839–861. doi:10.1194/jlr.E400004-JLR200.
Fahy, E., Subramaniam, S., Murphy, R. C., Nishijima, M., Raetz, C. R., Shimizu, T., et al. (2009). Update of the LIPID MAPS comprehensive classification system for lipids. Journal of Lipid Research, 50(Suppl), S9–S14. doi:10.1194/jlr.R800095-JLR200.
Felding-Habermann, B., Igarashi, Y., Fenderson, B. A., Park, L. S., Radin, N. S., Inokuchi, J., et al. (1990). A ceramide analogue inhibits T cell proliferative response through inhibition of glycosphingolipid synthesis and enhancement of N,N-dimethylsphingosine synthesis. Biochemistry, 29(26), 6314–6322.
Finley, K. D., Edeen, P. T., Cumming, R. C., Mardahl-Dumesnil, M. D., Taylor, B. J., Rodriguez, M. H., et al. (2003). blue cheese mutations define a novel, conserved gene involved in progressive neural degeneration. Journal of Neuroscience, 23(4), 1254–1264.
Fleischmann, R. (2012). Novel small-molecular therapeutics for rheumatoid arthritis. Current Opinion in Rheumatology, 24(3), 335–341. doi:10.1097/BOR.0b013e32835190ef.
Fujino, M., Funeshima, N., Kitazawa, Y., Kimura, H., Amemiya, H., Suzuki, S., et al. (2003). Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. Journal of Pharmacology and Experimental Therapeutics, 305(1), 70–77.
Fujita, T., Inoue, K., Yamamoto, S., Ikumoto, T., Sasaki, S., Toyama, R., et al. (1994). Fungal metabolites. Part 12. Potent immunosuppressant, 14-deoxomyriocin, (2S,3R,4R)-(E)-2-amino-3,4-dihydroxy-2-hydroxymethyleicos-6-enoic acid and structure-activity relationships of myriocin derivatives. Journal of Antibiotics (Tokyo), 47(2), 216–224.
Fukushima, N., Ishii, I., Contos, J. J., Weiner, J. A., & Chun, J. (2001). Lysophospholipid receptors. Annual Review of Pharmacology and Toxicology, 41, 507–534.
Furukawa, A., Kita, K., Toyomoto, M., Fujii, S., Inoue, S., Hayashi, K., et al. (2007). Production of nerve growth factor enhanced in cultured mouse astrocytes by glycerophospholipids, sphingolipids, and their related compounds. Molecular and Cellular Biochemistry, 305(1–2), 27–34. doi:10.1007/s11010-007-9524-4.
Fyrst, H., Herr, D. R., Harris, G. L., & Saba, J. D. (2004). Characterization of free endogenous C14 and C16 sphingoid bases from Drosophila melanogaster. Journal of Lipid Research, 45(1), 54–62. doi:10.1194/jlr.M300005-JLR200.
Fyrst, H., Oskouian, B., Bandhuvula, P., Gong, Y. Q., Byun, H. S., Bittman, R., et al. (2009). Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Research, 69(24), 9457–9464. doi:10.1158/0008-5472.Can-09-2341.
Fyrst, H., & Saba, J. D. (2010). An update on sphingosine-1-phosphate and other sphingolipid mediators. Nature Chemical Biology, 6(7), 489–497. doi:10.1038/nchembio.392.
Fyrst, H., Zhang, X., Herr, D. R., Byun, H. S., Bittman, R., Phan, V. H., et al. (2008). Identification and characterization by electrospray mass spectrometry of endogenous Drosophila sphingadienes. Journal of Lipid Research, 49(3), 597–606. doi:10.1194/jlr.M700414-JLR200.
Garnier-Amblard, E. C., Mays, S. G., Arrendale, R. F., Baillie, M. T., Bushnev, A. S., Culver, D. G., et al. (2011). Novel synthesis and biological evaluation of enigmols as therapeutic agents for treating prostate cancer. ACS Medicinal Chemistry Letters, 2(6), 438–443. doi:10.1021/ml2000164.
Ghasemi, R., Dargahi, L., & Ahmadiani, A. (2016). Integrated sphingosine-1 phosphate signaling in the central nervous system: From physiological equilibrium to pathological damage. Pharmacological Research, 104, 156–164. doi:10.1016/j.phrs.2015.11.006.
Gibellini, F., & Smith, T. K. (2010). The Kennedy pathway—De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life, 62(6), 414–428. doi:10.1002/iub.337.
Godfrey, J., Jeanguenin, L., Castro, N., Olney, J. J., Dudley, J., Pipkin, J., et al. (2015). Chronic voluntary ethanol consumption induces favorable ceramide profiles in selectively bred alcohol-preferring (P) rats. PLoS ONE, 10(9), e0139012. doi:10.1371/journal.pone.0139012.
Gomez-Munoz, A., Presa, N., Gomez-Larrauri, A., Rivera, I. G., Trueba, M., & Ordonez, M. (2016). Control of inflammatory responses by ceramide, sphingosine 1-phosphate and ceramide 1-phosphate. Progress in Lipid Research, 61, 51–62. doi:10.1016/j.plipres.2015.09.002.
Groves, A., Kihara, Y., & Chun, J. (2013). Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. Journal of the Neurological Sciences, 328(1–2), 9–18. doi:10.1016/j.jns.2013.02.011.
Gulbins, E., Walter, S., Becker, K. A., Halmer, R., Liu, Y., Reichel, M., et al. (2015). A central role for the acid sphingomyelinase/ceramide system in neurogenesis and major depression. Journal of Neurochemistry, 134(2), 183–192. doi:10.1111/jnc.13145.
Han, G., Gupta, S. D., Gable, K., Niranjanakumari, S., Moitra, P., Eichler, F., et al. (2009). Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proceedings of the National Academy of Sciences of the United States of America, 106(20), 8186–8191. doi:10.1073/pnas.0811269106.
Hanson, M. A., Roth, C. B., Jo, E., Griffith, M. T., Scott, F. L., Reinhart, G., et al. (2012). Crystal structure of a lipid G protein-coupled receptor. Science, 335(6070), 851–855. doi:10.1126/science.1215904.
Hebbar, S., Sahoo, I., Matysik, A., Argudo Garcia, I., Osborne, K. A., Papan, C., et al. (2015). Ceramides and stress signalling intersect with autophagic defects in neurodegenerative Drosophila blue cheese (bchs) mutants. Scientific Reports, 5, 15926. doi:10.1038/srep15926.
Hecht, J. H., Weiner, J. A., Post, S. R., & Chun, J. (1996). Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. Journal of Cell Biology, 135(4), 1071–1083.
Herr, D. R., & Chun, J. (2007). Effects of LPA and S1P on the nervous system and implications for their involvement in disease. Current Drug Targets, 8(1), 155–167.
Herr, D. R., Grillet, N., Schwander, M., Rivera, R., Muller, U., & Chun, J. (2007). Sphingosine 1-phosphate (S1P) signaling is required for maintenance of hair cells mainly via activation of S1P2. Journal of Neuroscience, 27(6), 1474–1478. doi:10.1523/JNEUROSCI.4245-06.2007.
Herr, D. R., Lee, C. W., Wang, W., Ware, A., Rivera, R., & Chun, J. (2013). Sphingosine 1-phosphate receptors are essential mediators of eyelid closure during embryonic development. Journal of Biological Chemistry, 288(41), 29882–29889. doi:10.1074/jbc.M113.510099.
Herr, D. R., Reolo, M. J., Peh, Y. X., Wang, W., Lee, C. W., Rivera, R., et al. (2016). Sphingosine 1-phosphate receptor 2 (S1P2) attenuates reactive oxygen species formation and inhibits cell death: Implications for otoprotective therapy. Scientific Reports, 6, 24541. doi:10.1038/srep24541.
Heusler, K., & Pletscher, A. (2001). The controversial early history of cyclosporin. Swiss Medical Weekly, 131(21–22), 299–302. doi:2001/21/smw-09702.
Hoeferlin, L. A., Wijesinghe, D. S., & Chalfant, C. E. (2013). The role of ceramide-1-phosphate in biological functions. Handbook of Experimental Pharmacology, 215, 153–166. doi:10.1007/978-3-7091-1368-4_8.
Hopson, K. P., Truelove, J., Chun, J., Wang, Y., & Waeber, C. (2011). S1P activates store-operated calcium entry via receptor- and non-receptor-mediated pathways in vascular smooth muscle cells. American Journal of Physiology. Cell Physiology, 300(4), C919–C926. doi:10.1152/ajpcell.00350.2010.
Hornemann, T., Penno, A., Rutti, M. F., Ernst, D., Kivrak-Pfiffner, F., Rohrer, L., et al. (2009). The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. Journal of Biological Chemistry, 284(39), 26322–26330. doi:10.1074/jbc.M109.023192.
Hornemann, T., Wei, Y., & von Eckardstein, A. (2007). Is the mammalian serine palmitoyltransferase a high-molecular-mass complex? Biochemical Journal, 405(1), 157–164. doi:10.1042/BJ20070025.
Hoshino, Y., Suzuki, C., Ohtsuki, M., Masubuchi, Y., Amano, Y., & Chiba, K. (1996). FTY720, a novel immunosuppressant possessing unique mechanisms. II. Long-term graft survival induction in rat heterotopic cardiac allografts and synergistic effect in combination with cyclosporine A. Transplantation Proceedings, 28(2), 1060–1061.
Hulette, C. M., Earl, N. L., Anthony, D. C., & Crain, B. J. (1992). Adult onset Niemann-Pick disease type C presenting with dementia and absent organomegaly. Clinical Neuropathology, 11(6), 293–297.
Igarashi, Y., Kitamura, K., Toyokuni, T., Dean, B., Fenderson, B., Ogawass, T., et al. (1990). A specific enhancing effect of N,N-dimethylsphingosine on epidermal growth factor receptor autophosphorylation. Demonstration of its endogenous occurrence (and the virtual absence of unsubstituted sphingosine) in human epidermoid carcinoma A431 cells. Journal of Biological Chemistry, 265(10), 5385–5389.
Ikeda, K., & Taguchi, R. (2010). Highly sensitive localization analysis of gangliosides and sulfatides including structural isomers in mouse cerebellum sections by combination of laser microdissection and hydrophilic interaction liquid chromatography/electrospray ionization mass spectrometry with theoretically expanded multiple reaction monitoring. Rapid Communications in Mass Spectrometry, 24(20), 2957–2965. doi:10.1002/rcm.4716.
Imai, H., Ohnishi, M., Hotsubo, K., Kojima, M., & Ito, S. (1997). Sphingoid base composition of cerebrosides from plant leaves. Bioscience, Biotechnology, and Biochemistry, 61(2), 351–353.
Inamine, M., Suzui, M., Morioka, T., Kinjo, T., Kaneshiro, T., Sugishita, T., et al. (2005). Inhibitory effect of dietary monoglucosylceramide 1-O-beta-glucosyl-N-2′-hydroxyarachidoyl-4,8-sphingadienine on two different categories of colon preneoplastic lesions induced by 1,2-dimethylhydrazine in F344 rats. Cancer Science, 96(12), 876–881. doi:10.1111/j.1349-7006.2005.00127.x.
Ishii, I., Friedman, B., Ye, X., Kawamura, S., McGiffert, C., Contos, J. J., et al. (2001). Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. Journal of Biological Chemistry, 276(36), 33697–33704. Epub 32001 Jul 33696.
Ishii, I., Ye, X., Friedman, B., Kawamura, S., Contos, J. J., Kingsbury, M. A., et al. (2002). Marked perinatal lethality and cellular signaling deficits in mice null for the two sphingosine 1-phosphate (S1P) receptors, S1P(2)/LP(B2)/EDG-5 and S1P(3)/LP(B3)/EDG-3. Journal of Biological Chemistry, 277(28), 25152–25159. Epub 22002 May 25152.
Jaillard, C., Harrison, S., Stankoff, B., Aigrot, M. S., Calver, A. R., Duddy, G., et al. (2005). Edg8/S1P5: An oligodendroglial receptor with dual function on process retraction and cell survival. Journal of Neuroscience, 25(6), 1459–1469. doi:10.1523/JNEUROSCI.4645-04.2005.
Jana, A., Hogan, E. L., & Pahan, K. (2009). Ceramide and neurodegeneration: Susceptibility of neurons and oligodendrocytes to cell damage and death. Journal of the Neurological Sciences, 278(1–2), 5–15. doi:10.1016/j.jns.2008.12.010.
Kappos, L., Antel, J., Comi, G., Montalban, X., O’Connor, P., Polman, C. H., et al. (2006). Oral fingolimod (FTY720) for relapsing multiple sclerosis [Multicenter Study Randomized Controlled Trial Research Support, Non-U.S. Gov’t]. New England Journal of Medicine, 355(11), 1124–1140. doi:10.1056/NEJMoa052643.
Kawaguchi, T., Hoshino, Y., Rahman, F., Amano, Y., Higashi, H., Kataoka, H., et al. (1996). FTY720, a novel immunosuppressant possessing unique mechanisms. III. Synergistic prolongation of canine renal allograft survival in combination with cyclosporine A. Transplantation Proceedings, 28(2), 1062–1063.
Kawamori, T., Kaneshiro, T., Okumura, M., Maalouf, S., Uflacker, A., Bielawski, J., et al. (2009). Role for sphingosine kinase 1 in colon carcinogenesis. FASEB Journal, 23(2), 405–414. doi:10.1096/fj.08-117572.
Kawamori, T., Osta, W., Johnson, K. R., Pettus, B. J., Bielawski, J., Tanaka, T., et al. (2006). Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB Journal, 20(2), 386–388. doi:10.1096/fj.05-4331fje.
Keller, C. D., Rivera Gil, P., Tolle, M., van der Giet, M., Chun, J., Radeke, H. H., et al. (2007). Immunomodulator FTY720 induces myofibroblast differentiation via the lysophospholipid receptor S1P3 and Smad3 signaling. American Journal of Pathology, 170(1), 281–292.
Keranen, A. (1976). Fatty acids and long-chain bases of gangliosides of human gastrointestinal mucosa. Chemistry and Physics of Lipids, 17(1), 14–21.
Kihara, Y., Maceyka, M., Spiegel, S., & Chun, J. (2014). Lysophospholipid receptor nomenclature review: IUPHAR Review 8. British Journal of Pharmacology, 171(15), 3575–3594. doi:10.1111/bph.12678.
Kihara, Y., Mizuno, H., & Chun, J. (2015). Lysophospholipid receptors in drug discovery. Experimental Cell Research, 333(2), 171–177. doi:10.1016/j.yexcr.2014.11.020.
Kim, G. S., Yang, L., Zhang, G., Zhao, H., Selim, M., McCullough, L. D., et al. (2015). Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nature Communications, 6, 7893. doi:10.1038/ncomms8893.
Kitatani, K., Idkowiak-Baldys, J., & Hannun, Y. A. (2008). The sphingolipid salvage pathway in ceramide metabolism and signaling. Cellular Signalling, 20(6), 1010–1018. doi:10.1016/j.cellsig.2007.12.006.
Kluepfel, D., Bagli, J., Baker, H., Charest, M. P., & Kudelski, A. (1972). Myriocin, a new antifungal antibiotic from Myriococcum albomyces. Journal of Antibiotics (Tokyo), 25(2), 109–115.
Kornhuber, J., Muller, C. P., Becker, K. A., Reichel, M., & Gulbins, E. (2014). The ceramide system as a novel antidepressant target. Trends in Pharmacological Sciences, 35(6), 293–304. doi:10.1016/j.tips.2014.04.003.
Kumar, A., Pandurangan, A. K., Lu, F., Fyrst, H., Zhang, M., Byun, H. S., et al. (2012). Chemopreventive sphingadienes downregulate Wnt signaling via a PP2A/Akt/GSK3beta pathway in colon cancer. Carcinogenesis, 33(9), 1726–1735. doi:10.1093/carcin/bgs174.
Kunkel, G. T., Maceyka, M., Milstien, S., & Spiegel, S. (2013). Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nature Reviews Drug Discovery, 12(9), 688–702. doi:10.1038/nrd4099.
Kwon, H. C., Lee, K. C., Cho, O. R., Jung, I. Y., Cho, S. Y., Kim, S. Y., et al. (2003). Sphingolipids from Bombycis Corpus 101A and their neurotrophic effects. Journal of Natural Products, 66(4), 466–469. doi:10.1021/np0204491.
Lamour, N. F., & Chalfant, C. E. (2005). Ceramide-1-phosphate: The “missing” link in eicosanoid biosynthesis and inflammation. Molecular Interventions, 5(6), 358–367. doi:10.1124/mi.5.6.8.
Lee, H., Deng, J., Kujawski, M., Yang, C., Liu, Y., Herrmann, A., et al. (2010). STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Nature Medicine, 16(12), 1421–1428. doi:10.1038/nm.2250.
Lee, T. C., Ou, M. C., Shinozaki, K., Malone, B., & Snyder, F. (1996). Biosynthesis of N-acetylsphingosine by platelet-activating factor: Sphingosine CoA-independent transacetylase in HL-60 cells. Journal of Biological Chemistry, 271(1), 209–217.
Lee, M. J., Thangada, S., Claffey, K. P., Ancellin, N., Liu, C. H., Kluk, M., et al. (1999). Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell, 99(3), 301–312.
Lee, M. J., Van Brocklyn, J. R., Thangada, S., Liu, C. H., Hand, A. R., Menzeleev, R., et al. (1998). Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science, 279(5356), 1552–1555.
Levy, M., & Futerman, A. H. (2010). Mammalian ceramide synthases. IUBMB Life, 62(5), 347–356. doi:10.1002/iub.319.
Liu, J., Zhang, C., Tao, W., & Liu, M. (2013). Systematic review and meta-analysis of the efficacy of sphingosine-1-phosphate (S1P) receptor agonist FTY720 (fingolimod) in animal models of stroke. International Journal of Neuroscience, 123(3), 163–169. doi:10.3109/00207454.2012.749255.
Lynch, D. V., & Dunn, T. M. (2004). An introduction to plant sphingolipids and a review of recent advances in understanding their metabolism and function. New Phytologist, 161(3), 677–702. doi:10.1111/j.1469-8137.2004.00992.x.
Macauley, S. L., Sidman, R. L., Schuchman, E. H., Taksir, T., & Stewart, G. R. (2008). Neuropathology of the acid sphingomyelinase knockout mouse model of Niemann-Pick A disease including structure-function studies associated with cerebellar Purkinje cell degeneration. Experimental Neurology, 214(2), 181–192. doi:10.1016/j.expneurol.2008.07.026.
MacLennan, A. J., Carney, P. R., Zhu, W. J., Chaves, A. H., Garcia, J., Grimes, J. R., et al. (2001). An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. European Journal of Neuroscience, 14(2), 203–209.
Malchinkhuu, E., Sato, K., Muraki, T., Ishikawa, K., Kuwabara, A., & Okajima, F. (2003). Assessment of the role of sphingosine 1-phosphate and its receptors in high-density lipoprotein-induced stimulation of astroglial cell function. Biochemical Journal, 370(Pt 3), 817–827.
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J., et al. (2002). Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science, 296(5566), 346–349.
Martinez, T. N., Chen, X., Bandyopadhyay, S., Merrill, A. H., & Tansey, M. G. (2012). Ceramide sphingolipid signaling mediates tumor necrosis factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Molecular Neurodegeneration, 7, 45. doi:10.1186/1750-1326-7-45.
Masubuchi, Y., Kawaguchi, T., Ohtsuki, M., Suzuki, C., Amano, Y., Hoshino, Y., et al. (1996). FTY720, a novel immunosuppressant, possessing unique mechanisms. IV. Prevention of graft versus host reactions in rats. Transplantation Proceedings, 28(2), 1064–1065.
Matloubian, M., Lo, C. G., Cinamon, G., Lesneski, M. J., Xu, Y., Brinkmann, V., et al. (2004). Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature, 427(6972), 355–360.
Matsuoka, Y., Nagahara, Y., Ikekita, M., & Shinomiya, T. (2003). A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. British Journal of Pharmacology, 138(7), 1303–1312. doi:10.1038/sj.bjp.0705182.
McNaughton, M., Pitman, M., Pitson, S. M., Pyne, N. J., & Pyne, S. (2016). Proteasomal degradation of sphingosine kinase 1 and inhibition of dihydroceramide desaturase by the sphingosine kinase inhibitors, SKi or ABC294640, induces growth arrest in androgen-independent LNCaP-AI prostate cancer cells. Oncotarget, 7(13), 16663–16675. doi:10.18632/oncotarget.7693.
Means, C. K., Miyamoto, S., Chun, J., & Brown, J. H. (2008). S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. Journal of Biological Chemistry, 283(18), 11954–11963. doi:10.1074/jbc.M707422200.
Means, C. K., Xiao, C. Y., Li, Z., Zhang, T., Omens, J. H., Ishii, I., et al. (2007). Sphingosine 1-phosphate S1P2 and S1P3 receptor-mediated Akt activation protects against in vivo myocardial ischemia-reperfusion injury. American Journal of Physiology: Heart and Circulatory Physiology, 292(6), H2944–H2951. doi:10.1152/ajpheart.01331.2006.
Merrill, A. H, Jr. (2011). Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chemical Reviews, 111(10), 6387–6422. doi:10.1021/cr2002917.
Merrill, A. H, Jr., Stokes, T. H., Momin, A., Park, H., Portz, B. J., Kelly, S., et al. (2009). Sphingolipidomics: A valuable tool for understanding the roles of sphingolipids in biology and disease. Journal of Lipid Research, 50(Suppl), S97–S102. doi:10.1194/jlr.R800073-JLR200.
Merrill, A. H, Jr., Sullards, M. C., Allegood, J. C., Kelly, S., & Wang, E. (2005). Sphingolipidomics: High-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods, 36(2), 207–224. doi:10.1016/j.ymeth.2005.01.009.
Merrill, A. H, Jr., Wang, E., Mullins, R. E., Jamison, W. C., Nimkar, S., & Liotta, D. C. (1988). Quantitation of free sphingosine in liver by high-performance liquid chromatography. Analytical Biochemistry, 171(2), 373–381.
Mielke, M. M., & Haughey, N. J. (2012). Could plasma sphingolipids be diagnostic or prognostic biomarkers for Alzheimer’s disease? Clinical Lipidology, 7(5), 525–536. doi:10.2217/clp.12.59.
Missmer, S. A., Suarez, L., Felkner, M., Wang, E., Merrill, A. H, Jr., Rothman, K. J., et al. (2006). Exposure to fumonisins and the occurrence of neural tube defects along the Texas-Mexico border. Environmental Health Perspectives, 114(2), 237–241.
Mitsusada, M., Suzuki, S., Kobayashi, E., Enosawa, S., Kakefuda, T., & Miyata, M. (1997). Prevention of graft rejection and graft-versus-host reaction by a novel immunosuppressant, FTY720, in rat small bowel transplantation. Transplantation International, 10(5), 343–349.
Miyake, Y., Kozutsumi, Y., Nakamura, S., Fujita, T., & Kawasaki, T. (1995). Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochemical and Biophysical Research Communications, 211(2), 396–403.
Mizugishi, K., Yamashita, T., Olivera, A., Miller, G. F., Spiegel, S., & Proia, R. L. (2005). Essential role for sphingosine kinases in neural and vascular development. Molecular and Cellular Biology, 25(24), 11113–11121. doi:10.1128/MCB.25.24.11113-11121.2005.
Mizushina, Y., Hanashima, L., Yamaguchi, T., Takemura, M., Sugawara, F., Saneyoshi, M., et al. (1998). A mushroom fruiting body-inducing substance inhibits activities of replicative DNA polymerases. Biochemical and Biophysical Research Communications, 249(1), 17–22. doi:10.1006/bbrc.1998.9091.
Moon, E., Han, J. E., Jeon, S., Ryu, J. H., Choi, J. W., & Chun, J. (2015). Exogenous S1P exposure potentiates ischemic stroke damage that is reduced possibly by inhibiting S1P receptor signaling. Mediators of Inflammation, 2015, 492659. doi:10.1155/2015/492659.
Motta, S., Monti, M., Sesana, S., Mellesi, L., Ghidoni, R., & Caputo, R. (1994). Abnormality of water barrier function in psoriasis. Role of ceramide fractions. Archives of Dermatology, 130(4), 452–456.
Mutoh, T., Rivera, R., & Chun, J. (2012). Insights into the pharmacological relevance of lysophospholipid receptors. British Journal of Pharmacology, 165(4), 829–844. doi:10.1111/j.1476-5381.2011.01622.x.
Nagle, D. G., McClatchey, W. C., & Gerwick, W. H. (1992). New glycosphingolipids from the marine sponge Halichondria panicea. Journal of Natural Products, 55(7), 1013–1017.
Napoli, K. L. (2000). The FTY720 story. Therapeutic Drug Monitoring, 22(1), 47–51.
Narayanaswamy, P., Shinde, S., Sulc, R., Kraut, R., Staples, G., Thiam, C. H., et al. (2014). Lipidomic “deep profiling”: An enhanced workflow to reveal new molecular species of signaling lipids. Analytical Chemistry, 86(6), 3043–3047. doi:10.1021/ac4039652.
Nicholas, G. M., & Molinski, T. F. (2000). Enantiodivergent biosynthesis of the dimeric sphingolipid oceanapiside from the marine sponge Oceanapia phillipensis. Determination of remote stereochemistry. Journal of the American Chemical Society, 122(17), 4011–4019. doi:10.1021/ja994215o.
Noda, N., Tanaka, R., Miyahara, K., & Kawasaki, T. (1993). Isolation and characterization of a novel type of glycosphingolipid from Neanthes diversicolor. Biochimica et Biophysica Acta, 1169(1), 30–38.
Nofer, J. R., van der Giet, M., Tolle, M., Wolinska, I., von Wnuck Lipinski, K., Baba, H. A., et al. (2004). HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. Journal of Clinical Investigation, 113(4), 569–581.
Novotny, J., Hrabalek, A., & Vavrova, K. (2010). Synthesis and structure-activity relationships of skin ceramides. Current Medicinal Chemistry, 17(21), 2301–2324.
Nyberg, L., Nilsson, A., Lundgren, P., & Duan, R. D. (1997). Localization and capacity of sphingomyelin digestion in the rat intestinal tract. Journal of Nutritional Biochemistry, 8(3), 112–118. doi:10.1016/S0955-2863(97)00010-7.
Obenberger, J., Seidl, Z., Pavlu, H., & Elleder, M. (1999). MRI in an unusually protracted neuronopathic variant of acid sphingomyelinase deficiency. Neuroradiology, 41(3), 182–184.
O’Brien, J. S., & Sampson, E. L. (1965). Lipid composition of the normal human brain: Gray matter, white matter, and myelin. Journal of Lipid Research, 6(4), 537–544.
Ohlsson, L., Hertervig, E., Jonsson, B. A., Duan, R. D., Nyberg, L., Svernlov, R., et al. (2010). Sphingolipids in human ileostomy content after meals containing milk sphingomyelin. American Journal of Clinical Nutrition, 91(3), 672–678. doi:10.3945/ajcn.2009.28311.
Okuyama, E., & Yamazaki, M. (1983). The principles of Tetragonia tetragonoides having anti-ulcerogenic activity. II. Isolation and structure of cerebrosides. Chemical and Pharmaceutical Bulletin, 31(7), 2209–2219.
Olsen, I., & Jantzen, E. (2001). Sphingolipids in bacteria and fungi. Anaerobe, 7(2), 103–112. doi:10.1006/anae.2001.0376.
Ong, W. Y., Herr, D. R., Farooqui, T., Ling, E. A., & Farooqui, A. A. (2015). Role of sphingomyelinases in neurological disorders. Expert Opinion on Therapeutic Targets, 19(12), 1725–1742. doi:10.1517/14728222.2015.1071794.
Oo, M. L., Thangada, S., Wu, M. T., Liu, C. H., Macdonald, T. L., Lynch, K. R., et al. (2007). Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. Journal of Biological Chemistry, 282(12), 9082–9089. doi:10.1074/jbc.M610318200.
Othman, A., Bianchi, R., Alecu, I., Wei, Y., Porretta-Serapiglia, C., Lombardi, R., et al. (2015). Lowering plasma 1-deoxysphingolipids improves neuropathy in diabetic rats. Diabetes, 64(3), 1035–1045. doi:10.2337/db14-1325.
Panganamala, R. V., Geer, J. C., & Cornwell, D. G. (1969). Long-chain bases in the sphingolipids of atherosclerotic human aorta. Journal of Lipid Research, 10(4), 445–455.
Pappu, R., Schwab, S. R., Cornelissen, I., Pereira, J. P., Regard, J. B., Xu, Y., et al. (2007). Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science, 316(5822), 295–298.
Patmanathan, S. N., Johnson, S. P., Lai, S. L., Panja Bernam, S., Lopes, V., Wei, W., et al. (2016). Aberrant expression of the S1P regulating enzymes, SPHK1 and SGPL1, contributes to a migratory phenotype in OSCC mediated through S1PR2. Scientific Reports, 6, 25650. doi:10.1038/srep25650.
Patmanathan, S. N., Yap, L. F., Murray, P. G., & Paterson, I. C. (2015). The antineoplastic properties of FTY720: Evidence for the repurposing of fingolimod. Journal of Cellular and Molecular Medicine, 19(10), 2329–2340. doi:10.1111/jcmm.12635.
Pebay, A., Toutant, M., Premont, J., Calvo, C. F., Venance, L., Cordier, J., et al. (2001). Sphingosine-1-phosphate induces proliferation of astrocytes: Regulation by intracellular signalling cascades. European Journal of Neuroscience, 13(12), 2067–2076.
Penno, A., Reilly, M. M., Houlden, H., Laura, M., Rentsch, K., Niederkofler, V., et al. (2010). Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. Journal of Biological Chemistry, 285(15), 11178–11187. doi:10.1074/jbc.M109.092973.
Pruett, S. T., Bushnev, A., Hagedorn, K., Adiga, M., Haynes, C. A., Sullards, M. C., et al. (2008). Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. Journal of Lipid Research, 49(8), 1621–1639. doi:10.1194/jlr.R800012-JLR200.
Pulkoski-Gross, M. J., Donaldson, J. C., & Obeid, L. M. (2015). Sphingosine-1-phosphate metabolism: A structural perspective. Critical Reviews in Biochemistry and Molecular Biology, 50(4), 298–313. doi:10.3109/10409238.2015.1039115.
Pyne, S., & Pyne, N. J. (2000). Sphingosine 1-phosphate signalling in mammalian cells. Biochemical Journal, 349, 385–402.
Pyne, S., & Pyne, N. J. (2002). Sphingosine 1-phosphate signalling and termination at lipid phosphate receptors. Biochimica et Biophysica Acta, 1582(1–3), 121–131.
Quehenberger, O., Armando, A. M., Brown, A. H., Milne, S. B., Myers, D. S., Merrill, A. H., et al. (2010). Lipidomics reveals a remarkable diversity of lipids in human plasma. Journal of Lipid Research, 51(11), 3299–3305. doi:10.1194/jlr.M009449.
Rao, T. S., Lariosa-Willingham, K. D., Lin, F. F., Palfreyman, E. L., Yu, N., Chun, J., et al. (2003). Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Research, 990(1–2), 182–194.
Renkonen, O., & Hirvisalo, E. L. (1969). Structure of plasma sphingadienine. Journal of Lipid Research, 10(6), 687–693.
Reynolds, C. P., Maurer, B. J., & Kolesnick, R. N. (2004). Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Letters, 206(2), 169–180. doi:10.1016/j.canlet.2003.08.034.
Riley, R. T., An, N. H., Showker, J. L., Yoo, H. S., Norred, W. P., Chamberlain, W. J., et al. (1993). Alteration of tissue and serum sphinganine to sphingosine ratio: An early biomarker of exposure to fumonisin-containing feeds in pigs. Toxicology and Applied Pharmacology, 118(1), 105–112.
Row, L. C., Ho, J. C., & Chen, C. M. (2007). Cerebrosides and tocopherol trimers from the seeds of Euryale ferox. Journal of Natural Products, 70(7), 1214–1217. doi:10.1021/np070095j.
Rozema, E., Binder, M., Bulusu, M., Bochkov, V., Krupitza, G., & Kopp, B. (2012a). Effects on inflammatory responses by the sphingoid base 4,8-sphingadienine. International Journal of Molecular Medicine, 30(3), 703–707. doi:10.3892/ijmm.2012.1035.
Rozema, E., Popescu, R., Sonderegger, H., Huck, C. W., Winkler, J., Krupitza, G., et al. (2012b). Characterization of glucocerebrosides and the active metabolite 4,8-sphingadienine from Arisaema amurense and Pinellia ternata by NMR and CD spectroscopy and ESI-MS/CID-MS. Journal of Agricultural and Food Chemistry, 60(29), 7204–7210. doi:10.1021/jf302085u.
Russo, S. B., Tidhar, R., Futerman, A. H., & Cowart, L. A. (2013). Myristate-derived d16:0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties. Journal of Biological Chemistry, 288(19), 13397–13409. doi:10.1074/jbc.M112.428185.
Saba, J. D., Nara, F., Bielawska, A., Garrett, S., & Hannun, Y. A. (1997). The BST1 gene of Saccharomyces cerevisiae is the sphingosine-1-phosphate lyase. Journal of Biological Chemistry, 272(42), 26087–26090.
Sadler, T. W., Merrill, A. H., Stevens, V. L., Sullards, M. C., Wang, E., & Wang, P. (2002). Prevention of fumonisin B1-induced neural tube defects by folic acid. Teratology, 66(4), 169–176. doi:10.1002/tera.10089.
Saito, M., Chakraborty, G., Hegde, M., Ohsie, J., Paik, S. M., Vadasz, C., et al. (2010). Involvement of ceramide in ethanol-induced apoptotic neurodegeneration in the neonatal mouse brain. Journal of Neurochemistry, 115(1), 168–177. doi:10.1111/j.1471-4159.2010.06913.x.
Salcedo, M., Cuevas, C., Alonso, J. L., Otero, G., Faircloth, G., Fernandez-Sousa, J. M., et al. (2007). The marine sphingolipid-derived compound ES 285 triggers an atypical cell death pathway. Apoptosis, 12(2), 395–409. doi:10.1007/s10495-006-0573-z.
Sanchez, A. M., Malagarie-Cazenave, S., Olea, N., Vara, D., Cuevas, C., & Diaz-Laviada, I. (2008). Spisulosine (ES-285) induces prostate tumor PC-3 and LNCaP cell death by de novo synthesis of ceramide and PKCzeta activation. European Journal of Pharmacology, 584(2–3), 237–245. doi:10.1016/j.ejphar.2008.02.011.
Santos-Cortez, R. L., Faridi, R., Rehman, A. U., Lee, K., Ansar, M., Wang, X., et al. (2016). Autosomal-recessive hearing impairment due to rare missense variants within S1PR2. American Journal of Human Genetics,. doi:10.1016/j.ajhg.2015.12.004.
Sato, K., Ishikawa, K., Ui, M., & Okajima, F. (1999). Sphingosine 1-phosphate induces expression of early growth response-1 and fibroblast growth factor-2 through mechanism involving extracellular signal-regulated kinase in astroglial cells. Brain Research Molecular Brain Research, 74(1–2), 182–189.
Schroeder, J. J., Crane, H. M., Xia, J., Liotta, D. C., & Merrill, A. H, Jr. (1994). Disruption of sphingolipid metabolism and stimulation of DNA synthesis by fumonisin B1. A molecular mechanism for carcinogenesis associated with Fusarium moniliforme. Journal of Biological Chemistry, 269(5), 3475–3481.
Schwab, S. R., & Cyster, J. G. (2007). Finding a way out: Lymphocyte egress from lymphoid organs. Nature Immunology, 8(12), 1295–1301.
Schwartz, G. K., Ward, D., Saltz, L., Casper, E. S., Spiess, T., Mullen, E., et al. (1997). A pilot clinical/pharmacological study of the protein kinase C-specific inhibitor safingol alone and in combination with doxorubicin. Clinical Cancer Research, 3(4), 537–543.
Shibuya, H., Kawashima, K., Sakagami, M., Kawanishi, H., Shimomura, M., Ohashi, K., et al. (1990). Sphingolipids and glycerolipids. I. Chemical structures and ionophoretic activities of soya-cerebrosides I and II from soybean. Chemical & Pharmaceutical Bulletin, 38(11), 2933–2938.
Shui, G., Stebbins, J. W., Lam, B. D., Cheong, W. F., Lam, S. M., Gregoire, F., et al. (2011). Comparative plasma lipidome between human and cynomolgus monkey: Are plasma polar lipids good biomarkers for diabetic monkeys? PLoS ONE, 6(5), e19731. doi:10.1371/journal.pone.0019731.
Slotte, J. P. (2016). The importance of hydrogen bonding in sphingomyelin’s membrane interactions with co-lipids. Biochimica et Biophysica Acta, 1858(2), 304–310. doi:10.1016/j.bbamem.2015.12.008.
Sorensen, S. D., Nicole, O., Peavy, R. D., Montoya, L. M., Lee, C. J., Murphy, T. J., et al. (2003). Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Molecular Pharmacology, 64(5), 1199–1209. doi:10.1124/mol.64.5.1199.
Spohr, T. C., Dezonne, R. S., Nones, J., Dos Santos Souza, C., Einicker-Lamas, M., Gomes, F. C., et al. (2012). Sphingosine 1-phosphate-primed astrocytes enhance differentiation of neuronal progenitor cells. Journal of Neuroscience Research, 90(10), 1892–1902. doi:10.1002/jnr.23076.
Stahlberg, S., Skolova, B., Madhu, P. K., Vogel, A., Vavrova, K., & Huster, D. (2015). Probing the role of the ceramide acyl chain length and sphingosine unsaturation in model skin barrier lipid mixtures by (2)H solid-state NMR spectroscopy. Langmuir, 31(17), 4906–4915. doi:10.1021/acs.langmuir.5b00751.
St-Jacques, M. (1973). Elucidation of structure and stereochemistry of myriocin. A novel antifungal antibiotic. Journal of Organic Chemistry, 38(7), 1253–1260.
Stockmann-Juvala, H., & Savolainen, K. (2008). A review of the toxic effects and mechanisms of action of fumonisin B1. Human and Experimental Toxicology, 27(11), 799–809. doi:10.1177/0960327108099525.
Struckhoff, A. P., Bittman, R., Burow, M. E., Clejan, S., Elliott, S., Hammond, T., et al. (2004). Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. Journal of Pharmacology and Experimental Therapeutics, 309(2), 523–532. doi:10.1124/jpet.103.062760.
Sugawara, T., Kinoshita, M., Ohnishi, M., Nagata, J., & Saito, M. (2003). Digestion of maize sphingolipids in rats and uptake of sphingadienine by Caco-2 cells. Journal of Nutrition, 133(9), 2777–2782.
Sugawara, T., Zaima, N., Yamamoto, A., Sakai, S., Noguchi, R., & Hirata, T. (2006). Isolation of sphingoid bases of sea cucumber cerebrosides and their cytotoxicity against human colon cancer cells. Bioscience, Biotechnology, and Biochemistry, 70(12), 2906–2912. doi:10.1271/bbb.60318.
Sullards, M. C., Lynch, D. V., Merrill, A. H, Jr., & Adams, J. (2000). Structure determination of soybean and wheat glucosylceramides by tandem mass spectrometry. Journal of Mass Spectrometry, 35(3), 347–353. doi:10.1002/(SICI)1096-9888(200003)35:3<347:AID-JMS941>3.0.CO;2-3.
Suzuki, S., Enosawa, S., Kakefuda, T., Shinomiya, T., Amari, M., Naoe, S., et al. (1996). A novel immunosuppressant, FTY720, with a unique mechanism of action, induces long-term graft acceptance in rat and dog allotransplantation. Transplantation, 61(2), 200–205.
Symolon, H., Bushnev, A., Peng, Q., Ramaraju, H., Mays, S. G., Allegood, J. C., et al. (2011). Enigmol: A novel sphingolipid analogue with anticancer activity against cancer cell lines and in vivo models for intestinal and prostate cancer. Molecular Cancer Therapeutics, 10(4), 648–657. doi:10.1158/1535-7163.MCT-10-0754.
Symolon, H., Schmelz, E. M., Dillehay, D. L., & Merrill, A. H, Jr. (2004). Dietary soy sphingolipids suppress tumorigenesis and gene expression in 1,2-dimethylhydrazine-treated CF1 mice and ApcMin/+ mice. Journal of Nutrition, 134(5), 1157–1161.
Szepanowski, F., Derksen, A., Steiner, I., Meyer Zu Horste, G., Daldrup, T., Hartung, H. P., et al. (2016). Fingolimod promotes peripheral nerve regeneration via modulation of lysophospholipid signaling. Journal of Neuroinflammation, 13(1), 143. doi:10.1186/s12974-016-0612-9.
Tan, J. W., Dong, Z. J., & Liu, J. K. (2003). New cerebrosides from the basidiomycete Cortinarius tenuipes. Lipids, 38(1), 81–84.
Tedesco-Silva, H., Pescovitz, M. D., Cibrik, D., Rees, M. A., Mulgaonkar, S., Kahan, B. D., et al. (2006). Randomized controlled trial of FTY720 versus MMF in de novo renal transplantation. Transplantation, 82(12), 1689–1697. doi:10.1097/01.tp.0000251718.95622.b3.
Thudichum, J. L. W., Simon, J., Thudichum, J. L. W., & St. Thomas’s Hospital. Medical School Library. (1884). A treatise on the chemical constitution of the brain. London: Baillière, Tindall and Cox.
t’Kindt, R., Jorge, L., Dumont, E., Couturon, P., David, F., Sandra, P., et al. (2012). Profiling and characterizing skin ceramides using reversed-phase liquid chromatography-quadrupole time-of-flight mass spectrometry. Analytical Chemistry, 84(1), 403–411. doi:10.1021/ac202646v.
Tolle, M., Pawlak, A., Schuchardt, M., Kawamura, A., Tietge, U. J., Lorkowski, S., et al. (2008). HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(8), 1542–1548. doi:10.1161/ATVBAHA.107.161042.
Tonelli, F., Lim, K. G., Loveridge, C., Long, J., Pitson, S. M., Tigyi, G., et al. (2010). FTY720 and (S)-FTY720 vinylphosphonate inhibit sphingosine kinase 1 and promote its proteasomal degradation in human pulmonary artery smooth muscle, breast cancer and androgen-independent prostate cancer cells. Cellular Signalling, 22(10), 1536–1542. doi:10.1016/j.cellsig.2010.05.022.
Tornquist, K., Blom, T., Shariatmadari, R., & Pasternack, M. (2004). Ceramide 1-phosphate enhances calcium entry through voltage-operated calcium channels by a protein kinase C-dependent mechanism in GH4C1 rat pituitary cells. Biochemical Journal, 380(Pt 3), 661–668. doi:10.1042/BJ20031637.
Ueda, H., Takahara, S., Azuma, H., Kusaka, M., Suzuki, S., & Katsuoka, Y. (2000). Effect of a novel immunosuppressant, FTY720, on allograft survival after renal transplant in rats. European Surgical Research, 32(5), 279–283.
Valsecchi, M., Chigorno, V., Nicolini, M., & Sonnino, S. (1996). Changes of free long-chain bases in neuronal cells during differentiation and aging in culture. Journal of Neurochemistry, 67(5), 1866–1871.
Van Overloop, H., Denizot, Y., Baes, M., & Van Veldhoven, P. P. (2007). On the presence of C2-ceramide in mammalian tissues: Possible relationship to etherphospholipids and phosphorylation by ceramide kinase. Biological Chemistry, 388(3), 315–324. doi:10.1515/BC.2007.035.
Venkataraman, K., Riebeling, C., Bodennec, J., Riezman, H., Allegood, J. C., Sullards, M. C., et al. (2002). Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. Journal of Biological Chemistry, 277(38), 35642–35649. doi:10.1074/jbc.M205211200.
Wang, J., Cheng, A., Wakade, C., & Yu, R. K. (2014). Ganglioside GD3 is required for neurogenesis and long-term maintenance of neural stem cells in the postnatal mouse brain. Journal of Neuroscience, 34(41), 13790–13800. doi:10.1523/jneurosci.2275-14.2014.
Wang, E., Norred, W. P., Bacon, C. W., Riley, R. T., & Merrill, A. H, Jr. (1991). Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. Journal of Biological Chemistry, 266(22), 14486–14490.
Wang, E., Riley, R. T., Meredith, F. I., & Merrill, A. H, Jr. (1999). Fumonisin B1 consumption by rats causes reversible, dose-dependent increases in urinary sphinganine and sphingosine. Journal of Nutrition, 129(1), 214–220.
Webb, M., Tham, C. S., Lin, F. F., Lariosa-Willingham, K., Yu, N., Hale, J., et al. (2004). Sphingosine 1-phosphate receptor agonists attenuate relapsing-remitting experimental autoimmune encephalitis in SJL mice. Journal of Neuroimmunology, 153(1–2), 108–121.
Welsch, C. A., Hagiwara, S., Goetschy, J. F., & Movva, N. R. (2003). Ubiquitin pathway proteins influence the mechanism of action of the novel immunosuppressive drug FTY720 in Saccharomyces cerevisiae. Journal of Biological Chemistry, 278(29), 26976–26982.
Willis, M. A., & Cohen, J. A. (2013). Fingolimod therapy for multiple sclerosis. Seminars in Neurology, 33(1), 37–44. doi:10.1055/s-0033-1343794.
Woodcock, J. M., Ma, Y., Coolen, C., Pham, D., Jones, C., Lopez, A. F., et al. (2010). Sphingosine and FTY720 directly bind pro-survival 14-3-3 proteins to regulate their function. Cellular Signalling, 22(9), 1291–1299. doi:10.1016/j.cellsig.2010.04.004.
Xia, P., Gamble, J. R., Wang, L., Pitson, S. M., Moretti, P. A., Wattenberg, B. W., et al. (2000). An oncogenic role of sphingosine kinase. Current Biology, 10(23), 1527–1530.
Yamagata, K., Tagami, M., Torii, Y., Takenaga, F., Tsumagari, S., Itoh, S., et al. (2003). Sphingosine 1-phosphate induces the production of glial cell line-derived neurotrophic factor and cellular proliferation in astrocytes. Glia, 41(2), 199–206.
Yamashita, R., Tabata, Y., Iga, E., Nakao, M., Sano, S., Kogure, K., et al. (2016). Analysis of molecular species profiles of ceramide-1-phosphate and sphingomyelin using MALDI-TOF mass spectrometry. Lipids, 51(2), 263–270. doi:10.1007/s11745-015-4082-0.
Yang, A. H., Ishii, I., & Chun, J. (2002). In vivo roles of lysophospholipid receptors revealed by gene targeting studies in mice. Biochimica et Biophysica Acta, 1582(1–3), 197–203.
Yang, Y., Torta, F., Arai, K., Wenk, M. R., Herr, D. R., Wong, P. T., et al. (2016). Sphingosine kinase inhibition ameliorates chronic hypoperfusion-induced white matter lesions. Neurochemistry International,. doi:10.1016/j.neuint.2016.02.012.
Yatomi, Y., Ruan, F., Megidish, T., Toyokuni, T., Hakomori, S., & Igarashi, Y. (1996). N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry, 35(2), 626–633. doi:10.1021/bi9515533.
Yu, N., Lariosa-Willingham, K. D., Lin, F. F., Webb, M., & Rao, T. S. (2004). Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia, 45(1), 17–27. doi:10.1002/glia.10297.
Yuan, S., Wu, R., Latek, D., Trzaskowski, B., & Filipek, S. (2013). Lipid receptor S1P(1) activation scheme concluded from microsecond all-atom molecular dynamics simulations. PLoS Computational Biology, 9(10), e1003261. doi:10.1371/journal.pcbi.1003261.
Zhang, K., Pompey, J. M., Hsu, F. F., Key, P., Bandhuvula, P., Saba, J. D., et al. (2007). Redirection of sphingolipid metabolism toward de novo synthesis of ethanolamine in Leishmania. EMBO Journal, 26(4), 1094–1104. doi:10.1038/sj.emboj.7601565.
Zitomer, N. C., Mitchell, T., Voss, K. A., Bondy, G. S., Pruett, S. T., Garnier-Amblard, E. C., et al. (2009). Ceramide synthase inhibition by fumonisin B1 causes accumulation of 1-deoxysphinganine: A novel category of bioactive 1-deoxysphingoid bases and 1-deoxydihydroceramides biosynthesized by mammalian cell lines and animals. Journal of Biological Chemistry, 284(8), 4786–4795. doi:10.1074/jbc.M808798200.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D.R.H. and G.L.H. have received consulting fees or grant support from Expression Drug Designs, LLC and Bayer Healthcare. J.C. has received honoraria, consulting fees and/or grant support from: Abbott, Amira Pharmaceuticals, Biogen-Idec, Celgene, GlaxoSmithKline, Johnson and Johnson, Merck, Mitsubishi Tanabe Pharma Corporation, Novartis, Ono Pharmaceutical Co., Pfizer and Taisho Pharmaceutical Co. M.K.P.L., W.S.C., F.T., and A.R. have no financial relationships to declare.
Rights and permissions
About this article

Cite this article
Lai, M.K.P., Chew, W.S., Torta, F. et al. Biological Effects of Naturally Occurring Sphingolipids, Uncommon Variants, and Their Analogs. Neuromol Med 18, 396–414 (2016). https://doi.org/10.1007/s12017-016-8424-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-016-8424-8