
Abstract
Most biobutanol-producing Clostridium strains are unable to ferment polysaccharides such as cellulose and xylan due to the lack of hydrolyzing enzymes. In this study, we show that Clostridium beijerinckii G117, a newly isolated biobutanol-producing strain, expresses xylanase enzyme in the presence of 1 % beechwood xylan. The xylanase activity in the medium containing actively growing culture and 1 % of beechwood xylan can reach up to 2.66 U/ml after 14 h of fermentation. Using salting-out and size-exclusion chromatography, we purify the crude xylanase by 8.7-fold from the supernatant with a yield of 32.2 %. This purified xylanase has a molecular weight of 22.6 kDa, making it one of the smallest reported clostridial xylanases. Conserved domain analysis reveals that the xylanase belongs to glycoside hydrolase family 11 (GH11) but lacks a carbohydrate binding domain. When beechwood xylan is used as substrate for the xylanase, majority of the products are xylo-oligosaccharide (~98 %), suggesting that this is an endo-1,4-β-xylanase.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The recent energy crisis and soaring oil prices have rekindled interest in biofuel production from renewable feedstock. Currently, biofuels such as biodiesel and bioethanol have been produced from renewable biomass on a large scale. However, biobutanol, another class of biofuel, has several advantages over both biodiesel and bioethanol, such as higher energy content, lower volatility, less corrosion, and improved compatibility with car engines [1]. Therefore, biobutanol attracted considerable attention in the past few years as a promising biofuel. To produce biobutanol, one common route is through acetone-butanol-ethanol (ABE) fermentation using a solventogenic Clostridium sp., such as Clostridium acetobutylicum, Clostridium beijerinkii, Clostridium saccharobutylicum, or Clostridium saccharoperbutylacetonicum. These Clostridium strains are able to utilize feedstocks such as glucose, galactose, xylose, starch, and molasses to perform ABE fermentation [2]. However, due to rising costs, the choice of feedstock has gradually shifted to nonedible, low-cost lignocellulosic biomass. This biomass typically consists of cellulose (40–50 %), hemicellulose (30–40 %), and lignin (8–10 %). Because cellulose is the major component, it is not surprising that extensive work has been dedicated to the use of cellulose for ABE fermentation [3]. However, ABE fermentation from lignocellulosic biomass is not economical, unless hemicellulose is also utilized alongside cellulose [4, 5].
Xylan, a major component in hemicellulose, is a polymer of d-xylose cross-linked by β-1,4-glycosidic bonds. However, before its uptake by Clostridia, xylan must first be hydrolyzed to liberate the fermentable monosugars [6]. Thus, pretreatment steps such as steam explosion or chemical hydrolysis with a strong acid or base are often required. However, these pretreatments are typically energy intensive and strong inhibitors, such as furfural, are formed during the process [7]. Alternatively, xylan can be hydrolyzed enzymatically. Several hydrolyzing enzymes including endo-β-1,4-xylanase (EC 3.2.1.8), α-L-arabinofuranosidase (EC 3.2.1.55), xylan esterases (EC 3.1.1.6), α-D-glucuronidase (EC 3.2.1.139), and β-D-xylosidase (EC 3.2.1.37) are required for the complete hydrolysis of xylan [15, 16]. Among them, endo-β-1,4-xylanase plays an important role by cleaving the polysaccharide backbone to release xylo-oligosaccharides or xylose, which can then be fermented by Clostridium [8]. Although certain Clostridium strains such as Clostridium thermocellum are able to use xylan to produce ethanol, most biobutanol-producing Clostridium species do not express xylanase natively [9]. Thus, they are unable to ferment lignocellulose for butanol production. To date, the only two reported exceptions are C. acetobutylicum ATCC 824 and Clostridium. sp. BOH3 [10, 11].
In this study, we focus on a wild-type Clostridium beijerinckii strain G117 that was recently isolated in our lab [12, 13]. This strain is capable of producing 13.5 g/l of butanol (from 60 g/l of glucose), which is 47 % higher than that produced by C. beijerinckii NCIMB 8052. Another unique feature of G117 is the absence of ethanol in the fermentation products. Finally, this strain also shows xylanolytic activity when xylan is used as the sole substrate. To better understand this unique strain and its xylanolytic system, extensive studies were carried out on the production, purification, and characterization of the xylanase from G117. This information is important for the production of ABE solvents from lignocellulosic biomass using G117. This is also the first purification study of xylanase from C. beijerinckii.
Materials and Methods
Materials and Reagents
All carbon sources (except arabinose) and the components of the culture media were obtained from Sigma-Aldrich (U.S.A.). Arabinose was obtained from Alfa Aesar (U.K.). The beechwood xylan (X4252) used in this study contains more than 90 % xylose residues. Chemicals and the apparatus used in gel electrophoresis were obtained from Bio-Rad (U.S.A.) unless otherwise stated. All other reagents used were analytical grade.
Cell Culture Conditions
The minimum salt medium was first prepared based on a previously reported protocol [14] with an additional 0.3 % of yeast extract. This medium was dispensed into serum bottles (20 ml each), purged with nitrogen and sealed with rubber septa prior to autoclaving at 121 °C for 20 min. For fermentation involving solid substrates (beechwood xylan, cellulose, carboxymethyl cellulose, or lignin), the substrate was added into the medium-containing bottle before it was sealed. Alternatively, the liquid carbon sources (d-glucose, d-xylose and l-arabinose) were prepared as stock solutions and sterilized separately. Inoculum of C. beijerinckii G117 was prepared by diluting an active subculture to 0.5–0.6 g/l cell dry weight (approximately 1.9–2.2 × 107 cell/ml). After adding the inoculum to the medium in a 1:9 ratio, the bottles were incubated at 35 °C with 130-rpm shaking. Samples were drawn periodically, and after centrifugation at 10,000×g for 10 min, the supernatant was used for further analysis. The resulting cell pellet was washed with deionized water and resuspended in 0.1 M sodium hydroxide. The solution was then boiled for 30 min and the total cell protein was measured using Bradford reagent.
Xylanase Assay and Protein Quantification
The xylanase activity was assayed by measuring the reducing sugars released during xylan hydrolysis. Xylanase solution (100 μl) was added to 1 ml of 0.5 % (w/v) beechwood xylan in acetate buffer (50 mM, pH 5.0). The reaction mixture was subsequently incubated at 50 °C for 15 min and then the total reducing sugar was quantified using dinitrosalicylic acid (DNS) [15]. One unit of activity (U) is defined as the liberation of 1 μmol of reducing sugar from the enzymatic reaction in 1 min. Two blank controls (enzyme-blank and substrate-blank) were also prepared. The values from both blanks were subtracted from the measured xylanase activity. The protein was quantified using Bradford reagent (Sigma-Aldrich, U.S.A.), according to the protocol provided by the manufacturer.
Purification of Xylanase
The purification of xylanase was completed into two steps. The first step was to precipitate xylanase using the salting-out method [16]. After growing G117 in a minimum salt medium containing 1 % beechwood xylan for 48 h, the supernatant was collected after centrifugation (12,000×g, 10 min, 4 °C) and then filtered through a 0.22-μm membrane filtration module (Corning, U.S.A). Ammonium sulfate was added gradually to achieve 80 % saturation (0.52 g/ml of supernatant) and the solution was incubated overnight at 4 °C. The protein was collected through centrifugation (8,000×g, 15 min, 4 °C) and then resuspended in phosphate buffer (pH 6.0, 50 mM). After desalting with a PD-10 gel-filtration column (GE Healthcare, U.K.), the solution obtained was treated as the crude protein solution. The second purification step was to separate the proteins using size-exclusion chromatography (SEC). First, the crude protein solution was loaded into a column (Superdex 200 10/300 GL) mounted on an ÄKTA purifier (both from GE Healthcare, U.K.). The column was pre-equilibrated with phosphate buffer (50 mM, pH 6.0) and flushed with 36 ml (equivalent to 1.5 column volumes) of the same buffer after sample loading. The column elution was collected in 1-ml fractions and the fractions exhibiting xylanase activity were pooled together for further analysis.
Gel Electrophoresis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was conducted under the conditions reported previously [11] except for the running voltage which was set at 160 V. The pre-stained Page RulerTM Plus (Thermo Scientific, U.S.A.) protein marker was used as a molecular weight indicator. The gel was stained with Coomassie and the image was captured using a Gel Doc system.
Zymography of purified xylanase was conducted using the protocol reported previously [11] with minor modifications. After the electrophoresis step under native condition, the gel was incubated with 1 % beechwood xylan in acetate buffer (pH 5.0, 50 mM) for 1 h at 35 °C with agitation. After washing, the gel was stained with 0.5 % Congo red for 10 min. The excess dye was washed away using ample 1 M sodium chloride. Acetic acid (0.5 %) was added to turn the gel blue to facilitate visualization.
Mass Spectrometry and Peptide Mass Fingerprinting
Mass spectrometry was used to determine the molecular weight of the G117 xylanase. For matrix preparation, 10 mg/ml 2,5-dihydroxybenzoic acid (Sigma, U.S.A) was prepared in an aqueous solution containing 0.1 % trifluoroacetic acid and 50 % acetonitrile. The purified xylanase solution was further concentrated using a Vivaspin ultrafiltration module (5 kDa cut-off, GE Healthcare, U.K.). The concentrated xylanase solution was then mixed with the matrix in a 3:1 ratio. The sample was analyzed with an Autoflex II MALDI-TOF mass spectrometer (Bruker Daltonics, U.S.A.). For protein sequence confirmation, peptide mass fingerprinting (PMF) was performed. The purified xylanase band on SDS-PAGE was first cut and digested in-gel using trypsin (Promega, Germany). It was given to the Protein and Proteomics Centre at the National University of Singapore for subsequent PMF analysis.
Xylanase Specificity and Kinetics
To investigate the substrate specificity, assays using purified xylanase were carried out with either 0.5 % w/v beechwood xylan, carboxymethylcellulose (CMC), Avicel cellulose, cellobiose, or starch. The kinetic studies with approximately 10 U purified xylanase were performed by varying the beechwood xylan concentration in the assay from 0.1 to 1.0 % (w/v). All assays were conducted under the conditions described previously.
Effect of Metal Ions and Inhibitors/Promoters
To study the effects of the presence of metal ions and inhibitors/promoters on xylanase activity, assays were performed with purified xylanase in the presence of metal salts (1 and 10 mM AgNO3, CaCl2, CoCl2, CuSO4, FeSO4, MnSO4, NiCl2, ZnCl2, and MgCl2) or common enzyme inhibitors/promoters (1, 10, and 50 mM ethylenediaminetetraacetic acid, urea, cysteine, dithiothreitol, beta-mercaptoethanol, SDS, 4-aminobenzoic acid, and Tween 80). The result was normalized using the enzyme activity without metal ions or inhibitors/promoters.
Effect of Temperature and pH
Xylanase activity was measured by assaying purified enzyme at pH 5.0 with temperatures ranging from 30 to 60 °C. Upon completion of the enzymatic reaction, the solution was immediately quenched in an ice bath before DNS reagent was added to prevent further reaction. The optimum xylanase pH was discerned by dissolving beechwood xylan in 50 mM citrate (pH 3.0), acetate (pH 4.0–5.5), phosphate (pH 5.5–8.0) or carbonate (pH 8.0–10) buffer as the assay substrate. The activity was then assayed at 50 °C. For the study of enzyme temperature stability, purified xylanase was first incubated at different temperatures (30–60 °C) at pH 5.0. Samples were then taken every 15 min and assayed at 50 °C at pH 5.0. For the pH stability studies, the samples were first buffer-exchanged with buffers at different pH values, which was accomplished by desalting using Vivaspin concentrators (5 kDa cut-off, GE Healthcare, U.K.) to avoid diluting the enzyme solution. The buffer solutions used were the same as those used in the determination of optimum pH. After buffer-exchange, the xylanase solutions were incubated at 35 °C. The residual activity of the xylanase was assayed after 0, 12 and 24 h of incubation.
Enzymatic Hydrolysis of Xylan
Approximately 10 U of purified xylanase was mixed with 2 ml of acetate buffer (pH 5.0, 50 mM) containing 4 % (w/v) beechwood xylan. After incubation for 48 h at 40 °C, the solution was centrifuged (12,000 × g, 10 min, 4 °C) and filtered with a 0.22-μm membrane filtration module (Corning, U.S.A). Subsequently, the solution was concentrated approximately 4-fold using a freeze drier (Christ, Germany). A control experiment lacking xylanase was also performed in parallel. The composition of the hydrolysis products was analyzed with high performance liquid chromatography (HPLC) using a Zorbax column (Agilent, U.S.A) and a refractive index detector (RID). The mobile phase was 75 % acetonitrile/25 % water with a flow rate of 1.4 ml/min.
Results and Discussion
Production of Xylanase
The growth profile (in terms of total cell protein) of G117 in a minimum salt medium containing 1 % of beechwood xylan is shown in Fig. 1. Its growth starts with a lag phase for approximately 7 h and is followed by a relatively short exponential phase. The xylanase activity starts to increase at the beginning of the exponential phase and attains its highest activity when the growth enters stationary phase. This result implies that the production of xylanase is associated with growth, which is similar to C. beijerinckii LU-1, as reported by Marichamy and Mattiasson [17]. The short exponential phase also corresponds to an acidogenesis phase that leads to an abrupt drop in pH (Fig. 1). Utilizing the fermentable sugar liberated by the xylanase, organic acids (2.67 g/l butyric and 0.57 g/l acetic acid) are produced during the acidogenesis phase. The final amount of solvents is relatively low (0.37 g/l butanol and 0.064 g/l acetone). Notably, the time required for G117 to attain maximum xylanase activity (2.66 U/ml) is rather short (approximately 14 h) compared to other wild-type strains, such as C. beijerinckii LU-1 (21–24 h) [17], Clostridium celerecrescens (20 h) [18], Clostridium cellulovorans (36 h) [19], Clostridium sp. SAIV (60 h) [20], Clostridium absonum CFR-702 (72 h) [21], C. acetobutylicum ATCC 39236 (175 h) [22], and Clostridium sp. PXYL1 (6 days) [23].

The growth, xylanase production and pH profiles of G117 cultured with 1 % beechwood xylan in a minimum salt medium
To determine the most efficient xylanase inducer, the bacterium was cultured with various carbon sources for 48 h before the activity was quantified. Among all carbon sources (1 % w/v) tested, beechwood xylan is the best inducer and leads to the highest xylanase activity (2.6 U/ml). In contrast, arabinose and xylose only result in 0.28 and 0.20 U/ml of xylanase activity, respectively. Negligible activity is observed when glucose, sucrose, lignin, cellulose, or filter paper is used as the carbon source. This trend is similar to xylanase from other clostridial strains [17, 19–21, 24].
Repression of Xylanase Production by Monosaccharides
To investigate the repression of xylanase production by monosaccharides, G117 was grown in media containing 1 % xylan in the presence of either glucose, xylose, or arabinose in varying concentrations. Both the xylanase production and total cell protein concentration were measured after 48 h of growth. To take cell growth into account, the observed xylanase production is normalized against the corresponding total cell protein concentration, as presented in Fig. 2. The xylanase production is affected by both the type of monosaccharide and its concentration (Fig. 2). Both xylose and arabinose enhance xylanase production at concentrations below 2.5 g/l, but they repress the production at higher concentrations. In contrast, glucose represses xylanase production at all concentrations. The repression of xylanase production has been reported for many clostridial species, except C. beijerinckii [25]. Our study confirms that the xylanase production in C. beijerinckii, like the other clostridial species, is also repressed by monosaccharides. When both monosaccharides and polysaccharides are present, strain G117 prefers the former as the main carbon source, because they are easier to metabolize. As a result, less hydrolyzing enzymes are expressed, which is driven by a mechanism known as catabolite repression. In contrast, the enhancement of xylanase production at low monosaccharide concentrations can be explained through a regulatory mechanism for the expression of xylanase [8, 25]. In brief, either xylose or arabinose can enter the cell membrane and induce the expression of xylanase. However, this is only true at low monosaccharide concentrations when it is nutrient-limiting. At a high monosaccharide concentration, this process is outweighed by the effect of catabolite repression. Thus, xylanase production is again repressed. A similar regulation pattern was also reported by Stoppok et al. [26] when cellobiose is used as a substrate for endoglucanase expression in Cellulomonas uda. Because glucose is not part of the xylanase regulation mechanism, xylanase production is suppressed regardless of its concentration. This result is supported by Han et al. [27], who showed that glucose suppresses the production of both C. cellulovorans cellulase and hemicellulase at the transcriptional level. The repression of xylanase production by monosaccharides has important implications for enzymatic hydrolysis in the production of biofuel from lignocellulosic biomass. This is because monosaccharides are released in certain biomass pre-treatment processes [28].

Influence of monosaccharide concentration on G117 xylanase production in the presence of 1 % beechwood xylan. Normalized activity refers to the ratio of xylanase activity to the corresponding total cell protein concentration (both measured after 48 h). Xylanase production is repressed when the monosaccharide concentration is more than 5 g/l but enhanced by 2.5 g/l of either xylose or arabinose
Purification of Xylanase
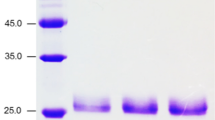
Crude xylanase was purified to homogeneity using the two-step method described in the experimental section. Xylanase is among the last proteins to elute, implying that it has a relatively small molecular weight compared to other proteins in the supernatant. The fractions showing xylanase activity were then pooled together and analyzed by SDS-PAGE. An image of the gel suggests that the fractions contain a single dominant protein and the non-denaturing zymogram of purified xylanase is also shown (Fig. 3). The highlighted clear zone represents the hydrolyzing activity of the purified xylanase when incubated in a 1 % beechwood xylan solution. The activity of the purified xylanase solution is 73.2 ± 1.8 U/mg, which is 8.7-fold higher than the crude protein solution. Based on total xylanase activity, the overall yield is 32.2 %. The detailed purification table can be found in the Electronic Supplementary Material. Compared to what is used for other bacterial xylanases, the purification scheme employed in this work is much simpler. Reasonable yield and purity can be achieved with just one chromatographic step, unlike the conventional two-step ion exchange-size exclusion strategy [16].

SDS-PAGE images of molecular weight marker (lane 1), the crude enzyme (lane 2) and purified enzyme (lane 3). The xylanase band on the gel is highlighted. Lane 4 shows the non-denaturing PAGE of the purified xylanase where a clear band signifies xylan-hydrolyzing activity
Using MALDI-TOF-MS, we further determined that the molecular weight of this xylanase is 22,599 Da, which is close to the theoretical molecular weight from the translated protein sequence (23.2 kDa). This small molecular weight makes this xylanase one of the smallest xylanases from Clostridium. For comparison, C. absonum CFR702, C. thermocellum ATCC 27405, Clostridium. sp. BOH3, and C. acetobutylicum ATCC 824 produce xylanases with molecular weights of 150, 74, 36, and 34 kDa, respectively [9, 11, 21, 29]. The only xylanase smaller than the one from G117 is xylanase B from Clostridium sp. SAIV, which has an estimated molecular weight of 20 kDa [30].
Gene Sequence of Xylanase
One xylanase gene is found in the whole genome of G117 [13] by subjecting each encoding gene to BLAST against the NCBI database. This xylanase gene sequence has been deposited in the NCBI GenBank database (accession number KM009141). The gene sequence is then translated into the corresponding amino acid sequence. To confirm the identity of the purified enzyme, PMF was performed. The resulting peptide fragments were aligned with the translated amino acid sequence and three exact matches were identified. Figure 4 shows the matching peptide sequences with their corresponding scores and E values. A protein conserved domain search was performed using protein-protein BLAST against the NCBI CDD database. The conserved catalytic domain of this xylanase belongs to glycoside hydrolase family 11. One interesting feature of this xylanase is the absence of carbohydrate-binding domain (CBD), which is a common feature of clostridial xylanases in the GH11 family [9].

The translated amino acid sequence from genomic data of the xylanase secreted by C. beijerickii G117. The peptide fragments obtained from peptide mass fingerprinting are aligned with the translated xylanase amino acid sequence. The alignment, where the matching sequences are boxed, reveals three exact matches between the two. The start and end of the GH11 family conserved domain are marked by asterisks. The only difference between G117 xylanase and C. beijerinckii NCIMB 8052 is underlined. The serine is replaced by a cysteine residue in the xylanase from NCIMB 8052
The 16S rRNA of G117 shows 99 % identity to C. beijerinckii NCIMB 8052 that has a genome size of 6.0 Mbp, which is 0.2 Mbp greater than G117. In addition, the xylanase gene sequence of Clostridium sp. G117 shows high homology (99 %) with xylanase from NCIMB 8052 (NCBI gene ID 5294226) [12, 13, 31]. In fact, the xylanases only differ by one amino acid (Fig. 4). Hence, it is expected that these two xylanases will have similar behaviors. However, further studies are needed to confirm the similarity of these two xylanases, because a detailed report of the xylanase from NCIMB 8052 was unavailable. In fact, any literature data regarding the xylanase from C. beijerinckii is rather scarce. In addition to the xylanase from NCIMB 8052, Marichamy and Mattiasson [17] described another xylanase from C. beijerinckii LU-1, but it is unclear whether this strain is solventogenic. The authors only described the production profile of xylanase and its physical properties but not protein purification or sequence.
Substrate Specificity and Reaction Kinetics
The specificity of xylanase was examined by incubating purified xylanase with different substrates. Among the substrates, the purified xylanase shows the highest activity with beechwood xylan but no activity for other substrates including carboxymethylcellulose (CMC), Avicel cellulose, cellobiose, and starch. This corroborates the conclusion reached by Biely et al. [32], who stated that high substrate specificity is one of the key features for the xylanases in the GH11 family. These authors also suggested that xylanases from this family generally have low molecular weights and lack the catalytic domains necessary for hydrolysis of substrates other than xylan. This high specificity is also observed in xylanases from C. beijerinckii LU-1 [17], C. absonum CFR-702 [33], Clostridium. sp. BOH3 [11], C. stercorarium F-9 [34], and C. thermocellum F1 [35]. The only exception is the xylanase from C. acetobutylicum ATCC 824 [24] that shows cross-reactivity towards both beechwood xylan and cellulose.
To determine the reaction kinetics of the xylanase, the activity was assayed with different concentrations of beechwood xylan as the substrate. Using a Lineweaver-Burk plot, we determined the K m and V max values to be 19.1 mg/ml and 2,766 U/mg, respectively. For comparison, the highest V max for a clostridial xylanase is 5,500 U/mg [36]. Moreover, this K m value is higher than the K m values from other bacterial xylanases, which typically fall below 10 mg/ml [25, 37]. The only exception is xyl-II from C. absonum CFR-702, which has a K m value of 14 mg/ml [33]. The lack of a CBD, which facilitates the binding of insoluble substrates with xylanase, is most likely the main reason the K m is high for the xylanase from C. beijerinckii G117 [38].
Effect of Temperature and pH
Figure 5a shows the relative xylanase activity at different temperatures, and the activity measured at 40 °C is defined as 100 % relative activity. Xylanase produces the highest activity between 40–50 °C (Fig. 5a), which quickly drops to approximately 10 % at 60 °C. Xylanase is also unstable and loses all activity at 50 °C and above after 15 min (Fig. 6a). However, this result is expected, because G117 is a mesophilic strain and its xylanase is likely denatured at 50 °C and above. For comparison, the optimal temperature of the xylanase from C. beijerinckii LU-1 is 60 °C [17]. Figure 5b shows the relative xylanase activity at different pH values. The optimum pH for xylanase activity is 5.0, which is the same as the xylanase from C. beijerinckii LU-1 [17], and activity loss is observed at extreme pH (Fig. 5b). At neutral and slightly acidic pH (5.0) values, the xylanase is relatively stable after 24 h. However, it loses its activity in basic (pH 9.0) or more acidic (pH 3.0) media (Fig. 6b), which substantiates a previous report that xylanases from Clostridium have a narrow optimal pH between 5 and 7 [9].

Influence of temperature (a) and pH (b) on xylanase activity. Activity is expressed as the percentage relative activity, where 100 % represents the highest recorded activity

Xylanase enzyme stability when incubated at different temperature (a) and pH (b) values. Initial activity is assigned as 100 % relative activity
Effect of Metal Ions and Common Enzyme Inhibitors/Promoters
The effects of various metal ions on the relative activity of xylanase are shown in Table 1. For comparison, the activity in the absence of any metal ion is defined as 100 %. Among all metal ions tested, Ag+ inhibits activity the most. The activity is reduced to 12 % after the addition of 10 mM Ag+. Similar behavior was reported for other clostridial xylanases [11, 33] and this is attributed to oxidization of the sulfhydryl moiety in xylanase by Ag+ [39]. In addition, xylanase is also inhibited by 10 mM Cu2+ and Fe2+, reducing the relative activities to 49.1 and 78.1 %, respectively. Only 10 mM Co2+ and 1 mM Mn2+ show activation effects, and the relative activities increase to 110 and 112 %, respectively. The remainder of the metal ions slightly reduces the activity. This information, in addition to the published data, does not elucidate a clear trend for the influence of metal ions on xylanases from Clostridium. One other study [11] reported activity enhancement by Co2+ and Mn2+, but the authors also observed an enhancement of xylanase activity by Cu2+ and Fe2+. Conversely, both Cu2+ and Fe3+ negatively affect the activity of xylanases from C. stercorarium [36] and C. sp. SAIV [30].
Table 1 also shows the effects of common enzyme inhibitors/promoters towards the xylanase from G117. The activities are slightly enhanced or inhibited (by less than 20 %) in the presence of up to 50 mM ethylenediaminetetraacetic acid (EDTA), urea, beta-mercaptoethanol, 4-aminobenzoic acid and Tween 80. However, 50 mM sodium dodecyl sulfate (SDS) leads to significant (65 %) activity loss. Similar inhibition effects of SDS on xylanase were also reported previously [11, 33]. Interestingly, in the presence of certain thiol compounds, such as 50 mM cysteine and dithiothreitol (DTT), the xylanase relative activities increase to 265 and 184 %, respectively. According to Knob and Carmona [40], DTT is able to prevent the oxidation of sulfhydryl groups, while cysteine helps retain the xylanase tertiary structure.
Xylanase Hydrolysis Products
Beechwood xylan was incubated with purified xylanase for 48 h and its hydrolysis product was then analyzed using HPLC. The hydrolysis product comprises 2 % xylose (X1), 27 % xylobiose (X2), 31 % xylotriose (X3), and 21 % xylotetraose (X4). The remainder of the products are xylo-oligosaccharides of 5 residues or more (>X5). No xylo-oligosaccharides were detected in the control experiment (without xylanase). All bacterial xylanases produce xylo-oligosaccharides as end-products, especially X2 and X3, but the release of xylose is not universal to all species [41]. Moreover, most xylanases from Clostridium reported in literature, such as C. acetobutylicum ATCC 824 [42], Clostridium. sp. BOH3 [11], C. cellulovorans [19], and C. sp. SAIV [20], do not produce xylose as a hydrolysis product. Biely et al. [32] postulated that this difference can be attributed to the family of the specific xylanase, and they empirically proved that GH11 xylanases show less affinity to xylo-oligosaccharides compared to the xylanases from the GH10 family. Hence, they do not show the beta-xylosidase type of activity.
Conclusions
In this study, a GH11 xylanase from mesophilic C. beijerinckii G117 was purified and characterized. The xylanase expression can be enhanced by adding low concentrations (<2.5 g/l) of xylose or arabinose in xylan-supplemented culture media. Xylanase purification from the supernatant can be achieved in two stages. Compared to the other clostridial xylanases, this xylanase features rapid expression (in 14 h), small size (23 kDa), and high specificity towards xylan. Because this xylanase is produced by a high biobutanol-producing Clostridium strain, it has the potential to be applied in the production of biobutanol, especially from hemicellulose-rich biomass, that would solely rely on one bacterial strain.
References
Jin, C., Yao, M., Liu, H., Lee, C. F., & Ji, J. (2011). Renewable and Sustainable Energy Reviews, 15, 4080–4106.
Ezeji, T. C., Qureshi, N., & Blaschek, H. P. (2007). Current Opinion in Biotechnology, 18, 220–227.
Sun, Y., & Cheng, J. (2002). Bioresource Technology, 83, 1–11.
Gírio, F. M., Fonseca, C., Carvalheiro, F., Duarte, L. C., Marques, S., & Bogel-Łukasik, R. (2010). Bioresource Technology, 101, 4775–4800.
Jeffries, T. W. (2006). Current Opinion in Biotechnology, 17, 320–326.
Menon, V., & Rao, M. (2012). Progress in Energy and Combustion Science, 38, 522–550.
Bobleter, O. (1994). Progress in Polymer Science, 19, 797–841.
Biely, P. (1985). Trends in Biotechnology, 3, 286–290.
Paes, G., Berrin, J. G., & Beaugrand, J. (2012). Biotechnology Advances, 30, 564–592.
Lee, S. F., Forsberg, C. W., & Gibbins, L. N. (1985). Applied and Environmental Microbiology, 50, 1068–1076.
Rajagopalan, G., Yew, K. W., He, J., & Yang, K. L. (2012). BioEnergy Research, 6, 448–457.
Chua, T. K., Liang, D. W., Qi, C., Yang, K. L., & He, J. (2013). Bioresource Technology, 135, 372–378.
Wu, Y. R., Li, Y., Yang, K. L., & He, J. (2012). Journal of Bacteriology, 194, 5470–5471.
He, J., Ritalahti, K. M., Yang, K.-L., Koenigsberg, S. S., & Loffler, F. E. (2003). Nature, 424, 62–65.
Miller, G. L. (1959). Analytical Chemistry, 31, 426–428.
Sa-Pereira, P., Paveia, H., Costa-Ferreira, M., & Aires-Barros, M. R. (2003). Molecular Biotechnology, 24, 257–281.
Marichamy, S., & Mattiasson, B. (2005). Enzyme and Microbial Technology, 37, 497–504.
Palop, M. L., Vallés, S., Piñaga, F., & Flors, A. (1991). Journal of Chemical Technology and Biotechnology, 51, 105–114.
Kosugi, A., Murashima, K., & Doi, R. H. (2001). Journal of Bacteriology, 183, 7037–7043.
Murty, M. V. S., & Chandra, T. S. (1991). Enzyme and Microbial Technology, 13, 430–435.
Rani, D. S., & Nand, K. (2000). Process Biochemistry, 36, 355–362.
Lemmel, S. A., Datta, R., & Frankiewicz, J. R. (1986). Enzyme and Microbial Technology, 8, 217–221.
Akila, G., & Chandra, T. S. (2003). FEMS Microbiology Letters, 219, 63–67.
Ali, M. K., Rudolph, F. B., & Bennett, G. N. (2005). Journal of Industrial Microbiology and Biotechnology, 32, 12–18.
Subramaniyan, S., & Prema, P. (2002). Critical Reviews in Biotechnology, 22, 33–64.
Stoppok, W., Rapp, P., & Wagner, F. (1982). Applied and Environmental Microbiology, 44, 44–53.
Han, S. O., Yukawa, H., Inui, M., & Doi, R. H. (2003). Journal of Bacteriology, 185, 6067–6075.
Narayanaswamy, N., Dheeran, P., Verma, S., & Kumar, S. (2013). Biological Pretreatment of Lignocellulosic Biomass for Enzymatic Saccharification. In Z. Fang (Ed.), Pretreatment Techniques for Biofuels and Biorefineries (pp. p. 3–p. 34). Berlin Heidelberg: Springer.
Ali, M. K., Rudolph, F. B., & Bennett, G. N. (2004). Journal of Industrial Microbiology and Biotechnology, 31, 229–234.
Murty, M. V., & Chandra, T. S. (1992). Antonie van Leeuwenhoek, 61, 35–41.
Wang, Y., Li, X., Mao, Y., & Blaschek, H. P. (2012). BMC Genomics, 13, 102.
Biely, P., Vrsanska, M., Tenkanen, M., & Kluepfel, D. (1997). Journal of Biotechnology, 57, 151–166.
Swaroopa Rani, D., & Nand, K. (2001). Anaerobe, 7, 45–53.
Sakka, K., Kojima, Y., Kondo, T., Karita, S., Shimada, K., & Ohmiya, K. (1994). Bioscience, Biotechnology, and Biochemistry, 58, 1496–1499.
Hayashi, H., Takehara, M., Hattori, T., Kimura, T., Karita, S., Sakka, K., & Ohmiya, K. (1999). Applied Microbiology and Biotechnology, 51, 348–357.
Bérenger, J.-F., Frixon, C., Bigliardi, J., & Creuzet, N. (1985). Canadian Journal of Microbiology, 31, 635–643.
Bastawde, K. B. (1992). World Journal of Microbiology and Biotechnology, 8, 353–368.
Ali, M. K., Hayashi, H., Karita, S., Goto, M., Kimura, T., Sakka, K., & Ohmiya, K. (2001). Bioscience, Biotechnology, and Biochemistry, 65, 41–47.
Nagarajan, D. R., Rajagopalan, G., & Krishnan, C. (2006). Applied Microbiology and Biotechnology, 73, 591–597.
Knob, A., & Carmona, E. C. (2010). Applied Biochemistry and Biotechnology, 162, 429–443.
Sunna, A., & Antranikian, G. (1997). Critical Reviews in Biotechnology, 17, 39–67.
Lee, S. F., Forsberg, C. W., & Rattray, J. B. (1987). Applied and Environmental Microbiology, 53, 644–650.
Acknowledgments
This work was supported by the Singapore National Research Foundation (NRF) under grant NRF 2009 NRF-CRP 001-039. The contributions of Dr. Wu Yirui to this work are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Table S1
Table summarizing the purification steps of G117 xylanase from the crude enzyme to the final purified product. (DOCX 12 kb)
Rights and permissions
About this article

Cite this article
Ng, C.H., He, J. & Yang, KL. Purification and Characterization of a GH11 Xylanase from Biobutanol-Producing Clostridium beijerinckii G117. Appl Biochem Biotechnol 175, 2832–2844 (2015). https://doi.org/10.1007/s12010-014-1470-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-014-1470-5