
Abstract
Purpose of the Review
The purpose of this review is to summarize the role of the osteocyte in muscle atrophy in cancer patients, sarcopenia, spinal cord injury, Duchenne’s muscular dystrophy, and other conditions associated with muscle deterioration.
Recent Findings
One type of bone cell, the osteocyte, appears to play a major role in muscle and bone crosstalk, whether physiological or pathological. Osteocytes are cells living within the bone-mineralized matrix. These cells are connected to each other by means of dendrites to create an intricately connected network. The osteocyte network has been shown to respond to different types of stimuli such as mechanical unloading, immobilization, aging, and cancer by producing osteocytes-derived factors. It is now becoming clear that some of these factors including sclerostin, RANKL, TGF-β, and TNF-α have detrimental effects on skeletal muscle.
Summary
Bone and muscle not only communicate mechanically but also biochemically. Osteocyte-derived factors appear to contribute to the pathogenesis of muscle disease and could be used as a cellular target for new therapeutic approaches.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The musculoskeletal system, primarily made up of bones, muscles, and joints to include cartilage, tendons, and ligaments, is essential for skeletal stability, movement, and metabolism. Both bone and skeletal muscle are versatile organs that interact with each other in both a mechanical and a biochemical manner to maintain not only musculoskeletal homeostasis but also overall health [1]. For skeletal integrity and homeostasis to be maintained, it is essential that the three bone cell types, osteoblasts, osteoclasts, and osteocytes, interact in tandem and synergistically. Osteoblasts are bone-forming cells, prodigious producers of collagen, the scaffold for hydroxyapatite, and other proteins necessary for mineralization to occur [2], whereas osteoclasts are bone-resorbing cells which function to remove this mineralized matrix [3]. Osteocytes, the most abundant and longest living cells, can regulate both osteoblast and osteoclast activities and when necessary act as osteoclasts to remove bone and bone matrix surrounding osteocyte cell bodies and dendrites and can act as osteoblasts to replace their surrounding bone matrix [4].
Osteocytes, comprising 90 to 95% of all bone cells in adult bone, are derived from terminally differentiated osteoblasts that become entombed within the bone matrix, occupying small chambers called lacunae. These cells form connections with other neighboring cells through dendrite-like processes in fluid-filled canals known as canaliculi. This lacuno-canalicular system is necessary and extensively used in cell-cell communication. In addition to communicating with each other, osteocytes are a major participant in bone-muscle crosstalk, utilizing both the lacuno-canalicular system and the bone vasculature [1, 4, 5].
Osteocytes are the primary bone cells to respond to mechanical stimulation such as loading, in the form of fluid-flow shear stress, and to unloading or the lack of shear stress [4]. They can sense changes in the mechanical environment and in the presence of loading release prostaglandin E2, PGE2, promoting bone formation [6] and in the absence of loading regulate osteoblast and osteoclast activities accordingly via production of different factors including sclerostin (Sost), a negative regulator of bone formation and Receptor activator of nuclear factor kappa β ligand (RANKL or Tnfsf11), a factor necessary for osteoclast formation and activation. Osteocytes can also function as immune cells, producing TNFa, IL-6, Il-1, and other cytokines [7].
RANKL is a type II membrane protein primarily produced by osteocytes [8,9,10]. The receptor of RANKL, RANK, is found on bone monocytes/macrophage osteoclast precursors. The RANK-RANKL interaction influences these precursors to fuse to become activated osteoclasts ready to resorb and remove bone. Osteoprotegerin (OPG or Tnfrsf11b), another member of the TNF family, is a decoy receptor for RANKL. OPG can bind to RANKL, thus preventing RANKL-RANK interaction, consequently inhibiting osteoclastogenic activity and bone resorption. The amounts and ratios of RANKL, RANK, and OPG determine the degree of bone formation or resorption [9]. In addition, tumor necrosis factor-α (TNFα), interleukin-6 (IL-6), and M-CSF are additional major osteocyte-derived factors that can play a role in osteoclast differentiation [5]. One study showed that TNFα/IL-6 alone could generate osteoclasts in vitro independent of RANKL or RANK [11].
Sclerostin, a glycoprotein encoded by Sost, is mainly expressed by mature osteocytes [12]. It acts as an antagonist of the Wnt/β-catenin signaling pathway and inhibits osteoblastic bone formation [13]. Sclerostin production is influenced by mechanical stress and loading. Loading and anabolic stimuli such as parathyroid hormone (PTH) decrease sclerostin expression, which ultimately results in increased osteoblast-mediated bone formation [5]. Conversely, unloading induces an increase in both sclerostin and RANKL [5, 14]. In addition to playing a major role in bone, sclerostin may also affect muscle function. Conditioned media from MLO‐Y4 osteocyte-like cell line can stimulate myogenic differentiation of C2C12 myoblasts, which can be inhibited by adding sclerostin [15].
In addition to mechanosensing, another important function of osteocytes is their ability to remove and replace the bone matrix surrounding their lacunae under calcium demanding conditions. Osteocyte removal of their perilacunar matrix during pathologic conditions is termed osteocytic osteolysis. This phenomenon was proposed more than a century ago [16], but could not be proven by subsequent studies [17, 18], primarily due to insufficient tools and technical advances. Recent years have seen an increase of interest in this osteocyte function, and studies are being performed to examine this function under both physiological conditions such as lactation [19, 20] and pathological conditions including hyperparathyroidism and hypophosphatemic rickets [16, 21]. During lactation, a calcium demanding condition marked by significant bone loss, osteocytes express cathepsin K (CtsK), tartrate-resistant acid phosphatase (TRAP, gene Apc5), carbonic anhydrase 1 (Car 1), and other genes previously only associated with osteoclasts [19]. Osteocyte lacunar area increases with lactation [19]. With weaning, at the end of lactation, this bone loss is reversed, and bone mass returns to previous normal levels partially due to osteocyte lacunar size returning to normal through matrix replacement by the osteocyte, in a process called perilacunar remodeling [22]. Moreover, recent studies have determined a sex difference in the osteocyte transcriptome. The female osteocyte transcriptome diverges from the male transcriptome at 4 weeks of age by elevating genes necessary for osteocytic perilacunar remodeling, most likely playing an essential role in delivering calcium to offspring, thereby essential for reproduction [23]. Therefore, osteocytes contribute to calcium homeostasis regulation.
Osteocytes Communicating with Muscle Under Physiologic Conditions
Osteocyte-secreted factors not only work on the other bone cells but can travel via the lacuno-canalicular network and the vasculature to reach nearby and distant organs, including muscle. Osteocytes secrete various factors that play important roles in myogenesis as well as muscle contraction, thus playing a vital role in bone-muscle crosstalk.
Studies have shown that conditioned media from osteocytes (both primary osteocytes and osteocyte-like cell lines) can enhance differentiation of myoblasts, via the production of Wnt3a [15] and PGE2 [24]. Wnt3a, an activator of β-catenin, can regulate intracellular Ca2+ signaling and thus trigger myogenesis and increase muscle contractility (9). A recent study conducted by Palla et al. has shown that in aging skeletal muscle tissue and with sarcopenia, the level of prostaglandin E2 degrading enzyme, 15-PGDH, is increased, indicating a critical role of PGE2 in myogenesis [25]. Mice with global deletion of 15-PGDH retained significant muscle mass with aging. However, it has not yet been determined the source of the PGE2 that is affecting muscle mass, whether it is coming from osteocytes or is strictly an autocrine effect on muscle.
Osteocytes produce factors such as sclerostin or fibroblast growth factor 9, FGF9, that can inhibit or reduce myogenesis. Conditioned media from the MLO‐Y4 osteocyte-like cell line can stimulate myogenic differentiation of C2C12 myoblasts, which is blocked by the addition of sclerostin [15]. This indicates a negative role of sclerostin on muscle differentiation. A recent study has found correlation between serum sclerostin and age, body fat, muscle mass, training activity, and multiple muscle-derived factors [26]. Another study found a negative correlation between serum sclerostin levels and skeletal muscle mass regardless of bone mineral content (BMC) and total body fat mass [27]. Unlike studies using MLO-Y4 and IDG-SW3 osteocyte cells and primary osteocytes [15], one study found that the Ocy454 osteocyte cell line subjected to fluid flow shear stress, produced conditioned media that decreased C2C12 differentiation due to a reduction in expression of both myogenic regulatory genes and cytokines involved in myogenesis [28]. The reason for this discrepancy is not clear but could be based on magnitude of stress, culture conditions, and other variables.
Osteocytes and Sarcopenia
Sarcopenia is defined as a geriatric condition characterized by the progressive loss of skeletal muscle mass and function [29] that is frequently associated with the presence of osteoporosis. Changes in body composition that occur during aging, such as the visceral or intramuscular increase of fat mass can make the diagnosis of sarcopenia difficult, a condition called “sarcopenic obesity” [30]. Related to sarcopenic obesity, “osteopenic obesity” is characterized by increased adiposity that results in increased bone loss [31]. It is unclear if muscle and bone decline simultaneously during aging or if sarcopenia precedes osteoporosis or vice versa and if these two processes have common causes and mechanisms. However, several lines of evidence suggest that osteocyte aging and muscle decline during aging are closely interconnected. Aged bone is characterized by a reduction in the number of osteocytes, their dendrites and their lacunae and canaliculi due to hypermineralization as well as by an increase in osteocyte cell death, which together increases bone brittleness and weakness [32, 33]. Moreover, several osteocyte-derived factors such as sclerostin and RANKL are increased with aging, potentially precipitating musculoskeletal decline.
Circulating levels of sclerostin markedly increase with age in both female and male subjects [34]. However, as the relationship between sclerostin levels and bone mass are clear in young animals, when low levels are associated with more bone, the relationship of sclerostin with muscle and aging is not clear. In three recent studies, serum levels of sclerostin were shown to be associated with changes in skeletal muscle mass with aging. Choi and co-workers described for the first time that high sclerostin levels were associated with reduced skeletal muscle mass independent of several factors such as age, sex, body mass index (BMI), and BMC [27]. In both males and females, elevated sclerostin had negative effects on lower appendicular skeletal muscle mass (ASM) and was associated with increased total body fat [27]. Another study analyzing a cohort of hemodialysis patients with diabetes reported a negative association between sclerostin levels and sarcopenia [35] where lower muscle mass index was associated with higher serum sclerostin levels in both diabetic and non-diabetic subjects [35]. In contrast, Ahn and co-workers showed that ASM and skeletal muscle mass index (SKI) had a positive correlation with serum sclerostin levels in a population of older adult with sarcopenia [36] and along with the reduced muscle mass, reduced muscle weakness was also associated with lower sclerostin levels [36]. Sost null mice have increased bone mass at 8 and 16 months of age, but only the older mice showed a tendency (p = 0.06) for an increase in total lean body mass [37]. However, in the same study, the authors showed that systemic hyperexpression of sclerostin by means of adenovirus infection with Sost is associated with a reduction of the lean body mass fraction in young mice [37]. These studies highlight the controversial role of osteocyte-derived sclerostin in muscle homeostasis during aging.
The RANKL-RANK-OPG triad is important not only for bone but also for muscle. Whereas osteocytes produce RANKL, skeletal muscle and fully differentiated myotubes express RANK receptor. The presence of RANK receptor in skeletal muscle indicates that this pathway could play a functional role in this tissue [38]. RANKL is increased in menopausal women and plays a critical role in driving bone loss that characterizes osteoporosis [39]. A recent study compared bone and skeletal muscle parameters in postmenopausal women affected by osteoporosis and treated with a bisphosphonate, a bone-targeting drug or treated with a neutralizing antibody targeting RANKL, called Denosumab [40]. Both treatments were able to improve spine areal bone mineral density when compared with the untreated group. However, only the Denosumab-treated group showed increased appendicular lean mass and handgrip strength [40]. This set of data shows that the beneficial effect of an osteoporosis treatment targeting RANKL on skeletal muscle is due to the blocking the RANK-RANKL signaling pathway in skeletal muscle.
Osteocytes, Muscle Wasting, and Cancer
The progressive loss of skeletal muscle mass associated with cancer burden is defined as cancer cachexia (CC) [41]. Skeletal muscle atrophy is the most relevant feature of this syndrome that negatively affects physical function and mobility as well as the efficacy of anticancer treatment and dramatically decreases quality of life and shortens survival [42]. However, many other organs and tissues are also affected and involved in the pathogenesis of CC making CC a multifactorial and complex syndrome. Even though we know that muscle and bone share common precursors [43] and that they are closely linked by mechanical and biochemical interactions, their interactions with CC are far from being understood.
The first study that showed that bone could contribute to muscle alteration during CC was reported by Waning and collaborators. Using bone metastatic breast, lung, and prostate cancer models, the authors showed that the release of transforming growth factor β (TGF-β) from bone affected by the metastatic cancer contributed to driving skeletal muscle atrophy and weakness [44]. Mechanistically, TGF-β increases oxidative stress levels in skeletal muscle, resulting in an altered intracellular Ca2+ signaling that initiates muscle decline [44]. It has also been shown that the levels of tumor-derived Sclerostin are increased in a breast cancer model [45] and that treatment with an anti-sclerostin antibody not only reduced bone metastatic burden and preserved bone mass but also was able to improve skeletal muscle atrophy and weakness. Interestingly, the phosphorylation levels of NF-kB and p38, two important mediators of skeletal muscle decline associated with cancer, were reduced after anti-sclerostin antibody administration [45]. These data clearly indicate that osteocyte-derived sclerostin could also be directly responsible of musculoskeletal deterioration. In contrast, in another study, plasma levels of sclerostin were found to be negatively correlated with the decline of weight loss in cachectic patients affected by pancreatic ductal adenocarcinoma [46].
Several studies have described trabecular and cortical bone loss in preclinical models of colorectal [47,48,49], lung [50], and ovarian [51] cancers even in the absence of bone metastasis. These non-bone metastatic models suggest that both tumor and host-derived factors can drive CC and could also be responsible for bone loss. Little is known regarding the cellular and molecular mechanisms responsible for cachexia and bone loss in these cancer models. Recently our group described that the bone loss and muscle atrophy observed in three different models of CC, Colon-26 adenocarcinoma (C26), ES-2 ovarian cancer (ES-2), and Lewis lung carcinoma (LLC) are accompanied by dramatic changes in osteocyte viability and secreted factors [52]. An increase in osteocyte expression of tartrate resistant acid phosphatase 5 and Cathepsin K was observed along with increased lacunar size in femurs of the C26, ES-2 and LLC tumor-bearing mice clearly indicating that these tumors are inducing osteocytic osteolysis. In addition, TUNEL staining showed a dramatic increase in osteocyte cell death in all three models of CC. Using co-cultures of osteocytes with cancer cells, we were able to show that the osteocytic osteolysis and osteocyte cell death are directly induced by tumor-derived factors [52]. Our hypothesis is that tumor factors can induce the secretion of osteoclast activating RANKL by the dying osteocyte and can induce osteolytic osteolysis at the same time along with newly elevated secretion of inflammatory cytokines creating the perfect storm to cause both bone and muscle losses. The increased expression of TNF-α and RANKL by osteocytes in response to the cancer factors could directly lead to the skeletal muscle atrophy and weakness present in these models of CC [52].
The role of TNF-α in driving skeletal muscle deterioration during CC is well understood, but less is known about the role played by RANKL. Using the ES-2 CC model, we showed increased expression of Tnfsf11, the mRNA that codes for RANKL, in the bone of tumor-bearing mice compared to controls. The mechanism responsible for this upregulation could be the same as what we previously described [52], i.e., that tumor-derived factors stimulate the osteocytes to produce and release RANKL. Consistent with the bone mRNA data, the circulating levels of RANKL were increased in the ES-2 bearing mice. However, the high RANKL plasma levels were also due to RANKL production by the ES-2 ovarian cancer cells, but the ratio of bone to tumor RANKL is not known. In this study, we showed that RANKL is sufficient to induce skeletal muscle atrophy and weakness in vivo, and that treatment with a neutralizing antibody against RANKL improved muscle mass and strength in the ES-2 tumor-bearing mice. Transcriptomic analysis performed on myotube cultures exposed to recombinant RANKL clearly showed upregulation of inflammatory and pro-atrophic pathways [53].
Zoledronic acid, a bone-targeting bisphosphonate that prevents osteoclast activation and bone loss, was shown to reduce circulating RANKL levels and improve the cachectic phenotype in the ovarian cancer model [53]. The expression of Tnfsf11 in the bone of the ES-2 bearers was reduced by zoledronic acid administration suggesting that this bisphosphonate could target the osteocyte to normalize RANKL production, thus partially protecting against muscle loss.
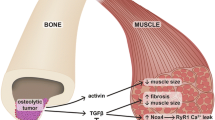
These data suggest that in response to cancer, the bone and specifically the osteocytes acquire a pathologic phenotype characterized by the increased production of RANKL and TNF-α that can act on skeletal muscle contributing to the detrimental effects of CC. In some of the tumor models, the tumors can produce factors normally made by osteocytes such as sclerostin, TNF-α, and RANKL. This makes it difficult to discriminate between the osteocyte and the tumor contribution of these factors and highlight the complexity of the bone-muscle interaction in a setting of non-metastatic bone cancer (Fig. 1).

Schematic representation of osteocyte-muscle interactions in a setting of cancer. A Cancer cells produce sclerostin, TNF-α, and RANKL that can directly induce skeletal muscle atrophy and weakness. B Other unknown tumor-derived factor(s) reprogram the osteocyte phenotype and stimulate the production of osteocyte-derived sclerostin, TNF-α, and RANKL that induce muscle alteration and osteoclast activation. C In metastatic bone disease, the release of TGF-β from bone due to osteoclast resorption directly affects muscle tissue inducing wasting and weakness. Image created with BioRender.com
Osteocytes and Other Muscle Disorders
Spinal cord injury (SCI) is a traumatic event of the central nervous system that can induce paralysis and severely affect quality of life. Patients affected by SCI have reduced mobility that leads to several detrimental musculoskeletal conditions such as skeletal muscle atrophy and bone loss [54, 55].
Invernizzi and collaborators showed significantly lower levels of BMD and higher levels of circulating sclerostin in SCI patients compared to healthy controls [56]. Also, the levels of myostatin, a negative regulator of skeletal muscle mass, were higher in chronic SCI patients and positively correlated with levels of sclerostin [56]. This suggests that osteocyte crosstalk with muscle is potentially involved in the pathogenesis of SCI. In contrast, a recent study has shown that individuals with SCI have significantly lower levels of circulating sclerostin than healthy controls [57]. This could be because these patients have lower bone mass and therefore fewer osteocytes to produce sclerostin. Several preclinical studies have shown that the administration of sclerostin-neutralizing antibody can improve bone loss after SCI. In particular, reduced bone loss and increased BMD as well as the maintenance of the osteocyte morphology were described [58, 59]. A recent study aimed to understand if sclerostin antibody treatment could protect against muscle wasting in SCI rats but could not show any effects on loss of soleus muscle mass [60]. This data suggests that there are also other mediators responsible for the muscle loss following SCI.
Duchenne muscular dystrophy (DMD) is an X-linked muscle disorder that occurs mainly in early childhood and leads to death in the late teens or early 20 s [61]. DMD patients develop progressive muscle weakness and reduced ambulatory movement that eventually leads to immobility and cardiovascular complications [61]. The inflammation that occurs in this condition is treated with corticosteroid, but when chronically administered, corticosteroids have dramatic negative effects on bone by reducing bone mass and increasing the risk of bone fracture [62].
A recent publication describes the musculoskeletal abnormalities associated with postnatal development of DMD using the dystrophin−/−/utrophin± murine model [63]. In this moue model, skeletal muscle changes appear before the first week of age and continue to persist during the course of the disease. Changes in bone do not appear until after muscle damage become more severe. The authors showed that after 4 weeks of age; the DMD mice have decreased numbers of osteocytes in association with reduced RANKL and increased sclerostin circulating levels [63]. These bone changes are then followed by osteopenia due to decreased osteoblastogenesis and increased osteoclastogenesis at 6 weeks of age [63].
After this publication, interest in the role of RANKL in the pathogenesis of skeletal muscle disease and in particular for the musculoskeletal defects associated with muscular dystrophy was increased. Dufresne and collaborators showed that skeletal muscle and fully differentiated C2C12 myotubes express RANK receptor and that muscle-specific RANK deletion in denervated fast-twitch EDL muscle decreased muscle mass, increased ratio of fast-twitch fibers, and modified sarco(endo)plasmic reticulum Ca (2+)-ATPase (SERCA) modulating Ca (2+) storage [38]. This shows an important role of RANK, and subsequently, RANKL, in muscle function. Therefore, full-length OPG-fc was used which appears to be more effective to improve the muscular dystrophy phenotype than the deletion of RANKL [64]. In this study, the authors using mdx dystrophic mice backcrossed with RANKmko (muscle specific KO mice) or mdx mice treated with OPG-fc to show that this treatment was able to prevent muscle eccentric contraction and increase SERCA activity in dystrophic muscle while the absence of RANK was ineffective [64]. These investigators also showed that the protein levels of RANK and RANKL were elevated in the muscle of utrophin haploinsufficient mdx (mdx/utrn±) mice [65]. Treatment with an anti-RANKL antibody was able to reduce muscle damage and fibrosis, improve function, and increase the mechanical property of the bone in this dystrophic muscle model. Mechanistically, the anti-RANKL treatment shifted the macrophage population toward an anti-inflammatory phenotype thus reducing the phosphorylation of NF-kB and the myofiber regeneration [65]. In a study enrolling 50 DMD patients and 50 controls from the age of 1 to 21 years of age, increased RANKL levels were observed only in the group of 6- to 10-year-old DMD patients compared to the controls [66]. This study showed reduced levels of both RANKL and OPG in the older DMD patients compared to controls [66]. This last data highlights the involvement of RANKL signaling in human DMD pathology.
Osteocytes and Muscle Atrophy due to Unloading
Prolonged periods of immobilization or unloading can severely affect the musculoskeletal system, by inducing bone loss and skeletal muscle atrophy. Unfortunately, the concept that osteocyte-derived factors can induce skeletal muscle atrophy during unloading is not well known.
Three different studies clearly show that prolonged bed rest causes an increase in circulating levels of sclerostin in healthy subjects [67,68,69]. However, none of those studies reports any information regarding muscle atrophy or weakness in these subjects. For these reasons, we can only speculate that is likely that the high levels of sclerostin observed in the subjects could be part of the pathogenetic mechanisms that induce skeletal muscle atrophy during immobilization. Another study using botulinum toxin to mimic hind limb disuse showed the inefficacy of the anti-sclerostin antibody romosozumab to protect skeletal muscle against atrophy [70]. Finally, a recent study aimed at investigating whether the exogenous injection of RANKL can exacerbate unloading-induced bone loss and muscle atrophy showed that only bone was negatively affected by the treatment [71]. We can only speculate that the differences with other studies demonstrating a negative effect of RANKL on skeletal muscle may be due to different levels of RANKL present into the circulation.
Conclusion
In summary, understanding both bone and muscle and the function of their individual cells in the context of whole organ physiology has led to novel findings with implications for treatment of musculoskeletal disorders and diseases. Investigators from different fields should be encouraged to learn from their colleagues and work together to develop novel hypotheses to test. This has certainly proved useful for investigators in the bone and in the muscle field. The future holds promise for additional interactions to take place.
References
Bonewald L. Use it or lose it to age: a review of bone and muscle communication. Bone. 2019;120:212–8. https://doi.org/10.1016/j.bone.2018.11.002.
Rosenberg N, Rosenberg O, Soudry M. Osteoblasts in bone physiology-mini review. Rambam Maimonides Med J. 2012;3(2):e0013. https://doi.org/10.5041/RMMJ.10080.
Feng X, Teitelbaum SL. Osteoclasts: new Insights. Bone Res. 2013;1(1):11–26. https://doi.org/10.4248/BR201301003.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26(2):229–38. https://doi.org/10.1002/jbmr.320.
Robling AG, Bonewald LF. The osteocyte: new insights. Annu Rev Physiol. 2020;82:485–506. https://doi.org/10.1146/annurev-physiol-021119-034332.
Siller-Jackson AJ, Burra S, Gu S, Xia X, Bonewald LF, Sprague E, et al. Adaptation of connexin 43-hemichannel prostaglandin release to mechanical loading. J Biol Chem. 2008;283(39):26374–82. https://doi.org/10.1074/jbc.M803136200.
Yoshimoto T, Kittaka M, Doan AAP, Urata R, Prideaux M, Rojas RE, et al. Osteocytes directly regulate osteolysis via MYD88 signaling in bacterial bone infection. Nat Commun. 2022;13(1):6648. https://doi.org/10.1038/s41467-022-34352-z.
Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the rankl required for osteoclast formation in remodeling bone. PLoS One. 2015;10(9):e0138189. https://doi.org/10.1371/journal.pone.0138189.
Ono T, Hayashi M, Sasaki F, Nakashima T. RANKL biology: bone metabolism, the immune system, and beyond. Inflamm Regen. 2020;40:2. https://doi.org/10.1186/s41232-019-0111-3.
Xiong J, O’Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. 2012;27(3):499–505. https://doi.org/10.1002/jbmr.1547.
O’Brien W, Fissel BM, Maeda Y, Yan J, Ge X, Gravallese EM, et al. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 2016;68(12):2889–900. https://doi.org/10.1002/art.39837.
Poole KE, van Bezooijen RL, Loveridge N, Hamersma H, Papapoulos SE, Lowik CW, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(13):1842–4. https://doi.org/10.1096/fj.05-4221fje.
Wang JS, Mazur CM, Wein MN. Sclerostin and osteocalcin: candidate bone-produced hormones. Front Endocrinol (Lausanne). 2021;12:584147. https://doi.org/10.3389/fendo.2021.584147.
Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283(9):5866–75. https://doi.org/10.1074/jbc.M705092200.
Huang J, Romero-Suarez S, Lara N, Mo C, Kaja S, Brotto L, et al. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/beta-catenin pathway. JBMR Plus. 2017;1(2):86–100. https://doi.org/10.1002/jbm4.10015.
Tsourdi E, Jahn K, Rauner M, Busse B, Bonewald LF. Physiological and pathological osteocytic osteolysis. J Musculoskelet Neuronal Interact. 2018;18(3):292–303.
Parfitt AM. The cellular basis of bone turnover and bone loss: a rebuttal of the osteocytic resorption–bone flow theory. Clin Orthop Relat Res. 1977;127:236–47.
van der Plas A, Aarden EM, Feijen JH, de Boer AH, Wiltink A, Alblas MJ, et al. Characteristics and properties of osteocytes in culture. J Bone Miner Res. 1994;9(11):1697–704. https://doi.org/10.1002/jbmr.5650091105.
Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, et al. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27(5):1018–29. https://doi.org/10.1002/jbmr.1567.
Jahn K, Kelkar S, Zhao H, Xie Y, Tiede-Lewis LM, Dusevich V, et al. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J Bone Miner Res. 2017;32(8):1761–72. https://doi.org/10.1002/jbmr.3167.
Yamazaki M, Michigami T. Osteocytes and the pathogenesis of hypophosphatemic rickets. Front Endocrinol (Lausanne). 2022;13:1005189. https://doi.org/10.3389/fendo.2022.1005189.
Qing H, Bonewald LF. Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int J Oral Sci. 2009;1(2):59–65. https://doi.org/10.4248/ijos.09019.
Youlten SE, Kemp JP, Logan JG, Ghirardello EJ, Sergio CM, Dack MRG, et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat Commun. 2021;12(1):2444. https://doi.org/10.1038/s41467-021-22517-1.
Mo C, Zhao R, Vallejo J, Igwe O, Bonewald L, Wetmore L, et al. Prostaglandin E2 promotes proliferation of skeletal muscle myoblasts via EP4 receptor activation. Cell Cycle. 2015;14(10):1507–16. https://doi.org/10.1080/15384101.2015.1026520.
Palla AR, Ravichandran M, Wang YX, Alexandrova L, Yang AV, Kraft P, et al. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. 2021;371(6528). https://doi.org/10.1126/science.abc8059.
Jurimae J, Karvelyte V, Remmel L, Tamm AL, Purge P, Gruodyte-Raciene R, et al. Serum sclerostin concentration is associated with specific adipose, muscle and bone tissue markers in lean adolescent females with increased physical activity. J Pediatr Endocrinol Metab. 2021;34(6):755–61. https://doi.org/10.1515/jpem-2020-0662.
Kim JA, Roh E, Hong SH, Lee YB, Kim NH, Yoo HJ, et al. Association of serum sclerostin levels with low skeletal muscle mass: The Korean Sarcopenic Obesity Study (KSOS). Bone. 2019;128:115053. https://doi.org/10.1016/j.bone.2019.115053.
Wood CL, Pajevic PD, Gooi JH. Osteocyte secreted factors inhibit skeletal muscle differentiation. Bone Rep. 2017;6:74–80. https://doi.org/10.1016/j.bonr.2017.02.007.
Traub J, Bergheim I, Eibisberger M, Stadlbauer V. Sarcopenia and liver cirrhosis-comparison of the European Working Group on Sarcopenia Criteria 2010 and 2019. Nutrients. 2020;12(2). https://doi.org/10.3390/nu12020547.
Batsis JA, Villareal DT. Sarcopenic obesity in older adults: aetiology, epidemiology and treatment strategies. Nat Rev Endocrinol. 2018;14(9):513–37. https://doi.org/10.1038/s41574-018-0062-9.
Keramidaki K, Tsagari A, Hiona M, Risvas G. Osteosarcopenic obesity, the coexistence of osteoporosis, sarcopenia and obesity and consequences in the quality of life in older adults >/=65 years-old in Greece. J Frailty Sarcopenia Falls. 2019;4(4):91–101. https://doi.org/10.22540/JFSF-04-091.
Busse B, Djonic D, Milovanovic P, Hahn M, Puschel K, Ritchie RO, et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. 2010;9(6):1065–75. https://doi.org/10.1111/j.1474-9726.2010.00633.x.
D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136(2):284–95. https://doi.org/10.1016/j.cell.2008.11.037.
Modder UI, Hoey KA, Amin S, McCready LK, Achenbach SJ, Riggs BL, et al. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J Bone Miner Res. 2011;26(2):373–9. https://doi.org/10.1002/jbmr.217.
Medeiros MC, Rocha N, Bandeira E, Dantas I, Chaves C, Oliveira M, et al. Serum sclerostin, body composition, and sarcopenia in hemodialysis patients with diabetes. Int J Nephrol. 2020;2020:4596920. https://doi.org/10.1155/2020/4596920.
Ahn SH, Jung HW, Lee E, Baek JY, Jang IY, Park SJ, et al. Decreased serum level of sclerostin in older adults with sarcopenia. Endocrinol Metab (Seoul). 2022. https://doi.org/10.3803/EnM.2022.1428.
Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci U S A. 2017;114(52):E11238–47. https://doi.org/10.1073/pnas.1707876115.
Dufresne SS, Dumont NA, Boulanger-Piette A, Fajardo VA, Gamu D, Kake-Guena SA, et al. Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am J Physiol Cell Physiol. 2016;310(8):C663–72. https://doi.org/10.1152/ajpcell.00285.2015.
Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111(8):1221–30. https://doi.org/10.1172/JCI17215.
Bonnet N, Bourgoin L, Biver E, Douni E, Ferrari S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest. 2019;129(8):3214–23. https://doi.org/10.1172/JCI125915.
Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12(5):489–95. https://doi.org/10.1016/S1470-2045(10)70218-7.
von Haehling S, Anker SD. Prevalence, incidence and clinical impact of cachexia: facts and numbers-update 2014. J Cachexia Sarcopenia Muscle. 2014;5(4):261–3. https://doi.org/10.1007/s13539-014-0164-8.
Pourquie O. Vertebrate somitogenesis. Annu Rev Cell Dev Biol. 2001;17:311–50. https://doi.org/10.1146/annurev.cellbio.17.1.311.
Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med. 2015;21(11):1262–71. https://doi.org/10.1038/nm.3961.
Hesse E, Schröder S, Brandt D, Pamperin J, Saito H, Taipaleenmäki H. Sclerostin inhibition alleviates breast cancer-induced bone metastases and muscle weakness. JCI Insight. 2019;5(9):e125543. https://doi.org/10.1172/jci.insight.125543.
Narasimhan A, Shahda S, Kays JK, Perkins SM, Cheng L, Schloss KNH, et al. Identification of potential serum protein biomarkers and pathways for pancreatic cancer cachexia using an aptamer-based discovery platform. Cancers (Basel). 2020;12(12):3787. https://doi.org/10.3390/cancers12123787.
Bonetto A, Kays JK, Parker VA, Matthews RR, Barreto R, Puppa MJ, et al. Differential bone loss in mouse models of colon cancer cachexia. Front Physiol. 2016;7:679. https://doi.org/10.3389/fphys.2016.00679.
Huot JR, Novinger LJ, Pin F, Narasimhan A, Zimmers TA, O’Connell TM, et al. Formation of colorectal liver metastases induces musculoskeletal and metabolic abnormalities consistent with exacerbated cachexia. JCI Insight. 2020;5(9):e136687. https://doi.org/10.1172/jci.insight.136687.
Huot JR, Pin F, Essex AL, Bonetto A. MC38 tumors induce musculoskeletal defects in colorectal cancer. Int J Mol Sci. 2021;22(3):1486. https://doi.org/10.3390/ijms22031486.
Berent TE, Dorschner JM, Craig TA, Drake MT, Westendorf JJ, Kumar R. Lung tumor cells inhibit bone mineralization and osteoblast activity. Biochem Biophys Res Commun. 2019;519(3):566–71. https://doi.org/10.1016/j.bbrc.2019.09.045.
Pin F, Barreto R, Kitase Y, Mitra S, Erne CE, Novinger LJ, et al. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: a new model for the study of cancer cachexia. J Cachexia Sarcopenia Muscle. 2018;9(4):685–700. https://doi.org/10.1002/jcsm.12311.
Pin F, Prideaux M, Huot JR, Essex AL, Plotkin LI, Bonetto A, et al. Non-bone metastatic cancers promote osteocyte-induced bone destruction. Cancer Lett. 2021;520:80–90. https://doi.org/10.1016/j.canlet.2021.06.030.
Pin F, Jones AJ, Huot JR, Narasimhan A, Zimmers TA, Bonewald LF, et al. RANKL blockade reduces cachexia and bone loss induced by non-metastatic ovarian cancer in mice. J Bone Miner Res. 2022;37(3):381–96. https://doi.org/10.1002/jbmr.4480.
Wang H, Xia Y, Li B, Li Y, Fu C. Reverse adverse immune microenvironments by biomaterials enhance the repair of spinal cord injury. Front Bioeng Biotechnol. 2022;10:812340. https://doi.org/10.3389/fbioe.2022.812340.
Giangregorio L, McCartney N. Bone loss and muscle atrophy in spinal cord injury: epidemiology, fracture prediction, and rehabilitation strategies. J Spinal Cord Med. 2006;29(5):489–500. https://doi.org/10.1080/10790268.2006.11753898.
Invernizzi M, Carda S, Rizzi M, Grana E, Squarzanti DF, Cisari C, et al. Evaluation of serum myostatin and sclerostin levels in chronic spinal cord injured patients. Spinal Cord. 2015;53(8):615–20. https://doi.org/10.1038/sc.2015.61.
Maimoun L, Ben Bouallegue F, Gelis A, Aouinti S, Mura T, Philibert P, et al. Periostin and sclerostin levels in individuals with spinal cord injury and their relationship with bone mass, bone turnover, fracture and osteoporosis status. Bone. 2019;127:612–9. https://doi.org/10.1016/j.bone.2019.07.019.
Qin W, Li X, Peng Y, Harlow LM, Ren Y, Wu Y, et al. Sclerostin antibody preserves the morphology and structure of osteocytes and blocks the severe skeletal deterioration after motor-complete spinal cord injury in rats. J Bone Miner Res. 2015;30(11):1994–2004. https://doi.org/10.1002/jbmr.2549.
Zhao W, Li X, Peng Y, Qin Y, Pan J, Li J, et al. Sclerostin antibody reverses the severe sublesional bone loss in rats after chronic spinal cord injury. Calcif Tissue Int. 2018;103(4):443–54. https://doi.org/10.1007/s00223-018-0439-8.
Phillips EG, Beggs LA, Ye F, Conover CF, Beck DT, Otzel DM, et al. Effects of pharmacologic sclerostin inhibition or testosterone administration on soleus muscle atrophy in rodents after spinal cord injury. PLoS ONE. 2018;13(3):e0194440. https://doi.org/10.1371/journal.pone.0194440.
Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51(8):759–64. https://doi.org/10.1111/jpc.12868.
Buckner JL, Bowden SA, Mahan JD. Optimizing bone health in duchenne muscular dystrophy. Int J Endocrinol. 2015;2015:928385. https://doi.org/10.1155/2015/928385.
Gao X, Tang Y, Amra S, Sun X, Cui Y, Cheng H, et al. Systemic investigation of bone and muscle abnormalities in dystrophin/utrophin double knockout mice during postnatal development and the mechanisms. Hum Mol Genet. 2019;28(10):1738–51. https://doi.org/10.1093/hmg/ddz012.
Dufresne SS, Boulanger-Piette A, Bosse S, Argaw A, Hamoudi D, Marcadet L, et al. Genetic deletion of muscle RANK or selective inhibition of RANKL is not as effective as full-length OPG-fc in mitigating muscular dystrophy. Acta Neuropathol Commun. 2018;6(1):31. https://doi.org/10.1186/s40478-018-0533-1.
Hamoudi D, Marcadet L, Piette Boulanger A, Yagita H, Bouredji Z, Argaw A, et al. An anti-RANKL treatment reduces muscle inflammation and dysfunction and strengthens bone in dystrophic mice. Hum Mol Genet. 2019;28(18):3101–12. https://doi.org/10.1093/hmg/ddz124.
Akhtar Ali S, Kang H, Olney R, Ramos-Platt L, Ryabets-Lienhard A, Cheung C, et al. Evaluating RANKL and OPG levels in patients with Duchenne muscular dystrophy. Osteoporos Int. 2019;30(11):2283–8. https://doi.org/10.1007/s00198-019-05077-5.
Belavy DL, Baecker N, Armbrecht G, Beller G, Buehlmeier J, Frings-Meuthen P, et al. Serum sclerostin and DKK1 in relation to exercise against bone loss in experimental bed rest. J Bone Miner Metab. 2016;34(3):354–65. https://doi.org/10.1007/s00774-015-0681-3.
Frings-Meuthen P, Boehme G, Liphardt AM, Baecker N, Heer M, Rittweger J. Sclerostin and DKK1 levels during 14 and 21 days of bed rest in healthy young men. J Musculoskelet Neuronal Interact. 2013;13(1):45–52.
Spatz JM, Fields EE, Yu EW, Divieti Pajevic P, Bouxsein ML, Sibonga JD, et al. Serum sclerostin increases in healthy adult men during bed rest. J Clin Endocrinol Metab. 2012;97(9):E1736–40. https://doi.org/10.1210/jc.2012-1579.
Brent MB, Bruel A, Thomsen JS. Anti-sclerostin antibodies and abaloparatide have additive effects when used as a countermeasure against disuse osteopenia in female rats. Bone. 2022;160:116417. https://doi.org/10.1016/j.bone.2022.116417.
Speacht TL, Lang CH, Donahue HJ. Soluble RANKL exaggerates hindlimb suspension-induced osteopenia but not muscle protein balance. J Orthop Res. 2021;39(9):1860–9. https://doi.org/10.1002/jor.24917.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shimonty, A., Bonewald, L.F. & Pin, F. Role of the Osteocyte in Musculoskeletal Disease. Curr Osteoporos Rep 21, 303–310 (2023). https://doi.org/10.1007/s11914-023-00788-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-023-00788-5