
Abstract
The potential for positron emission tomography (PET) to detect neuroinflammation in vivo has sparked a remarkable interest in various disciplines of neuroscience. Early PET radioligands, such as [11C]PK(R)-11195 for the 18-kDa translocator protein (TSPO) and [11C]L-deprenyl for monoamine oxidase B, have been used in studies designed to clarify the role of neuroinflammation in a variety of psychiatric and neurological disorders. Recent years have witnessed the development of several second-generation PET radioligands for TSPO and radioligands to measure endogenous targets that are active in various stages of the inflammatory cascade, such as cyclooxygenase and arachidonic acid. Here, we discuss some of the biomarkers for neuroinflammation that are available for quantification with PET, as well as recent findings from studies where neuroinflammation has been assessed in neurodegenerative disorders. In addition, we highlight the challenges to accurate interpretation of PET studies of neuroinflammation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation
Neuroinflammation—or more specifically, activation of the neuroimmune cells microglia and astrocytes into proinflammatory states—has been implicated as a pathological contributor in several neurodegenerative diseases. Many of these diseases are defined pathologically by abnormal accumulation of specific protein species (for instance, paired helical filamental tau-containing tangles in Alzheimer’s disease), hence the term “proteinopathy” to describe these disorders. In vitro studies and animal models have shown that many proteinopathies stimulate neuroimmune responses, and consequently, much work has been conducted to elucidate the role of neuroimmune activation in several disorders.
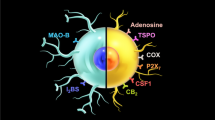
The brain has long been considered an “immunologically privileged” organ, as the peripheral immune cells are thought unable to penetrate the blood-brain barrier. Instead, the glial cells—microglia and astrocytes—are the primary constituents of a dedicated neuroimmune system, and its interaction with the peripheral immune system is poorly understood [1]. The glial cells provide pro- and anti-inflammatory functionality and participate in various functions under basal and disease conditions, including phagocytosis, steroid release, free radical reduction, and cellular repair. Proinflammatory functions, including release of cytokines and reactive oxygen species, may damage healthy neurons, causing synaptic dysfunction, loss of synapses, and neuronal death. Therefore, an imbalance between proinflammatory and reparatory functions of neuroimmune cells can result in CNS injury. While the damaging effects of such imbalance are recognized in classical neuroimmunological disease such as multiple sclerosis, growing evidence suggests that chronic low-level activation of glial cells may contribute to pathological changes found in many neurodegenerative diseases. The possibility to quantify the current inflammatory state in a living human brain has sparked a remarkable interest through various disciplines of neuroscience, as it provides means to measure disease severity, study pathophysiological mechanisms, and identify novel targets for treatment (Fig. 1).

Number of articles listed in PubMed that have listed as key words both “PET” and either “translocator protein (TSPO)” or “peripheral benzodiazepine receptor (PBR)”—the latter terms referring to a biomarker of inflammation
Positron Emission Tomography
Positron emission tomography (PET) is a molecular imaging modality capable of providing information of brain functionality. PET relies upon the employment of radioactively labeled pharmaceuticals (radioligands) that enter into the living organism. In PET studies of neuroinflammation, pharmaceuticals that bind specifically to 18-kDa translocator protein or monoaminoxidase B are often used, as the concentration of these proteins is believed to reflect ongoing neuroinflammation (discussed below). The PET system provides quantitative images reflecting the spatial and temporal distribution of the radioligand in vivo.
PET imaging comes with several caveats. First, the spatial and temporal resolution associated with emission tomography is limited; thus, small structures are typically not easily quantifiable [2]. Second, the PET system cannot distinguish between different sources of radioactivity. Enzymes in the liver and other tissues break down the pharmaceutical, leading to the generation of radioactive metabolites. If these metabolites enter the brain, they will contribute to the signal, potentially confounding accurate quantification. Another, and often complicated problem, is the presence of nonspecific binding sites. The PET image represents the sum of signals originating from different binding sites, and binding to the specific target of interest represents only a fraction of the signal [3, 4]. For instance, as radiopharmaceuticals must be lipophilic to passively cross the blood-brain barrier, they often bind nonspecifically to the lipid-rich myelin sheaths of white matter tracts. Since it is not always possible to estimate the fraction of specifically bound tracer, a high ratio of specific to nonspecific binding is favorable. Last, full quantification of PET data requires that the concentration of unchanged radioligand in arterial plasma (also known as the input function) is measured during the PET examination. Estimation of the input function necessitates arterial cannulation and specialized staff and instrumentation typically only available in dedicated PET research centers. Consequently, many PET studies rely on simplified acquisition and analysis procedures, often by calculating ratios between different brain regions.
For the subset of radioligands for which there exists a brain region with negligible specific binding to the target of interest (referred to as a reference region), these ratio calculations produce outcome measures that are well-correlated with those obtained from full quantification, omitting the need for arterial sampling [5, 6]. Because the targets currently used for studying neuroinflammation are expressed throughout the brain, no true reference region can be designated. Therefore, quantification using an arterial input function is considered the “gold standard” for PET studies of neuroinflammation.
Biomarkers for Neuroinflammation
The 18-kDa Translocator Protein
The 18-kDa translocator protein (TSPO) is a commonly targeted biomarker with PET [7]. TSPO is a transmembrane protein found mainly in the outer mitochondrial membrane. The protein was formerly called the peripheral benzodiazepine receptor (PBR) because it binds diazepam, and was first discovered as a high affinity receptor for Ro-4864 in kidney, liver, and lung [8]. The name PBR was chosen to distinguish it from the central benzodiazepine receptor. Results from subsequent studies showed that this protein binds to cholesterol and porphyrins and evidence supports a role in transporting substrates across membranes [9, 10]. Therefore, the name was changed to TSPO to avoid confusion with the central benzodiazepine receptor [11]. However, recent reports of viable mice genetically depleted of TSPO have cast doubts on its role in some of these functions [12,13,14,15,16]. TSPO is expressed in low levels in immune-competent cells, macrophages, and leukocytes in the periphery, as well as in microglia and astrocytes [17]. In response to cellular injury, the glial cells become activated and this morphological and functional change results in increased expression of TSPO [18].
Increased TSPO density has been observed in several neurological disorders. Not surprisingly, such increases are evident in classic neuroimmunological disorders such as multiple sclerosis and HIV encephalopathy [19]. Increased TSPO density has however also been demonstrated in brain tissue from patients with neurodegenerative diseases.
Monoamine Oxidase B
The monoamine oxidases (MAO) A and B are isoenzymes, functioning by oxidatively deaminating neurotransmitter and xenobiotic amines [20]. The subtype MAO-B hydrolyzes trace amine, phenylethylamine, and dopamine, and as a by-product, reactive oxygen species are excreted which in excessive concentrations have damaging effects [21]. MAO-B expression increases with age, which is thought to contribute to age-related neurodegeneration [22, 23].
There is evidence that astrocytes play an important role in regulating MAO-B activity under normal and pathological conditions [24,25,26,27]. Thus, it has been hypothesized that the concentration of MAO-B can reflect the current state of astrocytosis, which in turn serves as a biomarker for ongoing neuroinflammation.
Other Targets
Besides TSPO and MAO-B, a few other targets have been used in PET studies of neuroinflammation. Arachidonic acid (AA) is polyunsaturated omega-6 fatty acid, highly abundant in the phospholipid bilayer membranes in the brain, where it serves as a second messenger involved in the regulation of several signaling enzymes. The cascade by which regulatory compounds (prostaglandins) are formed from the breakdown of AA was awarded the Nobel Prize in 1982, and today, it is widely accepted that AA plays an important role in the inflammatory response. For the CNS specifically, binding of microglial derived cytokines to calcium channel coupled receptors on astrocytes results in activations of phospholipase enzymes that liberate AA from membrane lipoproteins. Thus, the mobilization of AA has been suggested to be a useful biomarker of neuroinflammation.
Cyclooxygenases (COX) are enzymes that catalyze the breakdown of AA into prostaglandins. The two isoforms (COX-1 and COX-2) are constitutively expressed in the mammalian brain but not co-localized. Under normal circumstances, COX-1 is predominantly found in microglia and some vascular tissue, whereas COX-2 is expressed postsynaptically, predominantly in neurons in the cortex, amygdala, and hippocampus [28]. COX is believed to be involved in the inflammatory cascade, and inhibition of the enzymes is often used for therapeutic anti-inflammatory treatment. Upregulation of COX has thus been suggested as a biomarker for neuroinflammation.
Radioligands for Neuroinflammation
Radioligands for TSPO
The prototypical PET radioligand for TSPO is [11C](R)-PK11195. [11C](R)-PK11195 has TSPO antagonist properties based on thermodynamic studies [29] and binds to TSPO within a tryptophan-rich pocket [30]. Autoradiography studies have shown an increase in [3H]PK11195 binding in Alzheimer’s disease (AD) patients, particularly in areas of reduced choline acetyltransferase activity [31, 32]. Radiolabeling with 11C has allowed in vivo detection of TSPO expression, and numerous clinical PET studies have been performed using this radioligand in neurodegenerative diseases.
While [11C](R)-PK11195 is the most highly represented TSPO radioligand in the literature, this radioligand has limitations. [11C](R)-PK11195 has high lipophilicity, which promotes nonspecific binding to lipids in the brain [33]. In addition, [11C](R)-PK11195 may bind to the acute phase reactant α1-acid glycoprotein [34]. Due to low amounts of TSPO under normal conditions, nonspecific binding can represent a significant contribution to the PET signal. A pharmacological blocking study using nonlabeled PK11195 showed that specific binding of [11C](R)-PK11195 in monkey brain is only 1.3 times the nonspecific binding [35], and thus, nonspecifically bound radioligand cannot be assumed to be negligible.
Several second-generation TSPO radioligands have been developed [36], and most have lower lipophilicity than [11C](R)-PK11195 and improved specific to nonspecific binding. However, a limitation shared by all tested second-generation radioligands is differential affinity to TSPO dependent on the polymorphism expressed. This was first discovered with [11C]PBR28, where a 40-fold difference in in vitro binding affinity was observed between “binders” and “nonbinders” [35]. Displacement assays later revealed a trimodal distribution of binding, classified as high, mixed, and low affinity binders (HABs, MABs, LABs) [37, 38]. Clear association between LAB and the rs6971 SNP led to the conclusion that this SNP is causing the differential binding [39]. Since the rs6971 SNP confers codominant expression, heterozygotes have reduced [11C]PBR28 binding, while homozygotes have negligible binding. Determination of binding affinity, through genotype analysis or in vitro binding assay, must therefore be performed. The differential affinity can be accounted for statistically, allowing inclusion of HABs and MABs in clinical studies [40]. However, requisite exclusion of LABs, which make up ∼9% of subjects of European and African American descent, is disadvantageous. To date, all tested second-generation radioligands are sensitive to this SNP [37].
In addition to the aforementioned radioligand [11C]PBR28, other second-generation radioligands include [18F]PBR06, [18F]PBR111, [18F]DPA-714, [18F]FEPPA, [11C]DAA1106, and [11C]ER176 (see [41] for a review).
Radioligands for MAO-B
Much work has been done to develop PET radioligands for MAOs [42]. To develop radioligands with affinity for only one isoform has proven challenging, and there currently exists only one reliable radioligand for MAO-B. L-deprenyl (selegiline) is a MAO-B inhibitor and its radiolabeled form [11C]L-deprenyl was first applied in clinical applications in humans in 1987 [43]. Irreversible [11C]L-deprenyl binding, however, induces problems when quantitating uptake [44], which motivated the development of a deuterium analogue, [11C]L-deprenyl-D2, with more favorable pharmacokinetics. Although other radioligands are being developed [45, 46], [11C]L-deprenyl-D2 is today the preferred radioligand for MAO-B [47].
Radioligands for Arachidonic Acid and COX
In contrast to radioligands for TSPO or MAO-B, in vivo PET studies targeting AA use radiolabeled AA itself as a marker of the compound turnover, rather than studying its binding sites [48, 49]. AA is involved in several brain functions [50], and thus, altered binding of [11C]AA is not easily interpretable.
The only applied PET radioligand for COX is [11C]ketoprofen and its methyl ester, which binds selectively to COX-1 [51]. Several preclinical studies in rats have demonstrated the utility of [11C]ketoprofen to quantify COX-1 levels [52, 53], but for humans, it has only been trialed in healthy volunteers [54] and in a small cohort of AD patients [55]. Unfortunately, this latter study revealed no differences between patients and controls, concluding that [11C]ketoprofen methyl ester is not a suitable diagnostic marker for AD.
With regard to COX-2, none of the evaluated tracers has been found to be useful for the study of neuroinflammation [56,57,58].
Neuroinflammation in Neurodegenerative Disorders
Alzheimer’s Disease
Several studies have implicated neuroimmune responses as a pathological contributor to AD pathophysiology [59,60,61,62,63]. In vitro and animal model studies have shown that β-amyloid and hyperphosphorylated tau aggregation induce proinflammatory conditions [64,65,66,67,68]. Activated microglia and reactive astrocytes are present in AD brain and have been shown to overexpress TSPO when proximal to β-amyloid plaques [19].
Most PET studies using [11C](R)-PK11195 have shown increased binding in patients with a clinical diagnosis of AD [69,70,71]. Studies have reported that cortical [11C](R)-PK11195 binding correlates with clinical severity [69,70,71]. While no association between [11C](R)-PK11195 binding and amyloid binding has been observed in cross-sectional studies [71, 72], a recent longitudinal study showed that an increase in [11C](R)-PK11195 binding correlated with an increase in amyloid burden over time [73•].
Some studies have reported no difference in [11C](R)-PK11195 binding between AD patients and controls [74,75,76]. In one such study, the “controls” included seven patients with unilateral gliomas, and the unaffected hemisphere was used as comparison data [74]. Another study showed no difference between controls and patients in the prodromal stage of AD—i.e., mild cognitive impairment (MCI)—regardless of whether the MCI patients progressed to dementia or remained clinically stable [76]. This study also found no correlation between [11C](R)-PK11195 binding and cognitive scores.
These conflicting results could be due to several factors. First, none of the above studies used absolute quantification of [11C](R)-PK11195 binding. Instead, clinical [11C](R)-PK11195 studies often identify a “pseudo-reference” region by extracting clusters of the subject’s PET voxels whose pharmacokinetics resembles that of nondisplaceable binding in normal gray matter. Although the technique appears useful in some cases, the reference region inevitably includes TSPO, and the resulting underestimation bias may reduce sensitivity to detect group differences. Second, the derivation of the reference cluster relies on predefined kinetic classes obtained from previous PET studies, sometimes even from other PET centers. Differences in instrumentation and injection protocol can influence the cluster pharmacokinetics, and it is therefore unclear how valid a representation of nonspecific binding is provided. Finally, the cost of PET often limits the number of subjects for imaging, and false negative results from underpowered studies are probable.
Some studies using second-generation TSPO radioligands have been conducted. First, [11C]PBR28 binding was found to be greater in amyloid-positive AD patients than amyloid-positive MCI patients or controls, particularly in temporo-parietal regions [77]. [11C]PBR28 binding correlated with volume loss and several cognitive indices, but not with amyloid load. These findings were later confirmed in a larger follow-up study that also indicated that the cerebellum could be useful as a pseudo-reference region [78•].
Autoradiography studies reported increased [3H]DAA-1106 binding in brain tissue from transgenic AD mice [79] and AD patients [80, 81], although no correlation with clinical severity was observed [81]. Further, increased [11C]DAA-1106 binding has been seen in the striatum and several cortical structures in MCI and AD patients [82]. Interestingly, only the MCI patient with the lowest level of [11C]DAA-1106 at baseline did not covert to dementia. While the [11C]DAA-1106 studies were performed without correction for TSPO genotype, the low prevalence of rs6971 SNP in Japanese suggests that these results may not have been confounded by physiological affinity differences.
In contrast to [11C]DAA-1106, [18F]FEDAA1106 did not detect differences between AD patients and controls [83]. These results could be due in part to (a) selection of relatively mildly affected patients and (b) an absence of correction for TSPO genotype. In contrast to the [11C]DAA-1106 study, subjects who had [18F]FEDAA PET were recruited from centers in Sweden and so more likely to carry the rs6971 SNP.
Three longitudinal studies using TSPO radioligands have been performed in AD to determine how neuroinflammation changes during disease progression. However, each study used a different radioligand. Studies using [11C](R)-PK11195 and [11C]PBR28 showed that TSPO binding increases with progression of AD [73•, 84]. Kreisl et al. [84] also found that patients who showed clinical progression at follow-up had greater increase in [11C]PBR28 binding than patients who remained clinically stable. A study using [18F]DPA-714 found that baseline TSPO binding was greater in both MCI and AD patients than in controls [85]. Somewhat contrary to studies using [11C]PBR28, the study of Hamelin et al. [85] found greater baseline binding of [18F]DPA-714 in the most mildly affected patients and in those who had slower progression of disease. Larger studies involving serial imaging with harmonized TSPO imaging methodology will likely be necessary to clarify the relationship between TSPO and progression of Alzheimer’s disease.
PET studies using [11C]L-deprenyl have repeatedly shown elevated binding in AD and MCI patients [86,88,89,89], indicating an early presence of astrocytosis in AD pathogenesis. [11C]L-deprenyl has also been used to assess the levels of MAO-B inhibitor binding by potential neuroprotective agents such as EVT301 [88] and sembragiline [90], in a bid to slow disease progression.
With the exception of the aforementioned study which concluded that [11C]ketoprofen methyl ester is not useful [55], no study has to our knowledge been conducted assessing COX in AD.
The only study where [11C]AA was used in humans [91] showed higher uptake in the eight AD patients than in nine age-matched controls. Although this indeed provides some support for upregulation of AA in AD, it is not evident how to interpret this finding based on the various roles of AA in the brain.
Synucleinopathies
Synucleinopathies are a collective group of neurodegenerative disorders that share a common proteinopathy. Abnormal accumulation of α-synuclein is associated with loss of synapses and neuronal death, with the location of the proteinopathy determining the clinical phenotype. In Parkinson’s disease (PD), α-synuclein aggregates target dopamine neurons in the midbrain, forming Lewy bodies [92]. In dementia with Lewy bodies (DLB), these aggregates are additionally found in neurons in the cerebral cortex where they are associated with cognitive impairment, hallucinations, and neuropsychiatric symptoms [93]. In multiple system atrophy (MSA), aggregates are found primarily in oligodendrocytes in varying proportions in the midbrain, brainstem, cerebellum, and basal ganglia [94, 95]. MSA patients therefore develop either a Parkinsonian or olivo-ponto-cerebellar disorder. In these disorders, α-synuclein aggregates may be found in additional structures such as olfactory bulb [96], ganglia of the autonomic nervous system [97], and gastrointestinal tract [98].
Increased [11C](R)-PK11195 binding was observed in PD patients without cognitive impairment in the pons, basal ganglia, and frontal and temporal cortices [99]. Of the 18 patients included, 8 were followed for 2 years and, at follow-up, had no change in [11C](R)-PK11195 binding despite their disability rating with the Unified Parkinson’s Disease Rating Scale worsening from 19 to 25.
In a study comparing [11C](R)-PK11195 binding in patients with PD and Parkinson’s disease dementia (PDD), both PD and PDD patients showed binding that was elevated in frontal, temporal, and occipital cortices, and inversely correlated with Mini Mental State Exam score among the PDD patients [100]. [18F]FDG imaging in the same cohort showed areas of hypometabolism that overlapped with [11C](R)-PK11195 binding.
When comparing patients with PD and DLB, [11C](R)-PK11195 binding was increased (compared to controls) in basal ganglia and substantia nigra for both patient groups [101]. DLB patients had additional increases in the cortex and cerebellum. All patients were within 1 year of symptom onset, suggesting that increases in TSPO density can be seen in the early stages of disease.
Increased [11C](R)-PK11195 binding has also been reported in MSA patients in the cortical, subcortical, and brainstem regions [102]. In a clinical trial, two out of three patients treated with minocycline for 24 weeks showed lower [11C](R)-PK11195 binding at follow-up, while elevated binding was observed in most placebo-treated patients [103].
[11C]PBR28 has been used in clinical drug trials to determine target engagement of novel anti-inflammatory therapeutics in patients with PD. In a phase 2 study [104•], 24 PD patients received either placebo or treatment with AZD4231, an irreversible myeloperoxidase inhibitor. In the treated patients, [11C]PBR28 binding was 13–16% lower at 4 and 8 weeks than at baseline, whereas it remained unchanged in patients given placebo, suggesting that myeloperoxidase inhibition reduces TSPO expression. Whether [11C]PBR28 binding is diffusely increased in the brain in PD is not yet known, although a study using [18F]FEPPA reported no difference [105].
To our knowledge, no PET studies using radioligands for MAO-B or COX-1 have been undertaken to study α-synucleinopathies.
Frontotemporal Lobar Degeneration and Related Tauopathies
The term frontotemporal lobar degeneration (FTLD) refers to a collection of diseases that cause synaptic dysfunction and neuronal loss in the frontal and temporal lobes and are pathologically distinct from AD and the α-synucleinopathies. Clinical designations include behavioral variant frontotemporal dementia, progressive nonfluent aphasia, and semantic dementia [106, 107]. Sporadic and familial forms of FTLD often, but not always, result from aggregation of abnormal tau filaments [108,110,111,112,113,113]. Primary progressive palsy and corticobasal gangiolonic degeneration, collectively labeled “Parkinson’s plus” disorders due to their shared nigrostriatal cell loss, are pathologically defined by tau aggregation and therefore often considered in the FTLD spectrum [114].
Increased [11C](R)-PK11195 binding was reported in five patients with a clinical diagnosis of FTLD [115], but the quantification was performed without either blood sampling or use of a valid reference region. In another study, four patients with CBD showed increased [11C](R)-PK11195 binding in the striatum, brainstem, and several cortical regions [116]. In a similarly powered study, four patients with progressive supranuclear palsy (PSP) showed increased [11C](R)-PK11195 in the striatum, thalamus, brainstem, cerebellum, and frontal lobe [117]. In these two studies, the primary visual cortex and occipital white matter were used as references for nonspecific binding, as the authors claimed that these regions are unaffected in CBD and PSP, albeit not free from TSPO. This methodology is prone to biased outcome measures. Nevertheless, the location of increased binding overlapped with distribution of tau pathology commonly seen in CBD [118]. To our knowledge, no PET study using second-generation radioligands for TSPO, or any radioligands for MAO-B or COX-1, has been conducted in patients with frontotemporal lobar degeneration or related taupathies.
Huntington’s Disease
Huntington’s disease (HD) is an autosomal dominantly inherited disorder, caused by a mutation in the IT15 gene. The mutation causes abnormal accumulation of the huntingtin protein, leading to gradual neuronal damage [119]. Activated microglia have been found in vitro to be upregulated in the striatal, hypothalamic, thalamic, and cortical brain regions in all grades of pathology [120]. All PET studies to date have been conducted using [11C](R)-PK11195, and the data acquired without arterial input functions. Instead, cluster-based approaches to identify a pseudo-reference tissue have been employed, resulting in semiquantitative outcomes.
Two different studies carried out by the same group showed increased levels of [11C](R)-PK11195 binding in both premanifest gene carriers and symptomatic patients [121, 122]. When the combined data from these studies were reanalyzed, the authors concluded that hypothalamic dysfunction may be related to the nonmotor-related symptoms of HD [123]. Using an independent cohort, the group later showed very little change in [11C](R)-PK11195 binding between presymptomatic gene carriers and symptomatic patients, suggesting that microglia activation is primarily an early contributor to the pathophysiology of HD and does not increase further during disease progression [124]. To our knowledge, no PET study in HD has been conducted using second-generation radioligands or arterial sampling to accurately quantify TSPO levels in vivo. Although the existing PET studies indeed corroborate the in vitro findings, the same group has conducted all studies.
Amytrophic Lateral Sclerosis
Amytrophic lateral sclerosis (ALS) is characterized by the gradual degeneration of motor neurons, commonly over up to 5 years. No effective treatment strategies exist; thus, the disease typically progresses until death occurs due to respiratory failure. Although the role of neuroinflammation in ALS is unknown, there is evidence for associated microglial activation [125].
The first published PET study in ALS showed greater [11C](R)-PK11195 binding in patients than controls in motor cortex, pons, dorsolateral prefrontal cortex, and thalamus [126]. Although quantification was conducted without blood samples, the binding correlated with upper motor neuron symptoms and supports a previous autoradiography study [127].
Second-generation TSPO radioligands have also been used in ALS. Increased [18F]DPA-714 binding was found in cortical regions of ten ALS patients, six of which had a bulbar presentation [21]. In another study, ten ALS patients showed increased [11C]PBR28 binding in their precentral gyrus [128]. When the cohort was stratified (limb onset vs. bulbar), the seven patients with limb-onset weakness were found to account for the increased binding. Also, [11C]PBR28 binding in the precentral gyrus correlated with upper motor neuron burden score and negatively correlated with functional status.
The [18F]DPA-714 study used a cluster-based approach to derive a reference region (similar to many [11C](R)-PK11195 studies). The [11C]PBR28 study normalized the uptake in precentral gyrus over a 60–90-min scan duration to the total brain uptake. Therefore, a global upregulation of activated microglia will reduce the effect size observed between the groups. Similarly, global downregulation of TSPO among ALS patients could result in erroneously elevated outcome measures among this group. Although reduction of TSPO can be deemed unlikely in ALS, the tendency to omit acquisition of arterial blood data obstructs clear interpretation of the results. To our knowledge, no studies targeting MAO-B, COX-1, or AA in ALS have been performed.
Conclusion
Evidence suggests that neuroimmune activation, defined as activation of microglia and astrocytes, occurs in a number of neurodegenerative diseases. Such responses do not necessarily reflect a primary role of these glial responses in pathogenesis or even a negative role as a downstream effect. However, increased densities of TSPO and MAO-B and, to some extent, turnover of AA appear to be valid markers of these responses, and measuring these biomarkers with PET is possible with several available radioligands. Continued use of PET to quantify neuroinflammation, particularly in longitudinal studies, promises to clarify the role of neuroimmune activation in the pathophysiology of several neurodegenerative diseases and the utility of improved anti-inflammatory treatments.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance
Kanegawa N, Collste K, Forsberg A, Schain M, Arakawa R, Jucaite A, et al. In vivo evidence of a functional association between immune cells in blood and brain in healthy human subjects. Brain Behav Immun. 2016;54:149–57. doi:10.1016/j.bbi.2016.01.019.
Moses WW. Fundamental limits of spatial resolution in PET. Nucl Instrum Methods Phys Res A. 2011;648(Supplement 1):S236–S40. doi:10.1016/j.nima.2010.11.092.
Slifstein M, Laruelle M. Models and methods for derivation of in vivo neuroreceptor parameters with PET and SPECT reversible radiotracers. Nucl Med Biol. 2001;28(5):595–608.
Varnas K, Varrone A, Farde L. Modeling of PET data in CNS drug discovery and development. J Pharmacokinet Pharmacodyn. 2013;40(3):267–79. doi:10.1007/s10928-013-9320-6.
Lammertsma AA, Hume SP. Simplified reference tissue model for PET receptor studies. Neuroimage. 1996;4(3 Pt 1):153–8. doi:10.1006/nimg.1996.0066.
Logan J, Fowler JS, Volkow ND, Wang GJ, Ding YS, Alexoff DL. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab. 1996;16(5):834–40. doi:10.1097/00004647-199609000-00008.
Chauveau F, Boutin H, Van Camp N, Dolle F, Tavitian B. Nuclear imaging of neuroinflammation: a comprehensive review of [11C]PK11195 challengers. Eur J Nucl Med Mol Imaging. 2008;35(12):2304–19. doi:10.1007/s00259-008-0908-9.
Braestrup C, Squires RF. Specific benzodiazepine receptors in rat brain characterized by high-affinity (3H)diazepam binding. Proc Natl Acad Sci U S A. 1977;74(9):3805–9.
Papadopoulos V, Aghazadeh Y, Fan J, Campioli E, Zirkin B, Midzak A. Translocator protein-mediated pharmacology of cholesterol transport and steroidogenesis. Mol Cell Endocrinol. 2015;408:90–8. doi:10.1016/j.mce.2015.03.014.
Papadopoulos V, Miller WL. Role of mitochondria in steroidogenesis. Best Pract Res Clin Endocrinol Metab. 2012;26(6):771–90. doi:10.1016/j.beem.2012.05.002.
Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, et al. Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27(8):402–9. doi:10.1016/j.tips.2006.06.005.
Banati RB, Middleton RJ, Chan R, Hatty CR, Kam WW, Quin C, et al. Positron emission tomography and functional characterization of a complete PBR/TSPO knockout. Nat Commun. 2014;5:5452. doi:10.1038/ncomms6452.
Morohaku K, Pelton SH, Daugherty DJ, Butler WR, Deng W, Selvaraj V. Translocator protein/peripheral benzodiazepine receptor is not required for steroid hormone biosynthesis. Endocrinology. 2014;155(1):89–97. doi:10.1210/en.2013-1556.
Selvaraj V, Stocco DM, Tu LN. Translocator protein (TSPO) and steroidogenesis: a reappraisal. Mol Endocrinol. 2015;29(4):490–501. doi:10.1210/me.2015-1033.
Tu LN, Morohaku K, Manna PR, Pelton SH, Butler WR, Stocco DM, et al. Peripheral benzodiazepine receptor/translocator protein global knock-out mice are viable with no effects on steroid hormone biosynthesis. J Biol Chem. 2014;289(40):27444–54. doi:10.1074/jbc.M114.578286.
Tu LN, Zhao AH, Stocco DM, Selvaraj V. PK11195 effect on steroidogenesis is not mediated through the translocator protein (TSPO). Endocrinology. 2015;156(3):1033–9. doi:10.1210/en.2014-1707.
Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int. 2002;40(6):475–86.
Kuhlmann AC, Guilarte TR. Cellular and subcellular localization of peripheral benzodiazepine receptors after trimethyltin neurotoxicity. J Neurochem. 2000;74(4):1694–704.
Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35(3):306–28. doi:10.1111/j.1365-2990.2008.01006.x.
Saura J, Luque JM, Cesura AM, Da Prada M, Chan-Palay V, Huber G, et al. Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience. 1994;62(1):15–30.
Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7(12):e52941. doi:10.1371/journal.pone.0052941.
Saura J, Richards JG, Mahy N. Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Neurobiol Aging. 1994;15(4):399–408.
Saura J, Richards JG, Mahy N. Age-related changes on MAO in Bl/C57 mouse tissues: a quantitative radioautographic study. J Neural Transm Suppl. 1994;41:89–94.
Aquilonius SM, Jossan SS, Ekblom JG, Askmark H, Gillberg PG. Increased binding of 3H-L-deprenyl in spinal cords from patients with amyotrophic lateral sclerosis as demonstrated by autoradiography. J Neural Transm Gen Sect. 1992;89(1–2):111–22.
Jossan SS, Gillberg PG, Gottfries CG, Karlsson I, Oreland L. Monoamine oxidase B in brains from patients with Alzheimer’s disease: a biochemical and autoradiographical study. Neuroscience. 1991;45(1):1–12.
Nakamura S, Kawamata T, Akiguchi I, Kameyama M, Nakamura N, Kimura H. Expression of monoamine oxidase B activity in astrocytes of senile plaques. Acta Neuropathol. 1990;80(4):419–25.
Oreland L. Monoamine oxidase activity and affective illness. Acta Psychiatr Scand Suppl. 1980;280:41–7.
Aid S, Bosetti F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: therapeutic implications. Biochimie. 2011;93(1):46–51. doi:10.1016/j.biochi.2010.09.009.
Le Fur G, Vaucher N, Perrier ML, Flamier A, Benavides J, Renault C, et al. Differentiation between two ligands for peripheral benzodiazepine binding sites, [3H]RO5-4864 and [3H]PK 11195, by thermodynamic studies. Life Sci. 1983;33(5):449–57.
Guo Y, Kalathur RC, Liu Q, Kloss B, Bruni R, Ginter C, et al. Protein structure. Structure and activity of tryptophan-rich TSPO proteins. Science. 2015;347(6221):551–5. doi:10.1126/science.aaa1534.
Diorio D, Welner SA, Butterworth RF, Meaney MJ, Suranyi-Cadotte BE. Peripheral benzodiazepine binding sites in Alzheimer’s disease frontal and temporal cortex. Neurobiol Aging. 1991;12(3):255–8.
Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, et al. PK11195 labels activated microglia in Alzheimer’s disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009;30(8):1217–26. doi:10.1016/j.neurobiolaging.2007.11.005.
Hatty CR, Le Brun AP, Lake V, Clifton LA, Liu GJ, James M, et al. Investigating the interactions of the 18 kDa translocator protein and its ligand PK11195 in planar lipid bilayers. Biochim Biophys Acta. 2014;1838(3):1019–30. doi:10.1016/j.bbamem.2013.12.013.
Lockhart A, Davis B, Matthews JC, Rahmoune H, Hong G, Gee A, et al. The peripheral benzodiazepine receptor ligand PK11195 binds with high affinity to the acute phase reactant alpha1-acid glycoprotein: implications for the use of the ligand as a CNS inflammatory marker. Nucl Med Biol. 2003;30(2):199–206.
Kreisl WC, Fujita M, Fujimura Y, Kimura N, Jenko KJ, Kannan P, et al. Comparison of [(11)C]-(R)-PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage. 2010;49(4):2924–32. doi:10.1016/j.neuroimage.2009.11.056.
Albrecht DS, Granziera C, Hooker JM, Loggia ML. In vivo imaging of human neuroinflammation. ACS Chem Neurosci. 2016;7(4):470–83. doi:10.1021/acschemneuro.6b00056.
Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011;52(1):24–32. doi:10.2967/jnumed.110.079459.
Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010;30(9):1608–18. doi:10.1038/jcbfm.2010.63.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32(1):1–5. doi:10.1038/jcbfm.2011.147.
Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL, et al. A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013;33(1):53–8. doi:10.1038/jcbfm.2012.131.
Herrera Rivero M, Heneka MT, Papadopoulos V. Translocator protein and new targets for neuroinflammation. Clin Transl Imaging. 2015;3(6):391–402. doi:10.1007/s40336-015-0151-x.
Fowler JS, Logan J, Shumay E, Alia-Klein N, Wang GJ, Volkow ND. Monoamine oxidase: radiotracer chemistry and human studies. J Labelled Comp Radiopharm. 2015;58(3):51–64. doi:10.1002/jlcr.3247.
Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, et al. Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987;235(4787):481–5.
Fowler JS, Logan J, Wang GJ, Volkow ND, Telang F, Ding YS, et al. Comparison of the binding of the irreversible monoamine oxidase tracers, [(11)C]clorgyline and [(11)C]l-deprenyl in brain and peripheral organs in humans. Nucl Med Biol. 2004;31(3):313–9. doi:10.1016/j.nucmedbio.2003.10.003.
Nag S, Lehmann L, Kettschau G, Toth M, Heinrich T, Thiele A, et al. Development of a novel fluorine-18 labeled deuterated fluororasagiline ([(18)F]fluororasagiline-D2) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg Med Chem. 2013;21(21):6634–41. doi:10.1016/j.bmc.2013.08.019.
Nag S, Kettschau G, Heinrich T, Varrone A, Lehmann L, Gulyas B, et al. Synthesis and biological evaluation of novel propargyl amines as potential fluorine-18 labeled radioligands for detection of MAO-B activity. Bioorg Med Chem. 2013;21(1):186–95. doi:10.1016/j.bmc.2012.10.050.
Fowler JS, Wang GJ, Logan J, Xie S, Volkow ND, MacGregor RR, et al. Selective reduction of radiotracer trapping by deuterium substitution: comparison of carbon-11-L-deprenyl and carbon-11-deprenyl-D2 for MAO B mapping. J Nucl Med. 1995;36(7):1255–62.
Chang MC, Arai T, Freed LM, Wakabayashi S, Channing MA, Dunn BB, et al. Brain incorporation of [1-11C]arachidonate in normocapnic and hypercapnic monkeys, measured with positron emission tomography. Brain Res. 1997;755(1):74–83.
Giovacchini G, Chang MC, Channing MA, Toczek M, Mason A, Bokde AL, et al. Brain incorporation of [11C]arachidonic acid in young healthy humans measured with positron emission tomography. J Cereb Blood Flow Metab. 2002;22(12):1453–62. doi:10.1097/00004647-200212000-00006.
Esposito G, Giovacchini G, Der M, Liow JS, Bhattacharjee AK, Ma K, et al. Imaging signal transduction via arachidonic acid in the human brain during visual stimulation, by means of positron emission tomography. Neuroimage. 2007;34(4):1342–51. doi:10.1016/j.neuroimage.2006.11.018.
Takashima-Hirano M, Shukuri M, Takashima T, Goto M, Wada Y, Watanabe Y, et al. General method for the (11)C-labeling of 2-arylpropionic acids and their esters: construction of a PET tracer library for a study of biological events involved in COXs expression. Chemistry. 2010;16(14):4250–8. doi:10.1002/chem.200903044.
Shukuri M, Takashima-Hirano M, Tokuda K, Takashima T, Matsumura K, Inoue O, et al. In vivo expression of cyclooxygenase-1 in activated microglia and macrophages during neuroinflammation visualized by PET with 11C-ketoprofen methyl ester. J Nucl Med. 2011;52(7):1094–101. doi:10.2967/jnumed.110.084046.
Shukuri M, Mawatari A, Ohno M, Suzuki M, Doi H, Watanabe Y, et al. Detection of cyclooxygenase-1 in activated microglia during amyloid plaque progression: PET studies in Alzheimer’s disease model mice. J Nucl Med. 2016;57(2):291–6. doi:10.2967/jnumed.115.166116.
Ohnishi A, Senda M, Yamane T, Sasaki M, Mikami T, Nishio T, et al. Human whole-body biodistribution and dosimetry of a new PET tracer, [(11)C]ketoprofen methyl ester, for imagings of neuroinflammation. Nucl Med Biol. 2014;41(7):594–9. doi:10.1016/j.nucmedbio.2014.04.008.
Ohnishi A, Senda M, Yamane T, Mikami T, Nishida H, Nishio T, et al. Exploratory human PET study of the effectiveness of (11)C-ketoprofen methyl ester, a potential biomarker of neuroinflammatory processes in Alzheimer’s disease. Nucl Med Biol. 2016;43(7):438–44. doi:10.1016/j.nucmedbio.2016.04.005.
Ji B, Kumata K, Onoe H, Kaneko H, Zhang MR, Seki C, et al. Assessment of radioligands for PET imaging of cyclooxygenase-2 in an ischemic neuronal injury model. Brain Res. 2013;1533:152–62. doi:10.1016/j.brainres.2013.08.026.
Pacelli A, Greenman J, Cawthorne C, Smith G. Imaging COX-2 expression in cancer using PET/SPECT radioligands: current status and future directions. J Labelled Comp Radiopharm. 2014;57(4):317–22. doi:10.1002/jlcr.3160.
Tietz O, Wuest M, Marshall A, Glubrecht D, Hamann I, Wang M, et al. PET imaging of cyclooxygenase-2 (COX-2) in a pre-clinical colorectal cancer model. EJNMMI Res. 2016;6(1):37. doi:10.1186/s13550-016-0192-9.
Baik SH, Kang S, Son SM, Mook-Jung I. Microglia contributes to plaque growth by cell death due to uptake of amyloid beta in the brain of Alzheimer’s disease mouse model. Glia. 2016;64(12):2274–90. doi:10.1002/glia.23074.
Jin SC, Carrasquillo MM, Benitez BA, Skorupa T, Carrell D, Patel D, et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol Neurodegener. 2015;10:19. doi:10.1186/s13024-015-0016-9.
Lee M, McGeer E, McGeer PL. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: implications for Alzheimer’s disease pathogenesis. Neurobiol Aging. 2015;36(1):42–52. doi:10.1016/j.neurobiolaging.2014.07.024.
McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126(4):479–97. doi:10.1007/s00401-013-1177-7.
Raha AA, Henderson JW, Stott SR, Vuono R, Foscarin S, Friedland RP, et al. Neuroprotective effect of TREM-2 in aging and Alzheimer’s disease model. J Alzheimers Dis. 2016. doi:10.3233/JAD-160663.
Asai H, Ikezu S, Woodbury ME, Yonemoto GM, Cui L, Ikezu T. Accelerated neurodegeneration and neuroinflammation in transgenic mice expressing P301L tau mutant and tau-tubulin kinase 1. Am J Pathol. 2014;184(3):808–18. doi:10.1016/j.ajpath.2013.11.026.
Maezawa I, Zimin PI, Wulff H, Jin LW. Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. 2011;286(5):3693–706. doi:10.1074/jbc.M110.135244.
Marlatt MW, Bauer J, Aronica E, van Haastert ES, Hoozemans JJ, Joels M, et al. Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia, and occurs at sites of amyloid deposition. Neural Plast. 2014;2014:693851. doi:10.1155/2014/693851.
Morales I, Jimenez JM, Mancilla M, Maccioni RB. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013;37(4):849–56. doi:10.3233/JAD-131843.
Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett. 2001;305(3):165–8.
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–7. doi:10.1016/S0140-6736(01)05625-2.
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32(3):412–9. doi:10.1016/j.nbd.2008.08.001.
Yokokura M, Mori N, Yagi S, Yoshikawa E, Kikuchi M, Yoshihara Y, et al. In vivo changes in microglial activation and amyloid deposits in brain regions with hypometabolism in Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2011;38(2):343–51. doi:10.1007/s00259-010-1612-0.
Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72(1):56–62. doi:10.1212/01.wnl.0000338622.27876.0d.
• Fan Z, Okello AA, Brooks DJ, Edison P. Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain. 2015;138(Pt 12):3685–98. doi:10.1093/brain/awv288. The first published longitudinal study to show that TSPO binding on PET increases over time in patients with Alzheimer’s disease.
Groom GN, Junck L, Foster NL, Frey KA, Kuhl DE. PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer’s disease. J Nucl Med. 1995;36(12):2207–10.
Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, et al. Carbon 11-labeled Pittsburgh compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009;66(1):60–7. doi:10.1001/archneurol.2008.511.
Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF, et al. Microglial activation in Alzheimer’s disease: an (R)-[(11)C]PK11195 positron emission tomography study. Neurobiol Aging. 2013;34(1):128–36. doi:10.1016/j.neurobiolaging.2012.04.021.
Kreisl WC, Lyoo CH, McGwier M, Snow J, Jenko KJ, Kimura N, et al. In vivo radioligand binding to translocator protein correlates with severity of Alzheimer’s disease. Brain. 2013;136(Pt 7):2228–38. doi:10.1093/brain/awt145.
• Lyoo CH, Ikawa M, Liow JS, Zoghbi SS, Morse C, Pike VW, et al. Cerebellum can serve as a pseudo-reference region in Alzheimer’s disease to detect neuroinflammation measured with PET radioligand binding to translocator protein (TSPO). J Nucl Med. 2015. doi:10.2967/jnumed.114.146027. This paper validated a reference region method in the second generation radioligand [ 11 C]PBR28 to avoid use of arterial catheterization in PET studies of patients with Alzheimer’s disease.
Maeda J, Zhang MR, Okauchi T, Ji B, Ono M, Hattori S, et al. In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. Australas J Neurosci. 2011;31(12):4720–30. doi:10.1523/JNEUROSCI.3076-10.2011.
Gulyas B, Makkai B, Kasa P, Gulya K, Bakota L, Varszegi S, et al. A comparative autoradiography study in post mortem whole hemisphere human brain slices taken from Alzheimer patients and age-matched controls using two radiolabelled DAA1106 analogues with high affinity to the peripheral benzodiazepine receptor (PBR) system. Neurochem Int. 2009;54(1):28–36. doi:10.1016/j.neuint.2008.10.001.
Yasuno F, Ota M, Kosaka J, Ito H, Higuchi M, Doronbekov TK, et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer’s disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiatry. 2008;64(10):835–41. doi:10.1016/j.biopsych.2008.04.021.
Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y, et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [(11)C]DAA1106. Psychiatry Res. 2012;203(1):67–74. doi:10.1016/j.pscychresns.2011.08.013.
Varrone A, Mattsson P, Forsberg A, Takano A, Nag S, Gulyas B, et al. In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer’s disease patients. Eur J Nucl Med Mol Imaging. 2013;40(6):921–31. doi:10.1007/s00259-013-2359-1.
Kreisl WC, Lyoo CH, Liow JS, Wei M, Snow J, Page E, et al. (11)C-PBR28 binding to translocator protein increases with progression of Alzheimer’s disease. Neurobiol Aging. 2016;44:53–61. doi:10.1016/j.neurobiolaging.2016.04.011.
Hamelin L, Lagarde J, Dorothee G, Leroy C, Labit M, Comley RA, et al. Early and protective microglial activation in Alzheimer’s disease: a prospective study using 18F-DPA-714 PET imaging. Brain. 2016;139(Pt 4):1252–64. doi:10.1093/brain/aww017.
Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, et al. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53(1):37–46. doi:10.2967/jnumed.110.087031.
Santillo AF, Gambini JP, Lannfelt L, Langstrom B, Ulla-Marja L, Kilander L, et al. In vivo imaging of astrocytosis in Alzheimer’s disease: an (11)C-L-deuteriodeprenyl and PIB PET study. Eur J Nucl Med Mol Imaging. 2011;38(12):2202–8. doi:10.1007/s00259-011-1895-9.
Hirvonen J, Kailajarvi M, Haltia T, Koskimies S, Nagren K, Virsu P, et al. Assessment of MAO-B occupancy in the brain with PET and [11C]-L-deprenyl-D2: a dose-finding study with a novel MAO-B inhibitor, EVT 301. Clin Pharmacol Ther. 2009;85(5):506–12. doi:10.1038/clpt.2008.241.
Choo IL, Carter SF, Scholl ML, Nordberg A. Astrocytosis measured by (11)C-deprenyl PET correlates with decrease in gray matter density in the parahippocampus of prodromal Alzheimer’s patients. Eur J Nucl Med Mol Imaging. 2014;41(11):2120–6. doi:10.1007/s00259-014-2859-7.
Sturm S, Forsberg A, Nave S, Stenkrona P, Seneca N, Varrone A, et al. Positron emission tomography measurement of brain MAO-B inhibition in patients with Alzheimer’s disease and elderly controls after oral administration of sembragiline. Eur J Nucl Med Mol Imaging. 2016. doi:10.1007/s00259-016-3510-6.
Esposito G, Giovacchini G, Liow JS, Bhattacharjee AK, Greenstein D, Schapiro M, et al. Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med. 2008;49(9):1414–21. doi:10.2967/jnumed.107.049619.
Ohama E, Ikuta F. Parkinson’s disease: distribution of Lewy bodies and monoamine neuron system. Acta Neuropathol. 1976;34(4):311–9.
McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3 Suppl):417–23.
Brettschneider J, Irwin DJ, Boluda S, Byrne MD, Fang L, Lee EB, et al. Progression of alpha-synuclein pathology in multiple system atrophy of the cerebellar type. Neuropathol Appl Neurobiol. 2016. doi:10.1111/nan.12362.
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 1998;251(3):205–8.
Taguchi K, Watanabe Y, Tsujimura A, Tanaka M. Brain region-dependent differential expression of alpha-synuclein. J Comp Neurol. 2016;524(6):1236–58. doi:10.1002/cne.23901.
Sone M, Yoshida M, Hashizume Y, Hishikawa N, Sobue G. alpha-Synuclein-immunoreactive structure formation is enhanced in sympathetic ganglia of patients with multiple system atrophy. Acta Neuropathol. 2005;110(1):19–26. doi:10.1007/s00401-005-1013-9.
Lebouvier T, Neunlist M, Bruley des Varannes S, Coron E, Drouard A, N’Guyen JM, et al. Colonic biopsies to assess the neuropathology of Parkinson’s disease and its relationship with symptoms. PLoS One. 2010;5(9):e12728. doi:10.1371/journal.pone.0012728.
Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis. 2006;21(2):404–12. doi:10.1016/j.nbd.2005.08.002.
Edison P, Ahmed I, Fan Z, Hinz R, Gelosa G, Ray Chaudhuri K, et al. Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology. 2013;38(6):938–49. doi:10.1038/npp.2012.255.
Iannaccone S, Cerami C, Alessio M, Garibotto V, Panzacchi A, Olivieri S, et al. In vivo microglia activation in very early dementia with Lewy bodies, comparison with Parkinson’s disease. Parkinsonism Relat Disord. 2013;19(1):47–52. doi:10.1016/j.parkreldis.2012.07.002.
Gerhard A, Banati RB, Goerres GB, Cagnin A, Myers R, Gunn RN, et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61(5):686–9.
Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord. 2010;25(1):97–107. doi:10.1002/mds.22732.
• Jucaite A, Svenningsson P, Rinne JO, Cselenyi Z, Varnas K, Johnstrom P, et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain. 2015;138(Pt 9):2687–700. doi:10.1093/brain/awv184. The authors demonstrated that TSPO binding can be reduced in patients with Parkinson’s disease using a novel myeloperoxidase inhibitor.
Koshimori Y, Ko JH, Mizrahi R, Rusjan P, Mabrouk R, Jacobs MF, et al. Imaging striatal microglial activation in patients with Parkinson’s disease. PLoS One. 2015;10(9):e0138721. doi:10.1371/journal.pone.0138721.
Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups. J Neurol Neurosurg Psychiatry. 1994;57(4):416–8.
Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JB, et al. Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 2014;13(7):686–99. doi:10.1016/S1474-4422(14)70065-1.
Ferrer I, Lopez-Gonzalez I, Carmona M, Arregui L, Dalfo E, Torrejon-Escribano B, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol. 2014;73(1):81–97. doi:10.1097/NEN.0000000000000030.
Laws SM, Friedrich P, Diehl-Schmid J, Muller J, Ibach B, Bauml J, et al. Genetic analysis of MAPT haplotype diversity in frontotemporal dementia. Neurobiol Aging. 2008;29(8):1276–8. doi:10.1016/j.neurobiolaging.2007.02.019.
Lindquist SG, Schwartz M, Batbayli M, Waldemar G, Nielsen JE. Genetic testing in familial AD and FTD: mutation and phenotype spectrum in a Danish cohort. Clin Genet. 2009;76(2):205–9. doi:10.1111/j.1399-0004.2009.01191.x.
Caroppo P, Camuzat A, Guillot-Noel L, Thomas-Anterion C, Couratier P, Wong TH, et al. Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol Genet. 2016;2(3):e80. doi:10.1212/NXG.0000000000000080.
He F, Jones JM, Figueroa-Romero C, Zhang D, Feldman EL, Goutman SA, et al. Screening for novel hexanucleotide repeat expansions at ALS- and FTD-associated loci. Neurol Genet. 2016;2(3):e71. doi:10.1212/NXG.0000000000000071.
Lee EB, Russ J, Jung H, Elman LB, Chahine LM, Kremens D, et al. Topography of FUS pathology distinguishes late-onset BIBD from aFTLD-U. Acta Neuropathol Commun. 2013;1(9):1–11. doi:10.1186/2051-5960-1-9.
Ioannidis P, Konstantinopoulou E, Maiovis P, Karacostas D. The frontotemporal dementias in a tertiary referral center: classification and demographic characteristics in a series of 232 cases. J Neurol Sci. 2012;318(1–2):171–3. doi:10.1016/j.jns.2012.04.002.
Cagnin A, Rossor M, Sampson EL, Mackinnon T, Banati RB. In vivo detection of microglial activation in frontotemporal dementia. Ann Neurol. 2004;56(6):894–7. doi:10.1002/ana.20332.
Gerhard A, Watts J, Trender-Gerhard I, Turkheimer F, Banati RB, Bhatia K, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in corticobasal degeneration. Mov Disord. 2004;19(10):1221–6. doi:10.1002/mds.20162.
Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006;21(1):89–93. doi:10.1002/mds.20668.
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002;61(11):935–46.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Imaging microglial activation in Huntington’s disease. Brain Res Bull. 2007;72(2–3):148–51. doi:10.1016/j.brainresbull.2006.10.029.
Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, et al. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol. 2001;60(2):161–72.
Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66(11):1638–43. doi:10.1212/01.wnl.0000222734.56412.17.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. 2007;130(Pt 7):1759–66. doi:10.1093/brain/awm044.
Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P. Hypothalamic involvement in Huntington’s disease: an in vivo PET study. Brain. 2008;131(Pt 11):2860–9. doi:10.1093/brain/awn244.
Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington’s disease: a multimodal imaging study. Hum Brain Mapp. 2011;32(2):258–70. doi:10.1002/hbm.21008.
Appel SH, Zhao W, Beers DR, Henkel JS. The microglial-motoneuron dialogue in ALS. Acta Myol. 2011;30(1):4–8.
Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15(3):601–9. doi:10.1016/j.nbd.2003.12.012.
Sitte HH, Wanschitz J, Budka H, Berger ML. Autoradiography with [3H]PK11195 of spinal tract degeneration in amyotrophic lateral sclerosis. Acta Neuropathol. 2001;101(2):75–8.
Zurcher NR, Loggia ML, Lawson R, Chonde DB, Izquierdo-Garcia D, Yasek JE, et al. Increased in vivo glial activation in patients with amyotrophic lateral sclerosis: assessed with [(11)C]-PBR28. NeuroImage Clinical. 2015;7:409–14. doi:10.1016/j.nicl.2015.01.009.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Martin Schain and William Charles Kreisl declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neuroimaging
Rights and permissions
About this article

Cite this article
Schain, M., Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders—a Review. Curr Neurol Neurosci Rep 17, 25 (2017). https://doi.org/10.1007/s11910-017-0733-2
Published:
DOI: https://doi.org/10.1007/s11910-017-0733-2