
Abstract
Purpose of Review
Here, we review the known relations between hypertension and obesity to inflammation and postulate the endogenous protective effect of melatonin and its potential as a therapeutic agent. We will describe the multiple effects of melatonin on blood pressure, adiposity, body weight, and focus on mitochondrial-related anti-inflammatory and antioxidant protective effects.
Recent Findings
Hypertension and obesity are usually associated with systemic and tissular inflammation. The progressive affection of target-organs involves multiple mediators of inflammation, most of them redundant, which make anti-inflammatory strategies ineffective. Melatonin reduces blood pressure, body weight, and inflammation. The mechanisms of action of this ancient molecule of protection involve multiple levels of action, from subcellular to intercellular. Mitochondria is a key inflammatory element in vascular and adipose tissue and a potential pharmacological target. Melatonin protects against mitochondrial dysfunction.
Summary
Melatonin reduces blood pressure and adipose tissue dysfunction by multiple anti-inflammatory/antioxidant actions and provides potent protection against mitochondria-mediated injury in hypertension and obesity. This inexpensive and multitarget molecule has great therapeutic potential against both epidemic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bringing Hypertension, Obesity, Inflammation, and Melatonin Altogether
The new definition of hypertension as a sustained systolic blood pressure higher than 130 mmHg or diastolic over 80 mmHg makes hypertensive half of the adult population [1••]. This preventive redefinition intends to protect vasculature, kidneys, heart, and brain of patients from progressive epidemic damage. The etiology of hypertension is not identifiable in 90% of cases and thus classified as “essential.” Essential hypertension often coexists with obesity, aging, and metabolic disorders [2]. Worldwide, body mass index over 25 kg/m2 corresponds to 38.0 and 36.9% of the female and male population, respectively [3]. Overweight redefinition for Asian population as body mass index > 23 kg/m2 raises the proportion the patients affected [4]. In the last time, inflammation took a key role in the pathophysiology of both hypertension and obesity [5, 6••]. There are controversies about whether inflammation is the primary cause or final consequence [7]. However, all agree that modulating the inflammatory condition is positive for the patients. Obesity and hypertension have systemic inflammatory components [8,9,, 9, 10••]. In vascular and adipose tissues, local inflammation is also pathogenic [8,9,, 9, 10••]. Vascular structures are more than blood conduits. From endothelium to adventitia, the endocrine, paracrine, and immune functions are extensively characterized [10••]. As well, adipose tissue is more than an energetic and a lipid reservoir. It has endocrine and immune properties [13,12,••, 14, 15]. Mitochondria, our main energetic organelle, is involved in oxidative stress and the inflammatory process in both tissues [13••]. Like a Trojan horse inside the cells, mitochondrial dysfunction inflames and kills [15,14,17]. Vascular and metabolic circadian responses led to postulate melatonin as a physiological regulator with pharmacological potential [18, 19]. Due to melatonin lipophilic properties, its protective action easily reaches all subcellular structures. Therefore, many protective effects of melatonin occur at the mitochondrial level [20•, 21, 22•]. Indeed, melatonin reduces mitochondria-triggered oxidative stress and inflammation [20•, 23].
The Intricate Essential Hypertension
It is well known that essential hypertension implicates alterations of the vasculature, heart, nervous system, and kidneys and usually coexist with obesity and metabolic syndrome [1••, 8, 24••, 25, 26]. However, the precise interactions between these systems remain under continued investigation, making essential a proper adjective to describe hypertension [2, 27,26,29].
Chronic hypertension usually increases either cardiac output or systemic vascular resistance. Cardiac output and fluid retention usually rise early in hypertension, and vascular adaptations occur later in the disease [8, 30]. Hypertension readapts adrenergic, and renin-angiotensin-aldosterone systems (RAAS) and progressively increases systemic vascular resistance [8, 31]. In addition, increased local production of potent mediators such as endothelin-1, angiotensin II, prostaglandin H2, and reactive oxygen species (ROS) enhance vasoconstriction [8, 9, 12]. Oxidative stress blunts endothelial nitric oxide availability, reducing endothelium-dependent vasodilatation in isolated vascular preparations and flow-mediated vasodilatation in humans [12]. With time, hypertension led to structural remodeling of vessels [8, 9, 12, 32]. Vascular lumen narrows and the media thickens. In this progressive process, inflammation takes part at many levels [6••, 9, 32].
What Is Inflamed During Hypertension?
During hypertension, we could have inflammation almost everywhere [6••, 34]. Inflammation in the kidney, arteries, brain, heart, adipose tissue, gut, and liver contribute to hypertension [34]. With more than the 50 years together, the relation of inflammation and hypertension deserves a golden gift. The first torch was carried by lymphocytes. In 1967, Okuda and Grollman demonstrated that lymphocytes from rats with unilateral renal infarction caused hypertension when transferred in recipient rats [33]. Subsequently, most of the innate and adaptive immunity has shown pathogenic involvement in hypertension [6••, 34]. For an extensive description of each actor roles in hypertension, we suggest the reviews of Caillon and Schiffrin, and the one of Norlander et al. [6••, 34]. Interestingly, once an organ becomes inflamed, the immune response could grow within and make it more dysfunctional, raising the blood pressure. Recently, it has been proposed that oxidative stress may contribute to inflammatory mechanisms of hypertension development [8, 35]. We will highlight those elements relative to melatonin effects, in particular at mitochondria level.
Fat and Furious
Obesity and hypertension frequently coexist [25, 36,35,36,37,38,39,42]. Classic studies showed that excess body weight accounts for more than a half of the risk of hypertension [43]. Overweight and obese patients present a higher prevalence of hypertension [44,43,46]. However, obesity-associated hypertension could be overestimated due to white coat hypertension [47]. Thus, not all obese patients have sustained high blood pressure [47, 48]. One explanation is that fat’s inflammation increases the vasoconstrictive responses [49]. However, both diseases have many causes. Therefore, the understanding of the several mechanisms that underlie this association remains incomplete [25, 47, 50,49,52]. Visceral fat relates more to hypertension than subcutaneous fat [42, 49, 53, 54]. Fat impairs pressure natriuresis by physical compression and contributes to higher RAAS and sympathetic activity in both animals and patients [50, 54]. Fat narrows and weakens the upper airways, increasing adrenergic responses due to obstructive sleep apnea episodes [55,54,57]. Beyond distributive or mechanical considerations, inflamed adipose tissue raises blood pressure. Adipose tissue dysfunction contributes systemically to hypertension by adiponectin deficiency and elevated leptin, resistin, free fatty acids, tumor necrosis factor-α (TNF-α), and interleukin-6 [54]. Lipids also contribute to macrophage infiltration [7]. Adipocytes secrete two aldosterone-releasing factors, the complement-C1q tumor necrosis factor-related protein 1 and the 12,13-epoxy-9-keto-10 (trans)-octadecenoic acid, a type of oxidized fatty acid [58, 59]. Adipokines also sensitizes adrenocortical cells to angiotensin II [60]. Local inflammatory-induced dysfunction of perivascular adipose tissue exerts vasoconstriction mediated by angiotensin peptides, reactive oxygen species, chemokines, and cytokines [38, 61]. This relationship is extensively covered in a dedicated issue of the British Journal of Pharmacology (vol 140:20; 2017). Remarkably, inflammation enhances mitochondrial ROS production from perivascular adipose tissue and makes vascular response dysfunctional [62]. Increased adipose production of TNF-α promotes mitochondrial oxidative stress and increases phenylephrine-induced vasoconstriction mediated by increased activity of the RhoA/Rho kinase pathway in vascular smooth muscle in obese mice [62]. Fat inflammation makes arteries more reactive (furious).
Antihypertensives: Uneasy Fatness
Antihypertensive treatment in obese patients is uneasy. Obese patients required more antihypertensive medications, and blood pressure remained elevated in approximately in one-third of the patients, despite treatment intensification [63]. However, in most of the patients with high body mass index, antihypertensive medications effectiveness persist reducing blood pressure and cardiovascular diseases [64]. In the megatrial ALLHAT—Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial—obese patients required more antihypertensive medications [65]. On the other hand, weight loss reduces blood pressure and the number of antihypertensive drugs to achieve blood pressure control [66,65,68]. The reduction in blood pressure by lifestyle interventions is greater for uncontrolled hypertensive patients [69, 70]. Unfortunately, weight loss programs are difficult to maintain, and high failure rates occur during the initial years of most programs [67, 68, 71, 72]. More sustained weigh reductions are achieved with pharmacological obesity treatments and with bariatric surgery [72,71,74]. In the GATEWAY trail, bariatric surgery reduces weight and blood pressure leading to antihypertensive medication reduction and even discontinuation in half of the patients [75]. These effects could be related to the significant improvements in fasting plasma glucose, HbA1c, LDL cholesterol, triglycerides, and high-sensitivity C-reactive protein in the bariatric group. However, since surgical interventions are not an option to millions of obese and hypertensive patients, the promising results look like a small gate and a long way. Despite being inefficient, we remark the effectiveness of this multitarget intervention that includes inflammation reduction.
In the Darkest Hour, an Ancient Protector Rises Hope
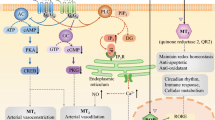
The melatonin is a ubiquitous molecule and widely distributed in nature, with functional activity occurring in unicellular organisms, plants, fungi, and animals [20••]. A primary biological function of melatonin in primitive unicellular organisms is in the antioxidant defense to protect against toxic free radical damage [20••]. During evolution, melatonin has been adopted by multicellular organisms to perform many other biological functions. These functions likely include the chemical expression of darkness in the vertebrates, environmental tolerance in fungi and plants, sexual signaling in birds and fish, seasonal reproductive regulation in photoperiodic mammals, and immunomodulation and anti-inflammatory activity in all vertebrates tested [76]. In most vertebrates, including humans, melatonin follows circadian rhythms. The natural regulator is the light during the day and the darkness of the night. The eyes detect the light and through a sympathetic pathway to the pineal gland inhibits melatonin secretion. The maximal melatonin plasma concentration usually occurs 3–5 h after darkness onset. René Descartes in the Renaissance period described the pineal gland as the “third eye.” Melatonin’s protective actions in many organs contribute to the so-called healing power of sleep. This mythical product of the “third eye” actively protects the cardiovascular system [77••].
Melatonin exerts physiological actions through receptor-dependent and receptor-independent effects [78]. Melatonin has membrane, cytoplasmic, and nuclear receptors. The MT1 and MT2 receptors are G-protein-coupled membrane receptors present in the cell membrane as dimers and heterodimers and act through many signal transduction mechanisms [79]. Numerous human cells express melatonin receptors, including cells in the nervous, cardiovascular, gastrointestinal, genitourinary, immune systems, and adipocytes [80]. Melatonin also displays an affinity for the cytoplasmic receptor called MT3 that is an enzyme quinone reductase 2 [78, 81]. Quinone reductases are antioxidants enzymes that prevent electron transfer reactions of quinones. Several mammalian tissues express the MT3 receptor [81]. The nuclear hormone receptor of melatonin belongs to the retinoid-related orphan receptor family (RZR/ROR), ubiquitously expressed in mammalian tissues [82]. Besides the action through receptors, melatonin easily crosses the cell membrane and interacts directly with other intracellular proteins, including calmodulin, calreticulin, and tubulin [83, 84••]. Additionally, melatonin is one of the most versatile free radical scavenging agents [20•]. As a direct scavenger, melatonin neutralizes different free radicals, including singlet oxygen, superoxide anion radical, hydroxyl radical, hydroperoxide, lipid peroxide radical, and peroxynitrite [85,84,87]. Melatonin reduces oxidative stress due to mitochondrial function improvement [20•], and stimulation of expressions and activation antioxidant enzymes, such as catalase, superoxide dismutase (SOD), and glutathione peroxidase [88••, 91]. Furthermore, melatonin potentiates other antioxidants, like glutathione, vitamin E, and vitamin C. All these actions contribute to better vascular functions and blood pressure regulation [84••, 88••].
Recent experiments in animals showed that melatonin prevents deaths caused by myocardial infarction and arrhythmias [89, 90]. The acute cardioprotection persists in hypertensive and dysmetabolic rat hearts [90]. Most scientists attribute its protection to its potent antioxidant effect. In addition, melatonin reduces inflammation and improves mitochondrial function [84••, 89, 91,90,91,94]. From a preventive point of view, melatonin’s pleiotropic mechanisms of protection could effectively limit the progression of both epidemics, obesity and hypertension.
Following the Inflammatory and Diffractive Path of Light
The light activates the retina and modulates the master circadian pacemaker of the organism, the suprachiasmatic nucleus [84••, 95, 96]. This hypothalamic structure subordinate most of other peripheral cellular clocks [84••, 95, 96]. One subordinate is the pineal gland, where the biological effect of light reduces melatonin secretion. During the daily light period, melatonin circulating levels are low or even undetectable [97]. Melatonin, as part of the light/dark cycles responses, also impact in the immune system [96]. The precise interactions are under intense research in a field called chrono-immunology. Melatonin inhibits the expression of cyclooxygenase synthase and reduces the excessive production of prostanoids, and leukotrienes [84••]. Melatonin also reduces pro-inflammatory cytokines, adipokines, chemokines, and adhesion molecules [84••]. Some of these actions respond to transcription factors modulation, such as nuclear factor kappa B (NF-κB), nuclear factor erythroid 2-related factor 2, hypoxia-inducible factor, heat shock proteins, and others [19, 98, 99]. Recently, melatonin included the regulatory effects over inflammasomes to its protective actions [94, 100]. Therefore, it is clear that this circadian hormone prevents inflammatory conditions.
Today, the environmental timers go beyond the photoperiod and include food, activities, and temperature among others [19, 84••, 96]. Concurrently, inflammation was recently redefined as a physiological homeostatic response to harmful stimuli such as pathogens, injury, and metabolic stress [94, 101]. Well-defined metabolic stressor like salt, sugar, and fat could be extended to include chronodisruption among them [84••, 96, 102]. The loss of circadian oscillation of melatonin reduces its antioxidant and anti-inflammatory effects [84••]. Chronodisruption was epidemiologically associated with higher incidence of cardiovascular diseases, obesity, diabetes, cognitive impairments, depression, aging, and cancer [103,102,105]. However, we suggest cautious interpretation because causal contributions will be hard to prove for these multifactorial diseases.
Our caution alert could be supported by at least two biological facts. First, other organs such as gastrointestinal tract, skin, salivary glands, platelets, and lymphocytes produce melatonin without a circadian pattern or contribution to circulating levels [76, 84••]. However, in the absence of suprachiasmatic signals, peripheral clock temporarily sustained rhythmicity but amplitude decrease and loss synchrony with other tissues [96, 99, 106]. Other elements respond to light in a clock-independent manner besides the conventional master circadian pacemaker [84••, 96]. Interpretation of this second fact is less clear.
Anti-Inflammatory Effect of Melatonin in Obesity
Obesity contributes as a risk factor for the development and maintenance of cardiovascular diseases [15, 47, 50, 107]. Complications are consistent with the development of hypertension, insulin resistance, metabolic syndrome, chronic kidney disease, and type 2 diabetes [15, 47, 50, 107,106,107,110]. Metabolic-triggered inflammatory process underlies obesity damages [5, 7, 102]. Melatonin levels correlate inversely with obesity [84••, 111, 112]. In humans, the reduction in melatonin levels is associated with insulin resistance and metabolic syndrome, due to the mitochondrial protection and metabolic regulation [20•, 84••, 111, 112]. Several mechanisms of the physiological and pharmacological beneficial actions of melatonin in obesity have been proposed, including antioxidant and anti-inflammatory [84••, 111, 112]. As a chronobiological agent, melatonin regulates the metabolic rate and the energy balance in the organism, partly by brown adipose tissue activation [77••, 113, 114]. In this section, we describe melatonin effects on adiposity, body weight, oxidative stress, and inflammation linked to mitochondrial function and dynamics in obesity.
Melatonin directly activates receptors in adipocytes [84••, 106]. Animals and humans express membrane receptor in white and brown adipose tissue. The cytoplasmic and nuclear actions of melatonin were proven in animals [80, 106]. Melatonin sensitizes adipocytes to insulin and leptin by a shared signaling pathway, specifically the phosphoinositide 3-kinase and the signal transducer and activator of transcription 3 [20•, 84, 115, 116]. Studies in pinealectomized animals demonstrated that circadian melatonin regulates internal biological clocks and energy metabolism, specifically in adipose tissue [84••, 106]. The lack of melatonin abolishes the daily pattern of the expression of clock genes (Clock, Cry1, and Per2) and PPARγ, leading to a decrease in leptin levels. Melatonin deficiency reduces the circadian expression of the lipogenic enzymes (ATP-citrate lyase, malic enzyme, fatty acid synthase, and glucose-6-phosphate dehydrogenase). However, some metabolic adipocyte functions, such as its ability to synthesize triacylglycerols from glucose along 24 h, are not compromised by pinealectomy [106]. Interestingly, a high-fat obesogenic diet causes chronodisruption of clock gene expression in the anterior pituitary of rats and decreases nocturnal melatonin levels [112]. Melatonin also exerts its effects on adipose tissue through sympathetic-induced lipolysis and modulates seasonal adiposity, at least in experimental animals [117]. High doses of melatonin supplementation induced the conversion of white to brown fat, by increasing thermogenic proteins in both Zucker diabetic fatty rats and lean controls [113].
Sleep deprivation contributes to the pathogenesis of obesity [84••, 114]. Epidemiologic studies related chronic sleep deprivation and long-term weight gain [84••, 118,117,120]. Experimental evidence showed circadian regulation of metabolism and energy balance [19, 102, 106]. Melatonin sensitizes hypothalamic leptin and insulin receptors and passes on information to correct energy balance in the organism [19, 84••]. Therefore, body weight is maintained by adjustments to the environmental inputs [19]. This adjustment could be reset to higher body weight or altered by chronodisruption [19]. Additionally, chronodisruption occurs through shift work, high-fat foods, and altered eating patterns. Some evidence suggests that obesity prevention and treatment should incorporate chrononutrition, a form of timing meals and macronutrient consumption, and sleep duration and quality improvement. [19]
The following paragraphs put the focus on molecular mechanisms of melatonin that reduces oxidation and inflammation, mainly in the mitochondria [20•, 22•, 94, 121]. Melatonin preserves complex I and III activity, inhibits mitochondrial permeability transition pore opening, and cytochrome c release [20•, 22•, 121]. To highlight, it has recently been confirmed that obesity and malnutrition grade also modulate mitochondrial dynamics in humans [15, 122]. In parallel were shown mitochondrial changes in a mice model of obesity (ob/ob), and melatonin treatment produced beneficial effects on mitochondrial morphology and dynamics by mitofusin-2 (Mtf2) and the intrinsic apoptotic cascade modulation [123]. The Mtf2 protein is involved in mitochondrial reduction after a high-fat diet; nonetheless, infliximab (a tumor necrosis factor TNF-alpha inhibitor) normalizes the Mtf2 levels. With these findings, Carraro et al. propose that inflammation is the critical mechanism activated after a high-fat diet and may play an essential role in the progress of mitochondrial abnormalities during obesity [124]. In close connection, mitochondrial dynamics modulated by mitofusins (Mtf1 and Mtf2) modulate the orexigenic agouti-related protein (Agrp) neuronal activity and diet-induced obesity [125]. Diminished Mfn2 expression in skeletal muscle has been observed in obese and type 2 diabetes individuals, which increase after weight loss. The Mfn2 expression is directly proportional to insulin sensitivity and is inversely proportional to the body mass index [122].
On the other hand, mitochondrial function is also a crucial point of interaction for circadian alterations in cardiovascular disease associated with renin-angiotensin-aldosterone system upregulation [18, 88•]. RAAS shows a tonic modulation of melatonin synthesis by inducing the tryptophan hydroxylase activity via angiotensin II type 1 receptors (AT1) [126]. Accordingly, a relationship between angiotensin II and melatonin has been suggested in the modulation of circadian rhythms; and since melatonin can improve metabolic abnormalities in diabetes mellitus and insulin resistance, the beneficial effects of RAAS blockade could be enhanced through combined melatonin therapy [127]. These studies are consistent and strengthen our unpublished finding in kidney from chronic kidney disease model about AT1 downregulation linked to the melatonin-cytoprotective effect. Specifically, melatonin restored vitamin D receptors/heat shock protein 70 (VDR/Hsp70) and decreased AT1 expressions, oxidative stress, fibrosis, and apoptosis, as well as conserved mitochondrial ultrastructure by prevented mitochondrial edema with dilated cristae and high NADPH oxidase activity. We postulate possible feedback (or reciprocal regulation) between AT1 and melatonin. The mitochondrial dysfunction by an over-activation of the RAAS linked to the NADPH oxidase activity is a consequence shared by obesity, diabetes, metabolic syndrome, impaired glucose tolerance, and dyslipidemias. In obesity patients, it has been described that lower vitamin D levels are linked to higher RAAS activity [128]. VDR activators antagonize RAAS effects, as well as modulate anti-inflammatory and antifibrotic actions [129, 130]. The high AT1 expression and NADPH activity were reverted in mitochondrial fractions [131]. Obesity is linked to inflammation with a substantial energetic cost. Recent reports suggest that vitamin D can act as a therapeutic agent for inflammation of chronic disease by cellular bioenergetics modulation [132].
Obesity induces the inflammasome response [11, 133]. The nucleotide-binding oligomerization domain-like receptor containing pyrin domain 3 (NLRP3) is a critical component of the inflammasome [54, 134, 135]. Particularly, pyroptosis (a pro-inflammatory programmed cell death) is mediated by NLRP3-dependent caspase-1 activation, and subsequent pro-inflammatory response in hypertrophic adipocytes [100, 136, 137]. Despite inflammasomes are cytoplasmic elements, activation is triggered by mitochondrial oxidative stress [93, 134, 138]. Exogenous melatonin ameliorates inflammation of adipose tissue by inhibiting the expression of inflammasome genes including NLRP3, an apoptotic-associated speck-like protein that contains a caspase recruitment domain, and thereby caspase-1 and IL-1β [100]. Furthermore, melatonin in the adipose tissue reduces the phosphorylation of NF-κB and inhibits the NLRP3 pathway [93, 138]. Melatonin also reverse systemic inflammation in obesity by other signals mediated by NF-κB [92, 94, 100].
There is a clear correlation between the global growth of kidney disease with diabetes, and recently also with obesity. Despite the fact that cardiovascular events increase in obese patients with chronic kidney disease, there is a lack of specific treatments for these related diseases. Nevertheless, there are promising experiments suggesting that the modulation of inflammation and oxidative stress could mitigate the progression of kidney disease in obesity and diabetes [139]. In this regard, endogenous melatonin secretion is impaired during chronic kidney disease [140]. Moreover, the impairment worsens with the degree and progression of kidney disease [91]. Therefore, melatonin supplementation in kidney disease and obesity could be of interest.
Several decades ago, it was proposed that melatonin acts as an antioxidant and anti-inflammatory molecule into of the central nervous system and suprarenal glands. However, specific action on renal tissue is quite recent. Conveniently, this was demonstrated in an animal model with obesity [141]. In the same way, melatonin exerts an antioxidant effect in kidneys of Zucker diabetic fatty rat, a model of diabetes type 2. The mechanism suggested includes the decrease of NADPH oxidase and glutathione peroxidase activities [142]. Uncoupling of ROS generation and the release from mitochondria to cytosol leads to inflammation, cell death, tissue damage, and disease progression. An adequate mitochondrial function maintains the structural and functional integrity of every tissue. Also, nitric oxide is associated with inflammatory pathologies when both impaired release and reduced bioavailability leading to inflammation. Moreover, a reduced nitric oxide release induces Hsp70 with effects against oxidative stress, inflammation, and apoptosis during insulin resistance [143]. Melatonin improves of insulin signal transduction by amplified insulin-induced Akt phosphorylation and nitric oxide levels in high-fat-diet-fed mice [144].
Melatonin effectiveness as an antioxidant factor includes the heat shock protein responses [98]. Particularly, melatonin decreases streptozotocin-induced diabetic renal injury by modulation of nitric oxide, oxidative markers, and Hsp70 expression [145]. Thus, clarifying the signaling pathways and the roles of Hsp70 is relevant to the application of new treatments. Fujimoto et al. reported that cytokine levels were not affected by Hsp70 in obese mice [146]. This finding could be somewhat controversial. However, we reviewed opposing actions of extracellular versus intracellular Hsp70 on NF-κB pathway activation [147].
The Hsp70 induction by melatonin also supports an additional mechanism linked to Tom70, translocase of the outer mitochondrial membrane. Hsp70 interaction with Tom70 is essential for the initiation of the mitochondrial import process [148], and required for the recognition, unfolding, and translocation of amino acids into the mitochondria. The Tom70 protein is a crucial member of the mitochondrial outer and inner membrane transport systems. In diabetes, Tom70 and inner mitochondrial membrane 44 (Timm44) were significantly decreased, and the binding of nuclear-encoded mitochondrial transcription factor A (TFAM) with Tom70, Timm44, and mitochondrial DNA was impaired. Hence, hyperglycemia affects the ability of TFAM to access into the mitochondria [149]. Similarly, Wang et al. demonstrated that Timm44 alters the mitochondrial fusion and fission dynamics and protects from type 2 diabetes [150]. Recently, melatonin protection on the mitochondrial function was associated with suppressing TNF-α expression, which is responsible for dynamin-related protein 1-mediated mitochondrial fission [151].
Anti-Inflammatory Effect of Melatonin in Hypertension
Melatonin regulates several parameters of the cardiovascular system, including blood pressure, and is a promising antihypertensive agent [77••, 80, 152, 153]. The mechanisms and pathways involved in its antihypertensive action are complex and some of them unclear. Melatonin receptor-dependent and receptor-independent actions involve autonomic, vascular, anti-inflammatory, and antioxidant effects. Not only melatonin, but its metabolites have antioxidant effects [85]. In this section, we will analyze the multiple antihypertensive actions of melatonin with the focus on mitochondrial effects.
The role of physiological melatonin level in blood pressure regulation comes from experimental and some clinical studies. Pinealectomy in rats causes a gradual and sustained rise in blood pressure [154]. When melatonin is administered chronically to pinealectomized rodents, there is no increase in blood pressure [155]. The loss of nocturnal blood pressure reduction was related to insufficient melatonin levels [156,156,158]. Light pollution may be one of the chornodisruptive factors that reduce melatonin level; metabolic and sleep changes the others [19, 99, 159]. Vascular effects of melatonin are not straightforward. Melatonin receptor actions depend on the cell type, and vascular territory and receptors activation induce vasoconstriction in some vasculatures and vasodilation in others. Activation of MT1 induces vasoconstriction and activation of MT2 produce vasodilation, but these responses change regarding circadian time, melatonin source, concentration, duration, and functional receptor sensitivity (homo and heterodimers) [160,160,161,162,164]. Melatonin has others, not less complex, receptor-independent effects that regulate the vascular tone. Melatonin decreases [Ca2+] in smooth muscle cells by interaction with Ca2+-calmodulin complex leading to relaxation, but in endothelial cells, the same reduction inhibits endothelial nitric oxide synthase, triggering vasoconstriction [165]. Melatonin increase antioxidant defenses like catalase and superoxide dismutase, leading to the reduction of oxidative blood pressure in patients [166, 167]. The mechanism of actions involves antioxidant enzyme expression by nuclear receptor and NF-κB [168]. In spontaneously hypertensive rats, melatonin decreases the NF-κB expression [169]. Melatonin action on the NF-κB pathway connects redox and inflammatory, both relevant in hypertension pathophysiology.
Besides its actions in the vasculature, melatonin regulates structures in the nervous system involved in vascular tone. Inflammation in the brainstem may contribute to hypertension [170]. Inflammatory molecules overexpressed in the endothelium of the surrounding solitary tract, the principal structure controlling arterial blood pressure (BP), increase cytokine release to the brainstem of hypertensive patients [171]. Mitochondria contribute to the oxidative stress-related neural mechanism of hypertension. Compromised mitochondrial biogenesis and bioenergetics in some forebrain circumventricular organs and the nucleus of the solitary tract contribute to neurogenic hypertension due to mitochondrial ROS accumulation under the conditions of systemic inflammation, angiotensin II stimulation, and metabolic syndrome. From the mechanistic point of view, the alterations in both neural mitochondrial dynamics and ROS production are associated hypertension development; in this sense, melatonin also acts as an antioxidant and as a regulator of mitochondrial bioenergetics function [172, 173].
Kidney oxidative stress and inflammation contribute to hypertension. Several studies have demonstrated that treatment with antioxidants improves the hypertensive conditions. In this sense, Nava et al. showed that melatonin improves hypertension in spontaneously hypertensive rats (SHR) with a decrease in renal inflammation, specifically to a decreased activation of NF-kB [174]. Following studies showed that melatonin reduces oxidative stress, renal inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction [175]. Melatonin exerts a renoprotective and antihypertensive effect through nitric oxide bioavailability improvement [176]. Qiao et al. demonstrated that melatonin decreases blood pressure and reduces infiltration of inflammatory cells of renal tubules. Also, melatonin attenuates urine protein excretion, serum creatinine, and reduces the oxidative stress in renal tissues [177]. The kidney improvements were correlated with the increases of iNOS and ICAM-1 expressions. Accordingly, the association of melatonin and exendin-4 was more efficient for reducing the deterioration of renal function in cardiorenal syndrome than each agent alone [178]. Particularly, the combined treatment produced—at renal level—a reduction of protein expressions of inflammation (TNF-α/NF-κB/MMP-9/iNOS/RANTES), oxidative stress (NOX-1/NOX-2/NOX-4/oxidized protein), apoptosis (cleaved caspase-3/cleaved PARP/Bax), DNA-damaged marker (γ-H2AX), and fibrosis (p-mad3/TFG-β). Moreover, melatonin regulates renal transcriptome and prevents prenatal L-NAME-induced fetal programming of hypertension. Interestingly, genes belonging to the RAAS and the arachidonic acid metabolism pathways were involved in hypertension induced by maternal nitric oxide depletion. However, maternal melatonin reprogrammed the RAAS and the arachidonic acid pathway via redox balance restoration in pregnancy and reducing the future burden of hypertension [179]. The preventive action on the kidney injury was confirmed in a high-salt diet-induced hypertension model. The positive effect was not mediated by blood pressure but by a direct antioxidative effect.
Kidney diseases are chronodisruptors. Chronic kidney diseases impair nighttime melatonin secretion Melatonin levels correlate negatively with intrarenal RAAS activity. Ishigaki et al. showed that melatonin ameliorates intrarenal RAAS in a 5/6 nephrectomy rat model and reduces blood pressure, oxidative stress, and interstitial fibrosis in the remnant kidneys [140]. In connection, AT1 receptor blockers have shown auspicious “preventive” intracellular anti-inflammatory/antioxidant activity in addition to their original pharmacological actions. Disruption of the AT1 promotes inflammation, cell growth, and proliferation [180]. Kidney protection was associated with an increased number of mitochondria and upregulation of the prosurvival genes such as nicotinamide phosphoribosyl transferase and sirtuin 3 (Sirt3) [181]. Sirt3 is a member of the family that is localized mostly to the mitochondria and protects against inflammation and oxidative stress-related diseases, including hypertension. Melatonin elevates Sirt3, stimulates superoxide dismutase activity, and suppresses mitochondrial oxidative stress through 5′ AMP-activated protein kinases [34]. Melatonin multiple actions as an anti-inflammatory/antioxidant provide potent protection against mitochondria-mediated injury [152].
Conclusions
Melatonin is a skilled rider to dressage the Trojan horses (dysfunctional mitochondria) that cause inflammation in obesity and hypertension. Melatonin reduces blood pressure and adipose dysfunction through its antioxidant and anti-inflammatory effects. The compelling evidence of its several protective actions and the apparent lack of adverse effects should promote the translational pathway as therapeutic options for millions of obese and hypertensive patients. Pharmacokinetics, safety, and efficacy studies oriented to these specific populations should be a reasonable next step. We hope that past evidence and future findings will stimulate large clinical trial to determine the efficacy of melatonin treatment for both epidemic diseases.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults. J Am Coll Cardiol. 2017;S0735–663 1097(17)41519–1. https://doi.org/10.1016/j.jacc.2017.11.006. Despite controversial, the new guidelines underlie the need to increase prevention. 10.1016/j.jacc.2017.11.006.
Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000;101(3):329–3325.
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden Of Disease Study 2013. Lancet. 2014;384(9945):766–81.
Jih J, Mukherjea A, Vittinghoff E, Nguyen TT, Tsoh JY, Fukuoka Y, et al. Using appropriate body mass index cut points for overweight and obesity among Asian Americans. Prev Med (Baltim). 2014;65:1–6.
Guida B, Cataldi M, Maresca ID, Germanò R, Trio R, Nastasi AM. Dietary intake as a link between obesity, systemic inflammation, and the assumption of multiple cardiovascular and antidiabetic drugs in renal transplant recipients. Biomed Res Int. 2013;2013:1–8.
•• Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and clinical evidence. Curr Hypertens Rep. 2016;18(3):21. A deep review of systemic and local inflammatory responses in hypertension. Cellular and humoral interactions in almost every physiological system that contribute to hypertension pathophysiology.
Bluher M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin Sci. 2016;130(18):1603–14.
Renna NF, Lembo C, Diez E, Miatello RM. Role of renin-angiotensin system and oxidative stress on vascular inflammation in insulin resistence model. Int J Hypertens. 2013;2013:420979. https://doi.org/10.1155/2013/420979.
Renna NF, Diez ER, Lembo C, Miatello RM. Role of Cox-2 in vascular inflammation: an experimental model of metabolic syndrome. Mediat Inflamm. 2013;2013:1–10.
•• Dorresteijn JAN, Visseren FLJ, Spiering W. Mechanisms linking obesity to hypertension. Obes Rev. 2012;13(1):17–26. This article describes the paracrine regulation of vascular response by mutual interactions between the endothelium, smooth muscle, and perivascular adipose tissue.
Stolarczyk E. Adipose tissue inflammation in obesity: a metabolic or immune response? Curr Opin Pharmacol. 2017;37:35–40.
Nava E, Llorens S. The paracrine control of vascular motion. A historical perspective. Pharmacol Res. 2016;113:125–45.
•• Vazquez-Prieto MA, Renna NF, Diez ER, Cacciamani V, Lembo C, Miatello RM. Effect of red wine on adipocytokine expression and vascular alterations in fructose-fed rats. Am J Hypertens. 2011;24(2):234–40. https://doi.org/10.1038/ajh.2010.214. This previous review from our group extensively covers the central role of mitochondrial dysfunction in feeding behavior, energy balance, autonomic response, oxidative stress, and inflammation.
Huby A-C, Antonova G, Groenendyk J, Gomez-Sanchez CE, Bollag WB, Filosa JA, et al. Adipocyte-derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. 2015;132(22):2134–45.
Lahera V, de las Heras N, López-Farré A, Manucha W, Ferder L. Role of mitochondrial dysfunction in hypertension and obesity. Curr Hypertens Rep. 2017;19(2):11.
Manfredi A, Rovere-Querini P. The mitochondrion—a Trojan horse that kicks off inflammation? N Engl J Med. 2010;362(22):2132–4.
Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. 2017;1(391)42–53. https://doi.org/10.1016/j.tox.2017.07.019.
Ohashi N, Isobe S, Ishigaki S, Yasuda H. Circadian rhythm of blood pressure and the renin–angiotensin system in the kidney. Hypertens Res. 2017;40(5):413–22.
Laermans J, Depoortere I. Chronobesity: role of the circadian system in the obesity epidemic. Obes Rev. 2016;17(2):108–25.
• Reiter RJ, Rosales-Corral S, Tan DX, Jou MJ, Galano A, Xu B. Melatonin as a mitochondria-targeted antioxidant: one of evolution’s best ideas. Cell Mol Life Sci. 2017;74(21):3863–3881. https://doi.org/10.1007/s00018-017-2609-7. This review covers most of the antioxidant mechanisms of melatonin with special attention to those relative to mitochondria.
Suofu Y, Li W, Jean-Alphonse FG, Jia J, Khattar NK, Li J, et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc Natl Acad Sci U S A. 2017;114(38):E7997–8006.
• Sharafati-Chaleshtori R, Shirzad H, Rafieian-Kopaei M, Soltani A. Melatonin and human mitochondrial diseases. J Res Med Sci. 2017;22:2. The mitochondrial pathological mechanisms and interactions with melatonin are covered by this review.
Volt H, García JA, Doerrier C, Díaz-Casado ME, Guerra-Librero A, López LC, et al. Same molecule but different expression: aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J Pineal Res. 2016;60(2):193–205.
•• Lüscher TF. Hypertension: detection, mechanisms, outcomes, and treatment. Eur Heart J. 2017;38(2):67–9. It is a well-illustrated review of hypertension. It addresses risk factor, pathogenic mechanisms, treatments, morbidity, and mortality.
Marco VG, Aroor AR, Sowers JR. The pathophysiology of hypertension in patients with obesity. Nat Rev Endocrinol 2014;10(6):364–76. https://doi.org/10.1038/nrendo.2014.44.
Oparil S, Acelajado MC, Bakris GL, Berlowitz DR, Cífková R, Dominiczak AF, et al. Hypertension. Nat Rev Dis Prim. 2018;4:18014.
Schiffrin EL. Inflammation, immunity and development of essential hypertension. J Hypertens. 2014;32(2):228–9.
Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med. 2011;17(11):1402–9.
Ferdinand KC, Nasser SA. Management of Essential Hypertension. Cardiol Clin. 2017 May;35(2):231–46.
Lee RM, Dickhout JG, Sandow SL. Vascular structural and functional changes: their association with causality in hypertension: models, remodeling and relevance. Hypertens Res. 2017;40(4):311–23.
Simko F, Pechanova O. Remodelling of the heart and vessels in experimental hypertension: advances in protection. J Hypertens 2010;28 Suppl 1:S1–6. https://doi.org/10.1097/01.hjh.0000388487.43460.db.
Renna NF, Diez EA, Miatello RM. Effects of dipeptidyl-peptidase 4 inhibitor about vascular inflammation in a metabolic syndrome model. PLoS One. 2014;9(9):e106563. https://doi.org/10.1371/journal.pone.0106563.
Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med. 1967;25(2):257–64.
Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. 2018;215(1):21–33.
Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol. 2017;70(2):212–29.
Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev. 2012;13:136–49.
D’Agati VD, Chagnac A, Vries AP, Levi M, Porrini E, Herman-Edelstein M. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol. 2016;12(8):453–71. https://doi.org/10.1038/nrneph.2016.75.
Xia N, Li H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br J Pharmacol. 2017;174(20):3425–42.
Imperatore R, Palomba L, Cristino L. Role of orexin-a in hypertension and obesity. Curr Hypertens Rep. 2017;19(4):34.
Ingaramo RA. Obesity, diabetes, and other cardiovascular risk factors in native populations of South America. Curr Hypertens Rep. 2016;18(1):9.
Soltani S, Shirani F, Chitsazi MJ, Salehi-Abargouei A. The effect of dietary approaches to stop hypertension (DASH) diet on weight and body composition in adults: a systematic review and meta-analysis of randomized controlled clinical trials. Obes Rev. 2016;17(5):442–54.
Scholz GH, Hanefeld M. Metabolic vascular syndrome: new insights into a multidimensional network of risk factors and diseases. Visc Med. 2016;32(5):319–26.
Kannel WB, Brand N, Skinner JJ, Dawber TR, McNamara PM. The relation of adiposity to blood pressure and development of hypertension. The Framingham study Ann Intern Med. 1967;67(1):48–59.
Bramlage P, Pittrow D, Wittchen HU, Kirch W, Boehler S, Lehnert H. Hypertension in overweight and obese primary care patients is highly prevalent and poorly controlled. Am J Hypertens. 2004;17:904–10.
Canning KL, Brown RE, Wharton S, Sharma AM, Kuk JL. Edmonton obesity staging system prevalence and association with weight loss in a publicly funded referral-based obesity clinic. J Obes. 2015;2015:1–7.
Hales CM, Carroll MD, Simon PA, Kuo T, Ogden CL. Hypertension prevalence, awareness, treatment, and control among adults aged ≥18 years—Los Angeles County, 1999-2006 and 2007-2014. MMWR Morb Mortal Wkly Rep. 2017;66(32):846–9.
Kotsis V, Nilsson P, Grassi G, Mancia G, Redon J, Luft F, et al. New developments in the pathogenesis of obesity-induced hypertension. J Hypertens. 2015;33(8):1499–508.
Movahed MR, Lee JZ, Lim WY, Hashemzadeh M. Strong independent association between obesity and essential hypertension. Clin Obes. 2016;6(3):189–92.
Ramirez JG, O’Malley EJ, Ho WSV. Pro-contractile effects of perivascular fat in health and disease. Br J Pharmacol. 2017;174(20):3482–95.
Niemann B, Rohrbach S, Miller MR, Newby DE, Fuster V, Kovacic JC. Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution: part 3 of a 3-part series. J Am Coll Cardiol. 2017;70(2):230–51.
do Carmo JM, da Silva AA, Wang Z, Fang T, Aberdein N, de Lara Rodriguez CEP, et al. Obesity-induced hypertension: brain signaling pathways. Curr Hypertens Rep. 2016;18(7):58.
Lim K, Jackson KL, Sata Y, Head GA. Factors responsible for obesity-related hypertension. Curr Hypertens Rep. 2017;19(7):53.
Ramos-Romero S, Hereu M, Atienza L, Casas J, Jáuregui O, Amézqueta S, et al. Mechanistically different effects of fat and sugar on insulin resistance, hypertension and gut microbiota in rats. Am J Physiol Metab. 2018; https://doi.org/10.1152/ajpendo.00323.2017.
Stafeev IS, Vorotnikov AV, Ratner EI, Menshikov MY, Parfyonova YV. Latent inflammation and insulin resistance in adipose tissue. Int J Endocrinol. 2017;2017:1–12.
Mansukhani MP, Kara T, Caples SM, Somers VK. Chemoreflexes, sleep apnea, and sympathetic dysregulation. Curr Hypertens Rep. 2014 Sep;16(9):476.
Konecny T, Kara T, Somers VK. Obstructive sleep apnea and hypertension: an update. Hypertension. 2014;63(2):203–9.
Lohmeier TE, Iliescu R, Tudorancea I, Cazan R, Cates AW, Georgakopoulos D, et al. Chronic interactions between carotid baroreceptors and chemoreceptors in obesity hypertension. Hypertension. 2016;68(1):227–35.
Goodfriend TL, Ball DL, Egan BM, Campbell WB, Nithipatikom K. Epoxy-keto derivative of linoleic acid stimulates aldosterone secretion. Hypertension. 2004;43(2):358–63.
Jeon JH, Kim K-Y, Kim JH, Baek A, Cho H, Lee YH, et al. A novel adipokine CTRP1 stimulates aldosterone production. FASEB J. 2007;22(5):1502–11.
Bentley-Lewis R, Adler GK, Perlstein T, Seely EW, Hopkins PN, Williams GH, et al. Body mass index predicts aldosterone production in normotensive adults on a high-salt diet. J Clin Endocrinol Metab. 2007;92(11):4472–5.
Lian X, Gollasch M. A clinical perspective: contribution of dysfunctional perivascular adipose tissue (PVAT) to cardiovascular risk. Curr Hypertens Rep. 2016;18(11):82.
da Costa RM, Fais RS, Dechandt CRP, Louzada-Junior P, Alberici LC, Lobato NS, et al. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br J Pharmacol. 2017;174(20):3527–41.
Jordan J, Nilsson PM, Kotsis V, Olsen MH, Grassi G, Yumuk V, et al. Joint scientific statement of the European Association for the Study of Obesity and the European Society of Hypertension: obesity and early vascular ageing. J Hypertens. 2015;33(3):425–34.
Owen JG, Reisin E. Anti-hypertensive drug treatment of patients with and the metabolic syndrome and obesity: a review of evidence, meta-analysis, post hoc and guidelines publications. Curr Hypertens Rep. 2015 Jun;17(6):558.
Shah RV, Abbasi SA, Yamal J-M, Davis BR, Barzilay J, Einhorn PT, et al. Impaired fasting glucose and body mass index as determinants of mortality in ALLHAT: is the obesity paradox real? J Clin Hypertens. 2014;16(6):451–8.
Siebenhofer A, Jeitler K, Horvath K, Berghold A, Siering U, Semlitsch T. Long-term effects of weight-reducing drugs in hypertensive patients. Cochrane Database Syst Rev. 2013;28(3):CD007654. https://doi.org/10.1002/14651858.CD007654.pub3.
Sabaka P, Dukat A, Gajdosik J, Bendzala M, Caprnda M, Simko F. The effects of body weight loss and gain on arterial hypertension control: an observational prospective study. Eur J Med Res. 2017;22(1):43.
Seimon RV, Espinoza D, Ivers L, Gebski V, Finer N, Legler UF, et al. Changes in body weight and blood pressure: paradoxical outcome events in overweight and obese subjects with cardiovascular disease. Int J Obes. 2014;38(9):1165–71.
Ndanuko RN, Tapsell LC, Charlton KE, Neale EP, Batterham MJ. Dietary patterns and blood pressure in adults: a systematic review and meta-analysis of randomized controlled trials. Adv Nutr. 2016;7(1):76–89.
Sorriento D, De Luca N, Trimarco B, Iaccarino G. The antioxidant therapy: new insights in the treatment of hypertension. Front Physiol. 2018;9:258.
Sjostrom CD, Peltonen M, Wedel H, Sjostrom L. Differential long-term effects of intentional weight loss on diabetes and hypertension. Hypertension. 2000;36(1):20–5.
Ho AK, Bartels CM, Thorpe CT, Pandhi N, Smith MA, Johnson HM. Achieving weight loss and hypertension control among obese adults: a US multidisciplinary group practice observational study. Am J Hypertens. 2016;29(8):984–91.
Khera R, Murad MH, Chandar AK, Dulai PS, Wang Z, Prokop LJ, et al. Association of pharmacological treatments for obesity with weight loss and adverse events. JAMA. 2016;315(22):2424–34.
Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and Management of Obesity. Longo DL, editor. N Engl J Med 2017;376(3):254–66.
Schiavon CA, Bersch-Ferreira AC, Santucci EV, Oliveira JD, Torreglosa CR, Bueno PT, et al. Effects of bariatric surgery in obese patients with hypertension: the GATEWAY Randomized Trial (gastric bypass to treat obese patients with steady hypertension). Circulation. 2018; 137(11):1132–1142. https://doi.org/10.1161/CIRCULATIONAHA.117.032130.
Tan DX, Hardeland R, Manchester LC, Paredes SD, Korkmaz A, Sainz RM, et al. The changing biological roles of melatonin during evolution: from an antioxidant to signals of darkness, sexual selection and fitness. Biol Rev. 2010;85(3):607–23.
•• Pandi-Perumal SR, BaHammam AS, Ojike NI, Akinseye OA, Kendzerska T, Buttoo K, et al. Melatonin and human cardiovascular disease. J Cardiovasc Pharmacol Ther. 2017;22(2):122–32. Update information about melatonin cardiovascular effects. Clear information that supports melatonin potential as a therapeutic option.
Dubocovich ML, Delagrange P, Krause DN, Sugden D, Cardinali DP, Olcese J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol Rev. 2010;62(3):343–80.
Jockers R, Maurice P, Boutin JA, Delagrange P. Melatonin receptors, heterodimerization, signal transduction and binding sites: what’s new? Br J Pharmacol. 2008;154(6):1182–95.
Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, Ostrom RS, Slominski AT. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol. 2012;351(2):152–66.
Nosjean O, Nicolas JP, Klupsch F, Delagrange P, Canet E, Boutin JA. Comparative pharmacological studies of melatonin receptors: MT1, MT2 and MT3/QR2. Tissue distribution of MT3/QR2. Biochem Pharmacol. 2001;61(11):1369–79.
Emet M, Ozcan H, Ozel L, Yayla M, Halici Z, Hacimuftuoglu A. A review of melatonin, its receptors and drugs. Eurasian J Med. 2016;48(2):135–41.
Benítez-King G. Melatonin as a cytoskeletal modulator: implications for cell physiology and disease. J Pineal Res. 2006 Jan;40(1):1–9.
•• Szewczyk-Golec K, Woźniak A, Reiter RJ. Inter-relationships of the chronobiotic, melatonin, with leptin and adiponectin: implications for obesity. J Pineal Res. 2015:277–91. This revision describes the endocrine interaction between melatonin and adipokines in obesity. Chornobiology is relevant for obesity.
Tan D-X, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res. 2007;42(1):28–42.
Dominguez-Rodriguez A, Abreu-Gonzalez P. Melatonin: still a forgotten antioxidant. Int J Cardiol. 2011;149(3):382.
Poeggeler B, Reiter RJ, Tan D-X, Chen L-D, Manchester LC. Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis. J Pineal Res. 1993;14(4):151–68.
•• Baltatu OC, Amaral FG, Campos LA, Cipolla-Neto J. Melatonin, mitochondria and hypertension. Cell Mol Life Sci. 2017;74(21):3955–64. This revision is of great relevance for the topic of the present review. It describes of the several mechanisms of action of melatonin on mitochondria and highlight those relevant to hypertension.
Dwaich KH, Al-Amran FGY, Al-Sheibani BIM, Al-Aubaidy HA. Melatonin effects on myocardial ischemia-reperfusion injury: impact on the outcome in patients undergoing coronary artery bypass grafting surgery. Int J Cardiol. 2016;221:977–86.
Diez ER, Renna NF, Prado NJ, Lembo C, Ponce Zumino AZ, Vazquez-Prieto M, et al. Melatonin, given at the time of reperfusion, prevents ventricular arrhythmias in isolated hearts from fructose-fed rats and spontaneously hypertensive rats. J Pineal Res. 2013;55(2):166–73.
Russcher M, Koch B, Nagtegaal E, van der Putten K, ter Wee P, Gaillard C. The role of melatonin treatment in chronic kidney disease. Front Biosci (Landmark Ed). 2012;17:2644–56.
Mauriz JL, Collado PS, Veneroso C, Reiter RJ, González-Gallego J. A review of the molecular aspects of melatonin’s anti-inflammatory actions: recent insights and new perspectives. J Pineal Res. 2013;54(1):1–14.
García JA, Volt H, Venegas C, Doerrier C, Escames G, López LC, et al. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-α and blocks the septic response in mice. FASEB J. 2015;29(9):3863–75.
Favero G, Franceschetti L, Bonomini F, Rodella LF, Rezzani R. Melatonin as an anti-inflammatory agent modulating Inflammasome activation. Int J Endocrinol. 2017;2017:1835195.
Touitou Y, Touitou D, Reinberg A. Disruption of adolescents’ circadian clock: the vicious circle of media use, exposure to light at night, sleep loss and risk behaviors. J Physiol. 2016;110(4):467–79.
Geiger SS, Fagundes CT, Siegel RM. Chrono-immunology: progress and challenges in understanding links between the circadian and immune systems. Immunology. 2015;146(3):349–58.
Yaprak M, Altun A, Vardar A, Aktoz M, Ciftci S, Ozbay G. Decreased nocturnal synthesis of melatonin in patients with coronary artery disease. Int J Cardiol. 2003;89(1):103–7.
Xu W, Cai SY, Zhang Y, Wang Y, Ahammed GJ, Xia XJ, et al. Melatonin enhances thermotolerance by promoting cellular protein protection in tomato plants. J Pineal Res. 2016;61(4):457–69.
Lemmer B. Signal transduction and chronopharmacology of regulation of circadian cardiovascular rhythms in animal models of human hypertension. Heart Fail Clin. 2017;13(4):739–57.
Liu Z, Gan L, Xu Y, Luo D, Ren Q, Wu S, et al. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J Pineal Res. 2017;63(1): e12414. https://doi.org/10.1111/jpi.12414.
Antonelli M, Kushner I. It’s time to redefine inflammation. FASEB J. 2017;31(5):1787–91.
Lanaspa MA, Kuwabara M, Andres-Hernando A, Li N, Cicerchi C, Jensen T, et al. High salt intake causes leptin resistance and obesity in mice by stimulating endogenous fructose production and metabolism. Proc Natl Acad Sci. 2018;115(12):3138–43.
Erren TC, Pape HG, Reiter RJ, Piekarski C. Chronodisruption and cancer. Naturwissenschaften. 2008;95(5):367–82.
Erren TC, Grob JV. Civil time ≠ biological time: recent options for empirically testing possible effects of chronodisruption. Chronobiol Int. 2015;32(5):697–8.
Bonmati-Carrion MA, Arguelles-Prieto R, Martinez-Madrid MJ, Reiter R, Hardeland R, Rol MA, et al. Protecting the melatonin rhythm through circadian healthy light exposure. Int J Mol Sci. 2014 Dec 17;15(12):23448–500.
De Farias TDSM, De Oliveira AC, Andreotti S, Do Amaral FG, Chimin P, De Proença ARA, et al. Pinealectomy interferes with the circadian clock genes expression in white adipose tissue. J Pineal Res. 2015;58(3):251–61.
Brady TM. The role of obesity in the development of left ventricular hypertrophy among children and adolescents. Curr Hypertens Rep. 2016;18(1):3.
Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116(6):991–1006.
Kawarazaki W, Fujita T. The role of aldosterone in obesity-related hypertension. Am J Hypertens. 2016;29(4):415–23.
Zamora E, Lupón J, de Antonio M, Urrutia A, Coll R, Díez C, et al. The obesity paradox in heart failure: is etiology a key factor? Int J Cardiol. 2013;166(3):601–5.
Ríos-Lugo MJ, Cano P, Jiménez-Ortega V, Fernández-Mateos MP, Scacchi PA, Cardinali DP, et al. Melatonin effect on plasma adiponectin, leptin, insulin, glucose, triglycerides and cholesterol in normal and high fat-fed rats. J Pineal Res. 2010;49(4):342–8.
Cardinali DP, Cano P, Jiménez-Ortega V, Esquifino AI. Melatonin and the metabolic syndrome: physiopathologic and therapeutical implications. Neuroendocrinology. 2011;93(3):133–42.
Jiménez-Aranda A, Fernández-Vázquez G, Campos D, Tassi M, Velasco-Perez L, Tan D-X, et al. Melatonin induces browning of inguinal white adipose tissue in Zucker diabetic fatty rats. J Pineal Res. 2013;55(4):416–23. https://doi.org/10.1111/jpi.12089.
Reiter RJ, Tan DX, Korkmaz A, Ma S. Obesity and metabolic syndrome: association with chronodisruption, sleep deprivation, and melatonin suppression. Ann Med. 2012;44(6):564–77.
Yang Y, Duan W, Jin Z, Yi W, Yan J, Zhang S, et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55(3):275–86.
Lim H-D, Kim Y-S, Ko S-H, Yoon I-J, Cho S-G, Chun Y-H, et al. Cytoprotective and anti-inflammatory effects of melatonin in hydrogen peroxide-stimulated CHON-001 human chondrocyte cell line and rabbit model of osteoarthritis via the SIRT1 pathway. J Pineal Res. 2012;53(3):225–37.
Bartness TJ, Liu Y, Shrestha YB, Ryu V. Neural innervation of white adipose tissue and the control of lipolysis. Front Neuroendocrinol. 2014 Oct;35(4):473–93.
Patel SR, Hu FB. Short sleep duration and weight gain: a systematic review. Obesity (Silver Spring). 2008;16(3):643–53.
Chaput JP. Is sleep deprivation a contributor to obesity in children? Eat Weight Disord. 2016 Mar;21(1):5–11.
Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc Natl Acad Sci. 2013;110(14):5695–700.
Ganie SA, Dar TA, Bhat AH, Dar KB, Anees S, Zargar MA, et al. Melatonin: a potential anti-oxidant therapeutic agent for mitochondrial dysfunctions and related disorders. Rejuvenation Res. 2016;19(1):21–40.
Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27(2):105–17.
Stacchiotti A, Favero G, Giugno L, Lavazza A, Reiter RJ, Rodella LF, et al. Mitochondrial and metabolic dysfunction in renal convoluted tubules of obese mice: protective role of melatonin. Ashton N, editor. PLoS One. 2014;9(10):e111141.
Carraro RS, Souza GF, Solon C, Razolli DS, Chausse B, Barbizan R, et al. Hypothalamic mitochondrial abnormalities occur downstream of inflammation in diet-induced obesity. Mol Cell Endocrinol. 2018;460:238–45.
Dietrich MO, Liu Z-W, Horvath TL. Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell. 2013;155(1):188–99.
Baltatu O, Afeche SC. José dos Santos SH, Campos LA, Barbosa R, Michelini LC, et al. locally synthesized angiotensin modulates pineal melatonin generation. J Neurochem. 2002;80(2):328–34.
Campos LA, Cipolla-Neto J, Amaral FG, Michelini LC, Bader M, Baltatu OC. The angiotensin-melatonin Axis. Int J Hypertens. 2013;2013:1–7.
Vaidya A, Sun B, Larson C, Forman JP, Williams JS. Vitamin D3 therapy corrects the tissue sensitivity to angiotensin II akin to the action of a converting enzyme inhibitor in obese hypertensives: an interventional study. J Clin Endocrinol Metab. 2012;97(7):2456–65.
Ferder M, Inserra F, Manucha W, Ferder L. The world pandemic of vitamin D deficiency could possibly be explained by cellular inflammatory response activity induced by the renin-angiotensin system. AJP Cell Physiol. 2013;304(11):C1027–39.
García IM, Altamirano L, Mazzei L, Fornés M, Cuello-Carrión FD, Ferder L, et al. Vitamin D receptor-modulated Hsp70/AT1 expression may protect the kidneys of SHRs AT the structural and functional levels. Cell Stress Chaperones. 2014;19(4):479–91.
Garcia IM, Altamirano L, Mazzei L, Fornes M, Molina MN, Ferder L, et al. Role of mitochondria in paricalcitol-mediated cytoprotection during obstructive nephropathy. AJP Ren Physiol. 2012;302(12):F1595–605.
Calton EK, Keane KN, Soares MJ. The potential regulatory role of vitamin D in the bioenergetics of inflammation. Curr Opin Clin Nutr Metab Care. 2015;18(4):367–73.
Haneklaus M, O’Neill LAJ. NLRP3 at the interface of metabolism and inflammation. Immunol Rev. 2015;265(1):53–62.
Kim M-J, Yoon J-H, Ryu J-H. Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep. 2016;49(10):529–35.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–89.
Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I, et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. 2013;54(9):2423–36.
Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7(2):99–109.
Salminen A, Ojala J, Kaarniranta K, Kauppinen A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci. 2012;69(18):2999–3013.
Whaley-Connell A, Sowers JR. Obesity and kidney disease: from population to basic science and the search for new therapeutic targets. Kidney Int. 2017 Aug;92(2):313–23.
Ishigaki S, Ohashi N, Isobe S, Tsuji N, Iwakura T, Ono M, et al. Impaired endogenous nighttime melatonin secretion relates to intrarenal renin-angiotensin system activation and renal damage in patients with chronic kidney disease. Clin Exp Nephrol. 2016;20(6):878–84.
Hussein MR, Ahmed OG, Hassan AF, Ahmed MA. Intake of melatonin is associated with amelioration of physiological changes, both metabolic and morphological pathologies associated with obesity: an animal model. Int J Exp Pathol. 2007;88(1):19–29.
Winiarska K, Focht D, Sierakowski B, Lewandowski K, Orlowska M, Usarek M. NADPH oxidase inhibitor, apocynin, improves renal glutathione status in Zucker diabetic fatty rats: a comparison with melatonin. Chem Biol Interact. 2014;218:12–9.
Molina MN, Ferder LL, Manucha W. Emerging role of nitric oxide and heat shock proteins in insulin resistance. Curr Hypertens Rep. 2016;18(1):1–13.
Sartori C, Dessen P, Mathieu C, Monney A, Bloch J, Nicod P, et al. Melatonin improves glucose homeostasis and endothelial vascular function in high-fat diet-fed insulin-resistant mice. Endocrinology. 2009;150(12):5311–7.
Motawi TK, Ahmed SA, AM Hamed, El-Maraghy SA, MW Aziz. Melatonin and/or rowatinex attenuate streptozotocin-induced diabetic renal injury in rats. J Biomed Res. 2017. https://doi.org/10.7555/JBR.31.20160028.
Fujimoto E, Imai A, Utsuyama M, Sato K. Effects of in vitro heat shock on immune cells in diet-induced obese mice. J Therm Biol. 2017;69:124–31.
Mazzei L, Docherty NG, Manucha W. Mediators and mechanisms of heat shock protein 70 based cytoprotection in obstructive nephropathy. Cell Stress Chaperones. 2015 Nov;20(6):893–906.
Fan ACY, Young JC. Function of cytosolic chaperones in Tom70-mediated mitochondrial import. Protein Pept Lett. 2011;18(2):122–31.
Santos JM, Kowluru RA. Impaired transport of mitochondrial transcription factor A (TFAM) and the metabolic memory phenomenon associated with the progression of diabetic retinopathy. Diabetes Metab Res Rev. 2013;29(3):204–13.
Wang Y, Katayama A, Terami T, Han X, Nunoue T, Zhang D, et al. Translocase of inner mitochondrial membrane 44 alters the mitochondrial fusion and fission dynamics and protects from type 2 diabetes. Metabolism. 2015;64(6):677–88.
Ding M, Ning J, Feng N, Li Z, Liu Z, Wang Y, et al. Dynamin-related protein 1-mediated mitochondrial fission contributes to post-traumatic cardiac dysfunction in rats and the protective effect of melatonin. J Pineal Res. 2018;64(1):e12447. https://doi.org/10.1111/jpi.12447.
Hrenak J, Paulis L, Repova K, Aziriova S, Nagtegaal EJ, Reiter RJ, et al. Melatonin and renal protection: novel perspectives from animal experiments and human studies (review). Curr Pharm Des. 2015;21(7):936–49.
Simko F. Chronobiology of blood pressure: emerging implications of melatonin. Eur J Clin Investig. 2012 Nov;42(11):1252–4.
Zanoboni A, Zanoboni-Muciaccia W. Experimental hypertension in pinealectomized rats. Life Sci. 1967;6(21):2327–31.
Reiter RJ, Tan DX, Korkmaz A. The circadian melatonin rhythm and its modulation: possible impact on hypertension. J Hypertens Suppl. 2009 Aug;27(6):S17–20.
Obayashi K, Saeki K, Iwamoto J, Okamoto N, Tomioka K, Nezu S, et al. Nocturnal urinary melatonin excretion is associated with non-dipper pattern in elderly hypertensives. Hypertens Res. 2013;36(8):736–40.
Fabbian F, Smolensky MH, Tiseo R, Pala M, Manfredini R, Portaluppi F. Dipper and non-dipper blood pressure 24-hour patterns: circadian rhythm-dependent physiologic and pathophysiologic mechanisms. Chronobiol Int. 2013;30(1–2):17–30.
Jonas M, Garfinkel D, Zisapel N, Laudon M, Grossman E. Impaired nocturnal melatonin secretion in non-dipper hypertensive patients. Blood Press. 2003;12(1):19–24.
Hermida RC, Ayala DE, Fernández JR, Mojón A, Crespo JJ, Ríos MT, et al. Bedtime blood pressure chronotherapy significantly improves hypertension management. Heart Fail Clin. 2017;13(4):759–73.
Benova M, Herichova I, Stebelova K, Paulis L, Krajcirovicova K, Simko F, et al. Effect of L-NAME-induced hypertension on melatonin receptors and melatonin levels in the pineal gland and the peripheral organs of rats. Hypertens Res. 2009;32(4):242–7.
Schepelmann M, Molcan L, Uhrova H, Zeman M, Ellinger I. The presence and localization of melatonin receptors in the rat aorta. Cell Mol Neurobiol. 2011;31(8):1257–65.
Masana MI, Doolen S, Ersahin C, Al-Ghoul WM, Duckles SP, Dubocovich ML, et al. MT(2) melatonin receptors are present and functional in rat caudal artery. J Pharmacol Exp Ther. 2002;302(3):1295–302.
Tunstall RR, Shukla P, Grazul-Bilska A, Sun C, O’Rourke ST. MT<inf>2</inf> receptors mediate the inhibitory effects of melatonin on nitric oxide-induced relaxation of porcine isolated coronary arteries. J Pharmacol Exp Ther. 2011;336(1):127-33. https://doi.org/10.1124/jpet.110.174482.
Yang Q, Scalbert E, Delagrange P, Vanhoutte PM, O’Rourke ST. Melatonin potentiates contractile responses to serotonin in isolated porcine coronary arteries. Am J Physiol Heart Circ Physiol. 2001;280(1):H76–82.
Pandi-Perumal SR, Trakht I, Srinivasan V, Spence DW, Maestroni GJM, Zisapel N, et al. Physiological effects of melatonin: role of melatonin receptors and signal transduction pathways. Prog Neurobiol. 2008;85(3):335–53.
Koziróg M, Poliwczak AR, Duchnowicz P, Koter-Michalak M, Sikora J, Broncel M. Melatonin treatment improves blood pressure, lipid profile, and parameters of oxidative stress in patients with metabolic syndrome. J Pineal Res. 2011;50(3):261–6.
van Marke de Lumen K, Kedziora-Kornatowska K, Czuczejko J, Szewczyk-Golec K, Pawluk H, Motyl J, et al. Time dependent effect of melatonin administration on lipid peroxidation, superoxide dismutase activity and melatonin concentration in the elderly patients with essential arterial hypertension. Przegl Lek. 2008;65(6):273–6.
Tomas-Zapico C, Coto-Montes A. A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J Pineal Res. 2005;39(2):99–104.
Pechánová O, Zicha J, Paulis L, Zenebe W, Dobesová Z, Kojsová S, et al. The effect of N-acetylcysteine and melatonin in adult spontaneously hypertensive rats with established hypertension. Eur J Pharmacol. 2007;561(1–3):129–36.
Waki H, Gouraud SS, Maeda M, Raizada MK, Paton JFR. Contributions of vascular inflammation in the brainstem for neurogenic hypertension. Respir Physiol Neurobiol. 2011;178(3):422–8.
Waki H, Gouraud SS, Maeda M, Paton JFR. Specific inflammatory condition in nucleus tractus solitarii of the SHR: novel insight for neurogenic hypertension? Auton Neurosci. 2008;142(1–2):25–31.
Hardeland R, Cardinali DP, Srinivasan V, Spence DW, Brown GM, Pandi-Perumal SR. Melatonin-a pleiotropic, orchestrating regulator molecule. Prog Neurobiol. 2011 Mar;93(3):350–84.
Pevet P, Challet E. Melatonin: both master clock output and internal time-giver in the circadian clocks network. J Physiol Paris. 2011 Dec;105(4–6):170–82.
Nava M, Quiroz Y, Vaziri N, Rodriguez-Iturbe B. Melatonin reduces renal interstitial inflammation and improves hypertension in spontaneously hypertensive rats. Am J Physiol Renal Physiol. 2003;284(3):F447–54.
Quiroz Y, Ferrebuz A, Romero F, Vaziri ND, Rodriguez-Iturbe B. Melatonin ameliorates oxidative stress, inflammation, proteinuria, and progression of renal damage in rats with renal mass reduction. Am J Physiol Ren Physiol. 2008;294(2):F336–44.
Cheng MC, Wu TH, Huang LT, Tain YL. Renoprotective effects of melatonin in young spontaneously hypertensive rats with L-NAME. Pediatr Neonatol. 2014;55(3):189–95.
Qiao YF, Guo WJ, Li L, Shao S, Qiao X, Shao JJ, et al. Melatonin attenuates hypertension-induced renal injury partially through inhibiting oxidative stress in rats. Mol Med Rep. 2016;13(1):21–6.
Chen KH, Chen CH, Wallace CG, Chen YT, Yang CC, Sung PH, et al. Combined therapy with melatonin and exendin-4 effectively attenuated the deterioration of renal function in rat cardiorenal syndrome. Am J Transl Res. 2017;9(2):214–29.
Tain YL, Lee CT, Chan JYH, Hsu CN. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal NG-Nitro-L-arginine-methyl ester (L-NAME)-induced fetal programming of hypertension in adult male offspring. Am J Obstet Gynecol. 2016;215(5):636.e1–636.e72.
Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Investig. 2009;119(3):524–30.
Chen Y, Qing W, Sun M, Lv L, Guo D, Jiang Y. Melatonin protects hepatocytes against bile acid-induced mitochondrial oxidative stress via the AMPK-SIRT3-SOD2 pathway. Free Radic Res. 2015;49(10):1275–84.
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript writing. The authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Natalia Prado, León Ferder, Walter Manucha, and Emiliano Diez declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of the Topical Collection on Hypertension and Obesity
Rights and permissions
About this article

Cite this article
Prado, N.J., Ferder, L., Manucha, W. et al. Anti-Inflammatory Effects of Melatonin in Obesity and Hypertension. Curr Hypertens Rep 20, 45 (2018). https://doi.org/10.1007/s11906-018-0842-6
Published:
DOI: https://doi.org/10.1007/s11906-018-0842-6