
Abstract
Purpose of Review
Conventional risk stratification algorithms that rely on age, clustered phenotypic traits, and biomarkers under-recognize the sizeable subgroup of individuals at high polygenic risk for atherosclerotic cardiovascular disease (ASCVD). This review provides perspective on the promising role of genetic testing in cardiovascular prevention through the lens of lipid metabolism.
Recent Findings
Recent advances in cardiovascular genetics identified a number of common and rare variants affecting ASCVD risk. This genetic susceptibility can be assessed by polygenic risk scores (PRS) which quantify risk conferred by the cumulative impact of common variants. This results in a normally distributed spectrum of risk for coronary artery disease that is present at birth and amplifies the effects of modifiable risk factors including lipids.
Summary
Polygenic risk is a significant determinant of ASCVD risk that is below the discrimination level of conventional guideline-based clinical frameworks. Genetic risk scores thus hold potential to refine phenotypic screening in cardiovascular prevention, identify subsets of the population that might derive particular benefit from early lifestyle and pharmaceutical interventions, and guide treatment eligibility. This might pave the way to personalized prevention aimed at reducing the unacceptable global burden of ASCVD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Despite major technological and pharmacological advances in cardiovascular medicine, atherosclerosis and its clinical manifestations remain the leading causes of death worldwide. Atherosclerosis is a multifactorial, systemic process that progresses at various rates in many vascular territories. It is a consequence of genetic predisposition and exogenous risk factors, whose cumulative effects become evident in the arterial wall over a lifetime and are best reflected by chronological aging, the strongest “risk factor” [1,2,3]. Acute clinical manifestations of atherosclerotic cardiovascular disease (ASCVD) are triggered by subsets of atherosclerotic lesions [4]. Such lesions are often found in individuals with a genetic disposition and in distinct metabolic phenotypes characterized by metabolic and inflammatory processes that are promoted by ectopic adiposity and associated high-risk traits [5,6,7]. Thus, numerous exposures, heritable [8] and acquired [2], and their interplay (epigenetics, i.e., genome-exposome interactions) [9••] modulate a given individual’s risk of developing atherosclerotic cardiovascular disease (ASCVD).
New technologies have significantly advanced our understanding of the underpinnings of the genetic architecture of ASCVD [10]. Common and rare genetic variants trigger causal pathways involving low-density lipoproteins (LDL), triglyceride-rich lipoproteins (TRL), lipoprotein(a) [Lp(a)], blood pressure, inflammation, and endothelial function including nitric oxide signaling [11,12,13]. Dyslipidemias are among the most extensively studied of the genetic exposures that underlie increased heritable susceptibility for ASCVD. In this regard, the retention and accumulation of apolipoproteinB-containing particles (apoB) within the arterial intima are the fundamental step that initiates the development of atherosclerotic lesions and in large part drive the inflammatory response [14,15,16,17]. The rate of progression of atherosclerosis is largely determined by the concentration of apoB particles within the arterial lumen over a given period of time, i.e., the cumulative exposure. In conjunction with insights from observational epidemiology, genetics [18], and clinical intervention studies [14, 15], current treatment guidelines strongly encourage lowering of apoB, including very low-density lipoproteins (VLDL), intermediate density lipoproteins (IDL), low-density lipoproteins (LDL), and Lp(a) as a major cornerstone in cardiovascular prevention [19].
While the paradigm “the lower, the better” has been broadly adopted for treating LDL particle-associated risk [19], the conceptual model of risk by lifetime exposure awaits clinical implication. Indeed, fatty streaks are initiated with apoB retention at a young age, suggesting that preventive measures should be advised prior to disease manifestation. Regarding the obvious request for “the earlier, the better,” it becomes the central question who benefits from such long-term intervention. Genetics, which are in place (and can be determined) already at birth, may help to guide such recommendations. Likewise, it is evident that acquired as well as inherited risk factors act multiplicatively [20, 21]. Specifically, a person at extreme risk for one, e.g., inherited risk factor experiences larger benefits from treatment of (another) modifiable risk factor, supporting the paradigm “the broader, the better” as depicted in Fig. 1 [22, 23]. The latter concepts, however, receive limited attention by our current risk stratification approaches which thereby fail to identify at young age a sizeable proportion of individuals at high subsequent risk for cardiovascular events [5]. This results in missed opportunities for primordial prevention by intensification of lifestyle intervention and/or pharmacotherapy in high-risk individuals.

Key concepts with respect to diagnosis and prevention of ASCVD risk. True prevention requires intervention before any disease manifestation. This may be achieved by implementing genetics as a diagnostic tool which allows “earlier” treatment. The multiplicative effects of CV risk factors and genetic disposition ask for a “broader” approach involving neutralization of as many factors as possible. Finally, signs of disease manifestation, e.g., by imaging, ask for intensive treatment leading to “lower” levels of apoB particles in conjunction with treatment of all other modifiable risk factors (The DNA-helix is reproduced from Servier Medical Art by Servier, which is licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com/)
In this review, we aim to provide perspective on the role of genetic risk scores in the determination of treatment strategies to control dyslipidemias. We discuss the potential of genetic testing as an adjunct to standard risk factor algorithms based on phenotypic screening for detection of high-risk patients who are candidates for interventions aimed at preventing major adverse cardiovascular events. Furthermore, we outline the challenges and unmet needs that remain to be addressed before these algorithms can be introduced in the clinical setting.
Dyslipidemias and Atherosclerotic Cardiovascular Disease Risk
Lifetime Exposure Model - the Concept of Cumulative Exposure
The lifetime exposure model supposes that atherosclerosis—and by extension ASCVD risk—is a function of absolute magnitude plus cumulative duration of exposure to apoB in conjunction with all other risk factors [14, 15, 24]. This model is based on evidence from clinical intervention trials, genome-wide association studies (GWAS), and natural experiments that collectively suggest that the clinical benefit is proportional to the absolute magnitude and the duration of apoB and/or LDL-C reduction, in context with other measures of ASCVD risk [24, 25].
Limitations of Current Risk Calculators
There are major limitations to the conventional paradigm of using calculated risk for assessing cardiovascular risk, and by extension, selection of individuals for preventive therapies.
The major determinant of risk to suffer from an event within 5–10 years in current risk calculators is age. Consistent with this notion, the majority of those eligible for primary prevention of ASCVD on the basis of risk calculators are aged > 60 years [26]. However, almost 50% of cardiovascular events in men and almost one-third in women occur before the age of 60 years [26], indicating disadvantages for those who are young. Second, interference with the chronic process leading to ASCVD implies the necessity for early—i.e., preventive—treatment, which is missed if young people fail to qualify because their short-term risk passes defined thresholds only later in life. Third, conventional strategies for assessing CAD risk and for guiding statin eligibility in cardiovascular prevention do not entirely capture genetic susceptibility to CAD [27]. Aragam and colleagues recently demonstrated that polygenic risk for ASCVD acts largely independently of the ACC/AHA Pooled Cohort Equations used to calculate 10-year risk. Thus, current guideline-based clinical risk assessment algorithms do not optimally reflect risk [27], particularly in those who are young and at high genetic risk.
Potential Role of Genetics in Lipid Metabolism
In the past years, large-scale genetic studies led to the discovery of more than two hundred genomic variants which are causally associated with CAD risk [28,29,30,31,32,33,34]. While approximately half of the variants are located within chromosomal regions that harbor genes which do not have a known role in the pathophysiology of the ASCVD, a considerable amount could be attributed to inflammatory pathways, NO-cGMP signaling, or traditional risk factors, e.g., blood pressure or lipid metabolism. For an overview, see [11,12,13]. Despite the development of novel therapeutic strategies, findings from such studies are expected to tailor treatment strategies and improve risk stratification [35, 36]. Indeed, risk stratification of individuals based on inherited DNA variation compared with phenotypic screening has demonstrated clinical superiority for more accurate risk estimation for the reasons outlined below [37, 38].
First, serial lipid measurements demonstrated in carriers of mutations typical for familial hypercholesterolemia (FH) a higher risk than in people with similar LDL-C levels lacking such mutations. An explanation may be a higher exposure to LDL-C in early age in mutation carriers as compared with non-carriers [39]. There is emerging evidence that this concept might also hold true for elevated levels of triglycerides as damaging mutations in the lipoprotein lipase (LPL) gene have been shown to be significantly associated with CAD risk [40].
Second, certain FH genotypes seem to confer a greater risk of CAD than others. Abul-Husn and colleagues found that increased odds of premature CAD in FH mutation carriers were most pronounced in carriers of LDL-receptor (LDLR) loss-of-function variants [41].
Third, the reliance on LDL-C levels for identification of high-risk patients and/or cascade screening may be inaccurate because there is substantial overlap between genotype-positive and genotype-negative individuals, and clinical criteria may no longer be present in patients treated with lipid lowering medication [41, 42].
Fourth, in certain jurisdictions, genetic testing is required for determining eligibility for insurance coverage of costly pharmaceutical agents such as PCSK9 inhibitors [43].
Genetics of Cardiovascular Diseases Through the Lens of Genetics of Lipid Metabolism
The Monogenic Model
The genetic basis of ASCVD risk has been largely constrained to carriers of rare monogenic mutations. Regarding lipids, truncating mutations in one of five genes confer a markedly increased risk for premature ASCVD: low-density lipoprotein receptor (LDLR) [44,45,46], lipoprotein lipase (LPL) [40], Apolipoprotein A5 (APOA5) [46], Apolipoprotein(a) (LPA) [47], and proprotein convertase subtilisin/kexin type 9 (PCSK9) [48]. These mutations are typically associated with a distinct lipid phenotype exposing markedly elevated LDL-C (LDLR, PCSK9), triglycerides (LPL, APOA5), and Lp(a) (LPA), respectively.
FH is the most common monogenic disease affecting humankind, with a prevalence of approximately 1 in 300 in the general population and an up to 10-, 20-, and 23-fold higher prevalence in certain subgroups with ischemic heart disease (IHD), premature IHD, and severe hypercholesterolemia [49]. This autosomal dominant genetic disorder is largely caused by pathogenic mutations in genes that encode proteins involved in the process of LDL particle clearance (LDLR, APOB, PCSK9) [50]. As a result of the molecular defects, carriers are exposed to lifelong high levels of apoB resulting in substantially elevated risk for premature ASCVD [39].
The Polygenic Model
Interestingly, in individuals with an LDL-C ≥ 190 mg/dl, FH mutations were found in less than 2% [39]. Similarly, only 4% of individuals with myocardial infarction (MI) at an early age (defined as ≤ 50 years in males and ≤ 60 years in females) carried a monogenic mutation [46]. Thus, from a population perspective, monogenic causes account for a relatively small proportion of genetic risk. This raises concerns that a meaningful number of individuals might not be identified by screening strategies focused on mutations in the protein coding sequence [51].
Genetic susceptibility to common complex diseases, including CAD, seems to be largely determined by many common DNA variants (i.e., single-nucleotide polymorphisms, SNPs) of small effect size [11, 32, 52].
While in monogenic diseases, usually a strong correlation between risk genotype and risk phenotype exists, blood biomarkers are typically unremarkable in polygenic disorders. As a consequence, while carriers of monogenic disorders are often detected clinically (based on their typical phenotype such as elevated LDL-C in, e.g., FH), individuals with high polygenic at risk remain unaware of their risk [11].
A high polygenic risk score (PRS) in contrast reflects the combined effect of various disease pathways rather than one single disease mechanism [52] and may thus more accurately reflect the complexity of common complex diseases. This genetic susceptibility risk can be assessed by PRS which quantify the risk conferred by the cumulative impact of common variants resulting in a single, normally distributed quantitative risk factor for CAD that is available at birth [11, 52]. Since the first genome-wide linkage [53] and association studies (GWAS) for CAD in 2002 and 2007 [29], technological advances have allowed for investigation of progressively larger sample sizes. This has paved the way to the discovery of more loci with genome-wide significant associative p < 5 × 10−8 for CAD [11, 12]. As mentioned above, about one in five of these loci is located near genes with confirmed roles in lipid metabolism such as LDL, TRL, or Lp(a), highlighting the key role for these pathways in the development of CAD [11].
First attempts to study genome-wide PRS were limited in their clinical utility by insufficient discriminative accuracy and were thus not considered to be of diagnostic or predictive value as an adjunct to clinical scores [54]. Recent technological advances and improved algorithms have made it possible to develop PRS that capture the full breadth of genetic data [55••]. Taking into account the full set of common polymorphisms for risk stratification better depicts the heritability of any given trait and thus had substantially better predictive value than previously used scores in genome-wide association study analyses restricted to, e.g., the 50 most significantly associated variants [56]. Furthermore, whole-genome sequencing, which captures the complete spectrum of genetic variation, both rare and common, via simultaneous ascertainment of monogenic mutations and polygenic score for each given individual, made it possible to compare the clinical relevance of monogenic mutations versus polygenic risk on premature CAD [10].
In participants of primarily European ancestry from UK Biobank genome-wide PRS for myocardial infarction, comprising a genome-wide set of 6.6 million common DNA variants identified 8% of the population at greater than threefold increased risk for CAD [55••]. It is worth noting that the PRS thus identified 20-fold more individuals at equal or greater risk compared with the carrier frequency of rare monogenic mutations such as FH [55••].
These data were validated by Khera and colleagues who simultaneously assessed the contribution of monogenic and polygenic models in a population encompassing four ethnic subgroups hospitalized in the USA with premature CAD. They reported that 1.7% of patients with early-onset myocardial infarction at an age of 55 years or younger displayed high monogenic risk but 17.3% presented a high PRS. The level of risk increase was comparable with monogenic and polygenic risk (3.8-fold and 3.7-fold, respectively). However, a tenfold higher number of individuals with premature CAD were identified by a high PRS compared with the monogenic model [10].
Complementing these findings, Aragam et al. reported that using a PRS as a CAD risk enhancer to inform statin eligibility in the subgroup in whom guideline-based recommendations were unclear would result in 1 in 25 primary prevention patients being newly suitable for a statin prescription [27].
The Genetic Basis for Resistance
In contrast to genetic variation leading to increased risk, protective mutations reduce susceptibility to a given disease. The discovery of protective mutations in the LDL [57, 58] and the TRL pathway [59, 60] has complemented preclinical prioritization strategies in developing lipid lowering therapies. This has guided the development of new pharmaceutical agents by providing confidence that these targets were causal for CAD. With regard to the LDL pathway, variants that permanently lower LDL-C confer protection against ASCVD, predicting that pharmaceutical agents which mimic those mutations would work in clinic [57, 58]. These genetic insights have been resembled by the development of ezetimibe, an inhibitor of the Niemann-Pick C1-like protein 1 (NPC1L1), and PCSK9 inhibitors which have proven to be effective in lowering LDL and patient-relevant endpoints in ASCVD [61,62,63]. Regarding TRL, their rate of lipolysis, which is largely determined by lipoprotein lipase (LPL), might be a key pathway associated with ASCVD. Rare mutations that disrupt functioning of ApoC3, ANGPTL3, and ANGPTL4—all three of which are endogenous regulators of LPL, the key enzyme that controls lipolysis and the ability to clear dietary triglycerides—are associated with lower levels of plasma triglycerides and carriers of these mutations expose a lower risk of ASCVD [40, 59, 64]. Pharmaceuticals that mimic these protective mutations are currently under way [65, 66].
Is Genetic Risk for ASCVD Modifiable by Early Intervention?
Directing clinical effort at early identification of individuals at risk is relevant if safe and efficacious interventions are to be offered that reduce risk. Encouragingly, data has consistently demonstrated that heritable risk for CAD can be substantially modified by either adherence to a healthy lifestyle and/or pharmacotherapy. Importantly, absolute risk reduction by these measures is more pronounced in individuals at high genetic risk.
Lifestyle Interventions
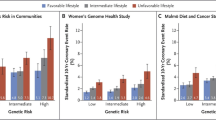
An analysis of data from 55,685 participants from the Atherosclerosis Risk in Communities study, the Women’s Genome Health Study, the Malmö Diet and Cancer Study, and cross-sectional BioImage Study demonstrated that a healthy lifestyle (defined as no current smoking, no obesity, regular physical activity, and a healthy diet) offsets about 50% of the inherited risk. More specifically, in individuals exposing a high polygenic risk for CAD and an unfavorable lifestyle, the 10-year event rate was 11%. Conversely, in individuals with a high polygenic risk and a favorable lifestyle, the 10-year event rate was only 5% [67••]. Taken together, these data provide evidence that genes are not destiny and that raising consciousness for heritable risk in subgroups at high genetic risk might open a huge therapeutic window for preventive measures aimed at attenuating ASCVD risk.
Pharmacotherapy
Randomized clinical trials demonstrated that statin therapy is an efficacious and safe strategy to lower ASCVD event rates [68]. In secondary prevention, individuals at high genetic risk, who were defined as the subgroup at the top quintile of a 27-single-nucleotide polymorphism (SNP) PRS for CHD, derived substantially greater relative risk reduction from statin therapy compared with the remaining 80% [20]. This finding was confirmed in three primary prevention trials: ASCOT-LLA [69], JUPITER [70], and WOSCOPS [71], where higher relative benefit from lipid lowering statin therapy was observed in those at high genetic risk compared with all others despite similar achieved LDL-C lowering. Moreover, a PRS—including more than 6 million SNPs—was strongly associated with CAD risk in a recent study: the quintile of individuals with the highest PRS was at 1.9-fold odds of CAD compared with the remainder of the population. Of note, this risk increase is comparable with that of risk increasing factors other than diabetes mellitus or hypercholesterolemia as described in cardiovascular disease prevention guidelines mandating initiation of statin therapy. However, the authors found that the 20% of individuals with high PRS did not display higher statin prescription rates indicating that the use of PRS in clinical practice might identify individuals at high risk which are not captured by current “traditional” risk increasing factors but might largely benefit from statin treatment [27].
Evidence from the two large clinical outcome trials with Alirocumab and Evolocumab (ODYSSEY OUTCOMES [22] and the FOURIER [23], respectively) paints a similar picture. In both trials, a high PRS for CAD, independent of clinical risk such as baseline LDL-C and other known risk factors, (i) identified higher-risk individuals and (ii) predicted a larger absolute and relative risk reduction with PCSK9-i treatment in this subgroup [22, 23].
Genetic Testing and Precision Medicine – Ready for Prime Time?
The rationale for genetic testing for informing decision-making has several layers of implications for preventive cardiology.
First, PRS identify approximately 10–20 times as many patients at elevated risk for CAD than rare monogenic mutations [10, 55]. Importantly, these individuals with risk comparable with that of a monogenic mutation cannot reliably be identified by any circulating biomarker. Second, PRS for CAD provide a quantitative number of risk available at birth, long before discriminative capacity emerges for risk factors currently used to predict CAD [55••]. Third, polygenic risk amplifies risk conferred by traditional risk factors such as hypertension or dyslipidemia. Collectively, PRS capture incremental information to current risk stratification algorithms. The introduction of PRS might thus pave the way to precision medicine approaches with broad relevance for improved identification and personalized treatment of high-risk subgroups [52]. If brought to attention of affected individuals early in life, awareness of their inherited susceptibility as well as information about possibilities to modify risk may guide lifestyle choices and enhance medication adherence in this subgroup [55••].
Challenges and Unmet Needs
The Introduction of PRS Is Not Without Challenges
First, lead SNPs conferring ASCVD risk are often specific to an ethnic group. Even within Western-European individuals, which have been studied most extensively in GWAS, the predictive value of polygenic risk score changes substantially across countries [72]. Thus, the additive diagnostic yield, clinical benefit, and cost-effectiveness of PRS need to be validated across different ethnicities [10] and in prospective clinical trials to determine if targeted prevention strategies will translate into a reduction of clinical endpoints in those at high polygenic risk [27].
Second, PRS reflect a continuum from very low to very high genetic burden. However, only few individuals are located at the extremes of the Gaussian curve, and there is little discrimination of risk for large parts of the population. Thus, it is challenging to determine actionable thresholds for therapeutic measures.
Third, identification of individuals at high risk yields clinical benefit only if strategies that modify risk are widely available. This includes the implementation of appropriate lifestyle measures and medical therapy if indicated.
Forth, a key issue is the allocation of financial resources to preventive cardiology. This should prompt the implementation of structured training programs that teach all dedicated skills necessary to assess and manage cardiovascular risk including (i) an understanding of all contributing factors including genetic, lifestyle, and environmental factors as well as their assessment and interpretation; (ii) the ability to communicate risk in a comprehensive way to the patient; and (iii) knowledge about risk modification via lifestyle modification or pharmacotherapy.
Fifth, ethical dilemmas between the physician’s “duty to inform” and the patient’s right “to not know” might arise. Furthermore, laws need to be passed to protect against employers and health insurance companies which might discriminate individuals due to their genetic information [43].
Sixth, even though data from natural experiments provide a strong and biologically plausible argument for the beneficial effect of lifelong low apoB [11], there are no data on the effect of lifelong lipid lowering therapy by means of pharmacotherapy. Whether lipid lowering therapy has similar effects like protective mutations is plausible but remains subject to speculation, particularly in view of the fact that off-target effects of pharmacotherapy need to be taken into account. This is even more an issue for interference of phenotypes like blood pressure or platelet function which come with a much smaller “therapeutic range” than lowering of LDL-C or Lp(a) levels. Such insecurities need to be discussed in an open and comprehensive manner with patients in a shared decision-making process by physicians with expertise in this field.
Concluding Remarks and Future Directions
The lifetime exposure model of atherosclerosis implies that even with modest reductions in cumulative exposure to a given risk factor, large absolute benefits accrue.
Polygenic risk is a prevalent, important, and significant determinant of ASCVD risk that mirrors the complex genetic architecture of common complex diseases like ASCVD. PRS provide a quantitative score for disease risk which is below the discrimination level of current risk assessment tools. It can be detected at the time of birth, long before risk factors, or clinical manifestations accrue. With regard to apoB and ASCVD risk, this serves as a conceptual model for the paradigm “the lower, the earlier, and the broader – the better” as depicted in Fig. 1 [25]. In this regard, the integration of genetic information into routine clinical management as an adjunct to current clinical risk assessment algorithms might refine clinical decision-making throughout a lifetime. Taken together, integration of genetic risk scores into conventional guideline-based clinical frameworks thus confers additive, complementary, and independent predictive information to clinical risk scores currently used in clinical practice, and early in life interpretation of the genome holds potential to identify subsets of the population that might derive particular benefit from early lifestyle and pharmaceutical interventions and guide treatment eligibility. This might pave the way to personalized prevention aimed at reducing the unacceptable global burden of ASCVD.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15(9):505–22. https://doi.org/10.1038/s41569-018-0064-2.
Lechner K, von Schacky C, McKenzie AL, Worm N, Nixdorff U, Lechner B, et al. Lifestyle factors and high-risk atherosclerosis: pathways and mechanisms beyond traditional risk factors. Eur J Prev Cardiol. 2019;27(4):394–406. https://doi.org/10.1177/2047487319869400.
Melzer D, Pilling LC, Ferrucci L. The genetics of human ageing. Nat Rev Genet. 2020;21(2):88–101. https://doi.org/10.1038/s41576-019-0183-6.
Otsuka F, Kramer MC, Woudstra P, Yahagi K, Ladich E, Finn AV, et al. Natural progression of atherosclerosis from pathologic intimal thickening to late fibroatheroma in human coronary arteries: a pathology study. Atherosclerosis. 2015;241(2):772–82. https://doi.org/10.1016/j.atherosclerosis.2015.05.011.
Lechner K, McKenzie AL, Kränkel N, Von Schacky C, Worm N, Nixdorff U, et al. High-risk atherosclerosis and metabolic phenotype: the roles of ectopic adiposity, atherogenic dyslipidemia, and inflammation. Metab Syndr Relat Disord. 2020;18(4):176–85. https://doi.org/10.1089/met.2019.0115.
Tomaniak M, Katagiri Y, Modolo R, Silva RD, Khamis RY, Bourantas CV, et al. Vulnerable plaques and patients: state-of-the-art. Eur Heart J. 2020. https://doi.org/10.1093/eurheartj/ehaa227.
Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. Jama. 2017;317(6):626–34. https://doi.org/10.1001/jama.2016.21042.
Schunkert H, von Scheidt M, Kessler T, Stiller B, Zeng L, Vilne B. Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol : official journal of the German Cardiac Society. 2018;107(Suppl 2):2–9. https://doi.org/10.1007/s00392-018-1324-1.
•• Zeng L, Talukdar HA, Koplev S, Giannarelli C, Ivert T, Gan LM, et al. Contribution of gene regulatory networks to heritability of coronary artery disease. J Am Coll Cardiol. 2019;73(23):2946–57. https://doi.org/10.1016/j.jacc.2019.03.520.
Khera Amit V, Chaffin M, Zekavat Seyedeh M, Collins Ryan L, Roselli C, Natarajan P, et al. Whole-genome sequencing to characterize monogenic and polygenic contributions in patients hospitalized with early-onset myocardial infarction. Circulation. 2019;139(13):1593–602. https://doi.org/10.1161/CIRCULATIONAHA.118.035658.
Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331–44. https://doi.org/10.1038/nrg.2016.160.
Erdmann J, Kessler T, Munoz Venegas L, Schunkert H. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018;114(9):1241–57. https://doi.org/10.1093/cvr/cvy084.
Kessler T, Vilne B, Schunkert H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med. 2016;8(7):688–701. https://doi.org/10.15252/emmm.201506174.
Boren J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2020;41:2313–30. https://doi.org/10.1093/eurheartj/ehz962.
Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38(32):2459–72. https://doi.org/10.1093/eurheartj/ehx144.
Kasikara C, Doran AC, Cai B, Tabas I. The role of non-resolving inflammation in atherosclerosis. J Clin Invest. 2018;128(7):2713–23. https://doi.org/10.1172/JCI97950.
Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. 2020;21(1):30–41. https://doi.org/10.1038/s41590-019-0548-1.
Linsel-Nitschke P, Götz A, Erdmann J, Braenne I, Braund P, Hengstenberg C, et al. Lifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease--a Mendelian randomisation study. PLoS One. 2008;3(8):e2986. https://doi.org/10.1371/journal.pone.0002986.
Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2019;41(1):111–88. https://doi.org/10.1093/eurheartj/ehz455.
Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield M, Devlin JJ, et al. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet (London, England). 2015;385(9984):2264–71. https://doi.org/10.1016/s0140-6736(14)61730-x.
Yusuf S, Hawken S, Ôunpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364(9438):937–52. https://doi.org/10.1016/S0140-6736(04)17018-9.
Damask A, Steg PG, Schwartz GG, Szarek M, Hagström E, Badimon L, et al. Patients with high genome-wide polygenic risk scores for coronary artery disease may receive greater clinical benefit from alirocumab treatment in the ODYSSEY OUTCOMES trial. Circulation. 2020;141(8):624–36. https://doi.org/10.1161/circulationaha.119.044434.
Marston NA, Kamanu FK, Nordio F, Gurmu Y, Roselli C, Sever PS, et al. Predicting benefit from evolocumab therapy in patients with atherosclerotic disease using a genetic risk score: results from the FOURIER trial. Circulation. 2020;141(8):616–23. https://doi.org/10.1161/circulationaha.119.043805.
Sniderman Allan D, Pencina M, Thanassoulis G. ApoB. Circ Res. 2019;124(10):1425–7. https://doi.org/10.1161/CIRCRESAHA.119.315019.
Ference BA, Bhatt DL, Catapano AL, Packard CJ, Graham I, Kaptoge S, et al. Association of genetic variants related to combined exposure to lower low-density lipoproteins and lower systolic blood pressure with lifetime risk of cardiovascular disease. Jama. 2019;322:1381. https://doi.org/10.1001/jama.2019.14120.
Sniderman Allan D, Thanassoulis G, Wilkins John T, Furberg Curt D, Pencina M. Sick individuals and sick populations by Geoffrey Rose: cardiovascular prevention updated. J Am Heart Assoc. 2018;7(19):e010049. https://doi.org/10.1161/JAHA.118.010049.
Aragam KG, Dobbyn A, Judy R, Chaffin M, Chaudhary K, Hindy G, et al. Limitations of contemporary guidelines for managing patients at high genetic risk of coronary artery disease. J Am Coll Cardiol. 2020;75(22):2769–80. https://doi.org/10.1016/j.jacc.2020.04.027.
Erdmann J, Grosshennig A, Braund PS, König IR, Hengstenberg C, Hall AS, et al. New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet. 2009;41(3):280–2. https://doi.org/10.1038/ng.307.
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357(5):443–53. https://doi.org/10.1056/NEJMoa072366.
Schunkert H, König IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8. https://doi.org/10.1038/ng.784.
Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, Thompson JR, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. https://doi.org/10.1038/ng.2480.
Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47(10):1121–30. https://doi.org/10.1038/ng.3396.
Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49(9):1385–91. https://doi.org/10.1038/ng.3913.
van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–43. https://doi.org/10.1161/CIRCRESAHA.117.312086.
Tragante V, Hemerich D, Alshabeeb M, Brænne I, Lempiäinen H, Patel Riyaz S, et al. Druggability of coronary artery disease risk loci. Circulation: Genomic and Precision Medicine. 2018;11(8):e001977. https://doi.org/10.1161/CIRCGEN.117.001977.
Lempiäinen H, Brænne I, Michoel T, Tragante V, Vilne B, Webb TR, et al. Network analysis of coronary artery disease risk genes elucidates disease mechanisms and druggable targets. Sci Rep. 2018;8(1):3434. https://doi.org/10.1038/s41598-018-20721-6.
•• Hall KT, Kessler T, Buring JE, Passow D, Sesso HD, Zee RYL, et al. Genetic variation at the coronary artery disease risk locus GUCY1A3 modifies cardiovascular disease prevention effects of aspirin. Eur Heart J. 2019;40(41):3385–92. https://doi.org/10.1093/eurheartj/ehz384Findings from this study suggest that in the setting of primary prevention of cardiovascular diseases, efficacy of aspirin is influenced by a common allele in guanylate cyclase GUCY1A3. While aspirin reduced CVD risk in individuals homozygous for the GUCY1A3 risk (G) allele, it increased risk in heterozygote individuals, calling for personalized prevention.
Kessler T, Wolf B, Eriksson N, Kofink D, Mahmoodi BK, Rai H, et al. Association of the coronary artery disease risk gene GUCY1A3 with ischaemic events after coronary intervention. Cardiovasc Res. 2019;115(10):1512–8. https://doi.org/10.1093/cvr/cvz015.
Khera AV, Won H-H, Peloso GM, Lawson KS, Bartz TM, Deng X, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67(22):2578–89. https://doi.org/10.1016/j.jacc.2016.03.520.
Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, König IR, et al. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374(12):1134–44. https://doi.org/10.1056/NEJMoa1507652.
Abul-Husn NS, Manickam K, Jones LK, Wright EA, Hartzel DN, Gonzaga-Jauregui C, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science (New York, NY). 2016;354(6319):aaf7000. https://doi.org/10.1126/science.aaf7000.
Besseling J, Huijgen R, Martin SS, Hutten BA, Kastelein JJ, Hovingh GK. Clinical phenotype in relation to the distance-to-index-patient in familial hypercholesterolemia. Atherosclerosis. 2016;246:1–6. https://doi.org/10.1016/j.atherosclerosis.2015.12.033.
Berberich AJ, Hegele RA. The role of genetic testing in dyslipidaemia. Pathology. 2019;51(2):184–92. https://doi.org/10.1016/j.pathol.2018.10.014.
Brown MS, Goldstein JL. Expression of the familial hypercholesterolemia gene in heterozygotes: mechanism for a dominant disorder in man. Science (New York, NY). 1974;185(4145):61–3. https://doi.org/10.1126/science.185.4145.61.
Tolleshaug H, Hobgood KK, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: multiple mutations disrupt transport and processing of a membrane receptor. Cell. 1983;32(3):941–51. https://doi.org/10.1016/0092-8674(83)90079-x.
Do R, Stitziel NO, Won HH, Jørgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–6. https://doi.org/10.1038/nature13917.
Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361(26):2518–28. https://doi.org/10.1056/NEJMoa0902604.
Abifadel M, Varret M, Rabès JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34(2):154–6. https://doi.org/10.1038/ng1161.
Beheshti SO, Madsen CM, Varbo A, Nordestgaard BG. Worldwide prevalence of familial hypercholesterolemia. J Am Coll Cardiol. 2020;75(20):2553–66. https://doi.org/10.1016/j.jacc.2020.03.057.
Kastelein JJP, Reeskamp LF, Hovingh GK. Familial hypercholesterolemia. J Am Coll Cardiol. 2020;75(20):2567–9. https://doi.org/10.1016/j.jacc.2020.03.058.
Brænne I, Kleinecke M, Reiz B, Graf E, Strom T, Wieland T, et al. Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet. 2016;24(2):191–7. https://doi.org/10.1038/ejhg.2015.100.
Khera AV, Kathiresan S. Is coronary atherosclerosis one disease or many? Setting realistic expectations for precision medicine. Circulation. 2017;135(11):1005–7. https://doi.org/10.1161/circulationaha.116.026479.
Broeckel U, Hengstenberg C, Mayer B, Holmer S, Martin LJ, Comuzzie AG, et al. A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet. 2002;30(2):210–4. https://doi.org/10.1038/ng827.
Ripatti S, Tikkanen E, Orho-Melander M, Havulinna AS, Silander K, Sharma A, et al. A multilocus genetic risk score for coronary heart disease: case-control and prospective cohort analyses. Lancet (London, England). 2010;376(9750):1393–400. https://doi.org/10.1016/s0140-6736(10)61267-6.
•• Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24. https://doi.org/10.1038/s41588-018-0183-z Findings from this study show that polygenic risk scores (PRS) identify approximately 10–20 times as many patients at comparable risk for CAD as rare monogenic mutations.
Evans DM, Visscher PM, Wray NR. Harnessing the information contained within genome-wide association studies to improve individual prediction of complex disease risk. Hum Mol Genet. 2009;18(18):3525–31. https://doi.org/10.1093/hmg/ddp295.
Stitziel NO, Won HH, Morrison AC, Peloso GM, Do R, Lange LA, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371(22):2072–82. https://doi.org/10.1056/NEJMoa1405386.
Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354(12):1264–72. https://doi.org/10.1056/NEJMoa054013.
Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. https://doi.org/10.1056/NEJMoa1307095.
Musunuru K, Kathiresan S. Cardiovascular endocrinology: is ANGPTL3 the next PCSK9? Nat Rev Endocrinol. 2017;13(9):503–4. https://doi.org/10.1038/nrendo.2017.88.
Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–22. https://doi.org/10.1056/NEJMoa1615664.
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379(22):2097–107. https://doi.org/10.1056/NEJMoa1801174.
Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372(25):2387–97. https://doi.org/10.1056/NEJMoa1410489.
Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374(12):1123–33. https://doi.org/10.1056/NEJMoa1510926.
Graham MJ, Lee RG, Brandt TA, Tai L-J, Fu W, Peralta R, et al. Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med. 2017;377(3):222–32. https://doi.org/10.1056/NEJMoa1701329.
Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373(5):438–47. https://doi.org/10.1056/NEJMoa1400283.
•• Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375(24):2349–58. https://doi.org/10.1056/NEJMoa1605086Findings from this study demonstrate that among participants at high genetic risk for CAD, a healthy lifestyle (defined as no current smoking, no obesity, regular physical activity, and a healthy diet) can offset about 50% of the inherited CAD risk.
Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet (London, England). 2016;388(10059):2532–61. https://doi.org/10.1016/s0140-6736(16)31357-5.
Sever PS, Dahlöf B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial--Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet (London, England). 2003;361(9364):1149–58. https://doi.org/10.1016/s0140-6736(03)12948-0.
Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195–207. https://doi.org/10.1056/NEJMoa0807646.
Natarajan P, Young R, Stitziel Nathan O, Padmanabhan S, Baber U, Mehran R, et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135(22):2091–101. https://doi.org/10.1161/CIRCULATIONAHA.116.024436.
Gola D, Erdmann J, Müller-Myhsok B, Schunkert H, König IR. Polygenic risk scores outperform machine learning methods in predicting coronary artery disease status. Genet Epidemiol. 2020;44(2):125–38. https://doi.org/10.1002/gepi.22279.
Funding
Open Access funding provided by Projekt DEAL. The authors acknowledge the support of the Bavarian State Ministry of Health and Care within the framework of DigiMed Bayern (grant No: DMB-1805-0001), the German Federal Ministry of Education and Research (BMBF) within the framework of ERA-NET on Cardiovascular Disease (Druggable-MI-genes: 01KL1802), within the scheme of target validation (BlockCAD: 16GW0198K), within the framework of the e:Med research and funding concept (AbCD-Net: 01ZX1706C), and within the British Heart Foundation (BHF)/German Centre of Cardiovascular Research (DZHK) collaboration. Further support was granted by the German Research Foundation (DFG) as part of the Sonderforschungsbereich SFB 1123 (B02) and the Sonderforschungsbereich SFB TRR 267 (B05) as well as the Corona-Foundation (Junior Research Group Translational Cardiovascular Genomics).
Author information
Authors and Affiliations
Contributions
K.L. did the literature search and drafted the manuscript. T.K. and H.S. critically revised and edited the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Katharina Lechner has received speaker’s honoraria from Goerlich Pharma, Novo Nordisk, Sanofi, and Amgen.
Thorsten Kessler declares no conflict of interest.
Heribert Schunkert has received an institutional grant and honorarium from AstraZeneca; a travel grant from Vifor Pharma; and honoraria from MSD Sharpe & Dohme, Sanofi-Aventis, Brahms, Boehringer-Ingelheim, Novartis, Amgen, Synlab, Daiichi-Sankyo, Servier, Bristol-Myers Squibb, Medtronic, Pfizer, and Bayer Vital.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topical Collection on Lipid Abnormalities and Cardiovascular Prevention
Rights and permissions
About this article

Cite this article
Lechner, K., Kessler, T. & Schunkert, H. Should We Use Genetic Scores in the Determination of Treatment Strategies to Control Dyslipidemias?. Curr Cardiol Rep 22, 146 (2020). https://doi.org/10.1007/s11886-020-01408-9
Published:
DOI: https://doi.org/10.1007/s11886-020-01408-9