
Abstract
As clinicians, we understand the development of atherosclerosis as a consequence of cholesterol deposition and inflammation in the arterial wall, both being triggered by traditional risk factors such as hypertension, hyperlipidaemia or diabetes mellitus. Another risk factor is genetic predisposition, as indicated by the predictive value of a positive family history. However, we had to wait until recently to appreciate the abundant contribution of genetic variation to the manifestation of atherosclerosis. Indeed, by now 164 chromosomal loci have been identified by genome-wide association studies (GWAS) to affect the risk of coronary artery disease. By design, practically all risk variants discovered by GWAS are frequently found in our population, resulting in the fact that principally every Western European individual carries between 130 and 190 risk alleles at the known, genome-wide significant loci (there are 0, 1, or 2 risk alleles per locus). One can assume that it is this widespread disposition that makes mankind susceptible to the detrimental effects of lifestyle factors, which likewise increase the risk of atherosclerosis. In this review, we summarize the recent genetic discoveries and attempt to group the multiple genetic risk variants in functional groups that may become actionable from a preventive or therapeutic perspective.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For the time being, a positive family history is the clinician’s only tool for estimating the impact of genetic disposition on the development of an atherosclerotic disease. For family members, a doubling of risk can be assumed, when myocardial infarction (MI) had been diagnosed before the 55th year of age in a male or before the 65th year in a female first-degree relative [1]. Specifically, a high rate for reoccurrence was found in identical twins of MI patients. For example, the chance to die from MI before the age of 55 years was found to be increased eightfold, when an identical twin was affected at an early age [2]. The inherited risk of coronary artery disease (CAD) is particularly evident in (rare) families with multiple affected family members [3]. However, with the exception of the LDL-receptor gene, the molecular causes underlying such risk remained elusive until very recently [3, 4]. Indeed, it was the emergence of GWAS that led to the discovery of multiple common variants which reproducibly affect CAD risk [5].
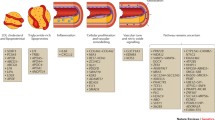
It all started in 2007 with the discovery of the 9p21 risk locus [6]. Subsequently, steadily growing GWAS consortia were formed, which unraveled by now 164 chromosomal loci reaching genome-wide significance (P < 5 × 10−8) (Fig. 1) [7,8,9,10,11], thus demonstrating a linear relationship between sample size and the number of loci (Fig. 2), implying that larger sample sizes will lead to new discoveries. However, the statistical power to detect associations between genetic variants and a trait clearly depends not only on sample size, but also on other factors, such as the effect size and allele frequency at those loci [12]. For example, when considering the so-called ultra-rare variants (i.e., those with a frequency of 1/100,000), whole-genome sequencing and a sample size of more than one million would be required to identify associations, but only when the effect sizes of the variants are very large [12]. Hence, it is not expected that we might reach a saturation (i.e., identification of nearly monogenic variants significantly associated with CAD) soon, at least not within the next 5 years to come. Currently, in fact, exome-wide studies have just started to identify rare variants in the coding regions additionally contributing to CAD risk [13]. Moreover, the discoveries regarding monogenic causes were so far limited to the genes causing familial hypercholesterolemia and disorders in the NO-cGMP signaling pathway [3, 4, 16]. However, this is now expected to change as the scientific community is increasingly moving to whole-genome sequencing, thus increasing the number of CAD risk variants that can be identified and, possibly even, revolutionizing the discovery and diagnosis of monogenic disorders [17].

The figure displays all genes that have achieved genome-wide significant association signals for CAD in GWAS studies as of today [10, 11]. Genes at the 164 loci were grouped into functional classes by gene ontology and canonical pathway maps, such as ConsensusPathDB (http://cpdb.molgen.mpg.de), including the Kyoto Encyclopedia of Genes and Genomes. Some genes have been assigned to multiple pathways. The figure indicates that most genetically influenced mechanisms leading to an increased risk of coronary artery disease are poorly defined and not addressed by current treatments

The number of individuals studied (x-axis) vs. the number of CAD loci reaching genome-wide significance (y-axis) since the first CAD GWAS in 2007, until today (taken from [11])
The genetic architecture of coronary disease
The chromosomal loci associated with atherosclerosis risk that were identified by GWAS analyses are remarkable for several reasons:
-
1.
Only few associated variants alter protein structure. Rather, most risk alleles appear to affect gene regulation.
-
2.
Only 30% of the chromosomal loci conferring CAD risk do so via modulating traditional risk factors like LDL cholesterol and blood pressure (Fig. 1). Thus, the mechanisms increasing atherosclerosis risk are vague for the majority of chromosomal loci [14].
-
3.
Almost all currently identified risk alleles—in part by design of GWAS analyses—are relatively frequent. For example, in Europeans, the probability to carry one or two risk alleles at the most prominent CAD risk locus, at chromosome 9p21.3, is 50% and 25%, respectively [6]. Thus, only 25% of our population is free of this specific genetic risk factor for MI—but there are at least 160 more risk loci in the genome! Given that we have two alleles at each locus, most Europeans carry overall between 130 and 190 of currently known risk alleles.
-
4.
Each risk allele increases the probability of atherosclerosis only by a small margin, i.e., 5–25 relative percent per allele. In other words, individuals who are homozygous for the risk allele on chromosome 9p21.3 carry a 50% relative risk increase to suffer from MI (since they carry this risk allele twice) as compared to the 25% of our population, who do not carry this allele. However, even if a subject does not carry one of the 9p21.3 risk alleles, he or she is likely to carry many other risk alleles at the other loci.
-
5.
The high number of loci carrying risk alleles, the high frequency of most of these risk alleles, and the even spread of risk alleles in the genome of subjects of a given population explain why the implications of the recently identified genetic factors are substantial at the population level, even though each individual risk allele confers only a relatively moderate effect.
-
6.
The genetic risk conferred by the newly discovered common risk variants is largely independent of the risk signaled by a positive family history [15]. By contrast, in heavily affected families cascade-screening for familial hypercholesterolemia should be initiated to provide early preventive treatment [16].
-
7.
Of note, however, each individual will also carry a number of alleles that may decrease the CAD risk, i.e., the so-called “protective alleles” (e.g., by disrupting certain protein functions, typically via loss-of-function effects of genes that go along with increased risk such as APOC3 [18] and ANGPTL4 [13]). Other alleles may specifically neutralize or diminish risk coming from environmental or endogenous factors.
-
8.
Finally, the overall genomic architecture has to be considered, as genetic variants associated with increased risk of complex diseases such as CAD may be also found in genomes of long-lived people, and do not seem to compromise their longevity [19].
Clinical utilization of GWAS findings
From a clinician´s perspective one may ask, how these discoveries may improve prevention and treatment of coronary artery disease? A first step is the conduction of Mendelian randomization studies, which aim to predict the beneficial effect of medications [20]. The principle is based on the fact that any given genetic variant, which exclusively affects a biomarker (e.g., a lipid or inflammatory molecule), can only be related to the outcome (e.g., coronary disease), if this biomarker plays a causal role in this condition [20]. In this respect, GWAS have provided compelling evidence that pharmacological interventions to increase HDL cholesterol are unlikely to lower coronary risk, since there is little evidence that genetic variants which increase HDL cholesterol levels decrease CAD risk [21]. By contrast, medications that lower LDL cholesterol or triglycerides may be good candidates, since multiple genetic variants which lower LDL cholesterol or triglycerides levels also lower CAD risk [13, 22].
Nowadays, pharmaceutical companies increasingly pay attention to the predictive value of such GWAS results in their decision-making, when they select novel agents for clinical evaluation. Indeed, genetic variants may mimic drug effects and thereby allow predicting the outcome of clinical studies [13, 23]. Moreover, it has become increasingly clear that genetic variation can actually affect drug responses in individual patients, including susceptibility to adverse drug reactions.
Finally, the prediction of premature atherosclerosis may be improved by consideration of a genetic risk score build on the hundreds if not thousands of genetic variants that all modulate the respective disease risk [24]. Moreover, in addition to the 164 CAD risk loci, GWAS have identified a large number of genetic variants associated with the traditional CAD risk factors such as hypertension [25], type 2 diabetes [26] and hypercholesterolemia [27]. Indeed, it has been demonstrated that genetic risk scores based on risk factor SNPs, e.g. for hypertension, are likewise associated with CAD [25]. Hence, these additional genetic variants may be considered when constructing genetic risk scores, as this information may lead to more precise risk estimation for CAD and, in some cases, also to specific lifestyle recommendations.
Genotyping arrays are able to yield such information at low cost (e.g., 40 € in a research setting) and assign each individual a percentile rank of a genetic risk score within a given population. The higher the rank, the higher the risk to develop CAD, in particular if the genetic risk score is beyond the 80th or 90th percentile [24]. The advantage of such testing is that the predictive CAD risk value can be obtained already at a young age and thus before any manifestation of atherosclerotic lesions. However, future studies first need to determine the clinical utility of such information before genetic testing can be recommended as a diagnostic tool. Finally, as the run time and costs of whole-genome sequencing drop rapidly, we might be entering a new era of next generation diagnostics, soon.
The growing spectrum of causal pathways
From a clinical point of view, it may be helpful to condensate the many loci (and genes) to a manageable number of functional groups and pathways that may need therapeutic attention [5]. Figure 1 offers such grouping of genes. All genes listed are genome-wide significantly associated with CAD risk [11]. The allocation of the genes to downstream effects was made by gene ontology and canonical pathway maps including, among others, the Kyoto Encyclopedia of Genes and Genomes. Given that a gene can play a role in multiple biological processes, some genes are found multiple times in the Figure such that the overall number of entries is much larger than the 164 loci that house these respective genes. As can be seen, only a few functional groups and pathways (or genes) are currently addressed by therapeutic interventions. Indeed, only genetic variants affecting LDL cholesterol, triglycerides, platelet function, blood pressure or inflammation can be addressed by pharmacological or lifestyle measures that may neutralize an unfavorable disposition.
Figure 3 exemplifies a hypothetical sub-network from such a functional group: cell migration and adhesion. All genes illustrated in the sub-network are genome-wide significantly associated with CAD [11]. Endothelin-1, its receptor type A and other downstream genes in the Figure are likely to play a role in the development of atherosclerosis by modulating cell migration and adhesion, most probably through their impact on the activation of integrins [31]. Databases on protein–protein interactions curated from scientific literature suggest that the respective gene products may interact, as these genes were also annotated to the respective categories in databases or found manual curation (e.g., EDNRA) to affect cell migration and adhesion. Thus, it is possible that these genetic variants, identified for their genome-wide significant association with CAD, disturb this cellular function (e.g., in monocytes or endothelial cells) and therefore increase MI risk. Future studies need to merge such in silico predictions with experimental validation to broaden our understanding of the mechanisms leading to coronary disease [28].

The figure displays a hypothetical sub-network affecting CAD risk. All genes shown in the figure were genome-wide significantly associated with CAD in GWAS studies [10, 11]. Interestingly, all these CAD GWAS hits are related to the term cell migration and adhesion in functional annotations retrieved from the Gene Ontology (http://www.geneontology.org/) and ConsensusPathDB (http://cpdb.molgen.mpg.de/database) databases. The latter database integrates 32 public resources, including biochemical pathway data and protein–protein interactions (PPI) curated from the literature. Querying the protein–protein interactions and searching for direct interactions among the CAD GWAS hits previously annotated to the cell migration and adhesion functional group constructed the sub-network. It illustrates a hypothetical cascade by which endothelin-1 (EDN1) via its receptor A (EDNRA) and activation of the insulin receptor substrate signaling protein (IRS1) could potentially influence these processes. EDN1 endothelin-1, EDNRA Endothelin Receptor Type A, RHOA Ras homolog gene family member A, PRKCE Protein kinase C epsilon type, ITGB5 integrin subunit beta 5, PLCG1 phospholipase C gamma 1, NCK1 NCK adaptor protein 1, IRS1 Insulin receptor substrate1
The successful discovery or multiple risk alleles by GWAS allows to explain an increasing proportion of overall CAD heritability (i.e., currently about 25%) [11]. However, there is still a substantial proportion of “missing heritability”. This is particularly eminent in subjects with a positive family history, suggesting either specific gene–gene interactions (epistasis) or rare (private) variants have a profound effect in such families or individuals with otherwise unexplained risk. Finally, as for most complex multi-factorial diseases, it is the interplay between genetic predisposition, as well as lifestyle and environmental factors that modulates each individual’s risk of developing CAD, suggesting that more efforts should be put on documenting and integrating the latter.
Moreover, there is a substantial need to explain the disease mechanisms both, at the chromosomal level, as well as the level of subsequently affected functional groups and pathways. Currently, large efforts address the systems biology affected by genome-wide significant risk alleles [28].
The first step in the elucidation of the pathophysiological pathway is to identify the casual variant at each locus, followed by the challenge to identify the target gene affected by that variant which is ultimately responsible for the GWAS signal [29]. Next, the downstream mechanisms that are disturbed by changes of the causal gene need to be determined [28]. In most cases unraveled so far, this happens via alteration of gene expression and subsequent protein abundance in the first place [30]. However, despite valid hypotheses regarding many genes and pathways, the exact mechanisms underlying the identified loci often remain unknown. Even the assignment of the loci to genes is mainly based on proximity.
Conclusion
The last decade of genomic research led to the identification of 164 common genetic loci, each of them conferring modest risk for CAD and MI [10, 11]. It is foreseeable that more variants will be identified with increasing sample sizes of GWAS. In addition, whole-exome and whole-genome sequencing studies have identified rare risk variants in families and large patients’ cohorts with stronger effects. Particularly GWAS expanded the understanding of genetic disease etiology, such that by now we have a much better picture of the underlying biology. Currently, functional studies investigating the mechanistic link between genetic variation and disease onset, aim at identifying novel treatment targets. Enormous progress has been made in this respect, as exemplified by GUCY1A3, PCSK9, ANGPTL4, and ANGPTL3, i.e., genes with genome-wide association to CAD and potential druggability. Indeed, these recent findings are excellent starting points for individualized treatment strategies in the future. Finally, despite all these advances, only a part of the heritable risk for CAD can be explained until now.
Abbreviations
- CAD:
-
Coronary artery disease
- GWAS:
-
Genome-wide association study/studies
- MI:
-
Myocardial infarction
- NO:
-
Nitric oxide
References
Myers RH, Kiely DK, Cupples LA, Kannel WB (1990) Parental history is an independent risk factor for coronary artery disease: the Framingham Study. Am Heart J 120:963–969
Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U (1994) Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med 330:1041–1046
Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, de Wit C, Kaiser FJ, Braunholz D, Medack A, Fischer M, Zimmermann ME, Tennstedt S, Graf E, Eck S, Aherrahrou Z, Nahrstaedt J, Willenborg C, Bruse P, Braenne I, Nothen MM, Hofmann P, Braund PS, Mergia E, Reinhard W, Burgdorf C, Schreiber S, Balmforth AJ, Hall AS, Bertram L, Steinhagen-Thiessen E, Li SC, Marz W, Reilly M, Kathiresan S, McPherson R, Walter U, Ott J, Samani NJ, Strom TM, Meitinger T, Hengstenberg C, Schunkert H, CardioGram (2013) Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature 504:432–436
Goldstein JL, Brown MS (2009) “History of discovery: the LDL receptor”. Arterioscler Thromb Vasc Biol 29:431–438
Kessler T, Vilne B, Schunkert H (2016) The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med 8:688–701
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, Konig IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H (2007) WTCCC, Cardiogenics. Genomewide association analysis of coronary artery disease. N Engl J Med 357:443–453
Erdmann J, Grosshennig A, Braund PS, Konig IR, Hengstenberg C, Hall AS, Linsel-Nitschke P, Kathiresan S, Wright B, Tregouet DA, Cambien F, Bruse P, Aherrahrou Z, Wagner AK, Stark K, Schwartz SM, Salomaa V, Elosua R, Melander O, Voight BF, O’Donnell CJ, Peltonen L, Siscovick DS, Altshuler D, Merlini PA, Peyvandi F, Bernardinelli L, Ardissino D, Schillert A, Blankenberg S, Zeller T, Wild P, Schwarz DF, Tiret L, Perret C, Schreiber S, El Mokhtari NE, Schafer A, Marz W, Renner W, Bugert P, Kluter H, Schrezenmeir J, Rubin D, Ball SG, Balmforth AJ, Wichmann HE, Meitinger T, Fischer M, Meisinger C, Baumert J, Peters A, Ouwehand WH, Deloukas P, Thompson JR, Ziegler A, Samani NJ, Schunkert H, Italian Atherosclerosis Vascular Biology Working Group, Myocardial Infarction Genetics Consortium, Wellcome Trust Case Control Consortium, Cardiogenics (2009) New susceptibility locus for coronary artery disease on chromosome 3q22.3. Nat Genet 41:280–282
Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, Preuss M, Stewart AF, Barbalic M, Gieger C, Absher D, Aherrahrou Z, Allayee H, Altshuler D, Anand SS, Andersen K, Anderson JL, Ardissino D, Ball SG, Balmforth AJ, Barnes TA, Becker DM, Becker LC, Berger K, Bis JC, Boekholdt SM, Boerwinkle E, Braund PS, Brown MJ, Burnett MS, Buysschaert I, Cardiogenics, Carlquist JF, Chen L, Cichon S, Codd V, Davies RW, Dedoussis G, Dehghan A, Demissie S, Devaney JM, Diemert P, Do R, Doering A, Eifert S, Mokhtari NE, Ellis SG, Elosua R, Engert JC, Epstein SE, de Faire U, Fischer M, Folsom AR, Freyer J, Gigante B, Girelli D, Gretarsdottir S, Gudnason V, Gulcher JR, Halperin E, Hammond N, Hazen SL, Hofman A, Horne BD, Illig T, Iribarren C, Jones GT, Jukema JW, Kaiser MA, Kaplan LM, Kastelein JJ, Khaw KT, Knowles JW, Kolovou G, Kong A, Laaksonen R, Lambrechts D, Leander K, Lettre G, Li M, Lieb W, Loley C, Lotery AJ, Mannucci PM, Maouche S, Martinelli N, McKeown PP, Meisinger C, Meitinger T, Melander O, Merlini PA, Mooser V, Morgan T, Muhleisen TW, Muhlestein JB, Munzel T, Musunuru K, Nahrstaedt J, Nelson CP, Nothen MM, Olivieri O, Patel RS, Patterson CC, Peters A, Peyvandi F, Qu L, Quyyumi AA, Rader DJ, Rallidis LS, Rice C, Rosendaal FR, Rubin D, Salomaa V, Sampietro ML, Sandhu MS, Schadt E, Schafer A, Schillert A, Schreiber S, Schrezenmeir J, Schwartz SM, Siscovick DS, Sivananthan M, Sivapalaratnam S, Smith A, Smith TB, Snoep JD, Soranzo N, Spertus JA, Stark K, Stirrups K, Stoll M, Tang WH, Tennstedt S, Thorgeirsson G, Thorleifsson G, Tomaszewski M, Uitterlinden AG, van Rij AM, Voight BF, Wareham NJ, Wells GA, Wichmann HE, Wild PS, Willenborg C, Witteman JC, Wright BJ, Ye S, Zeller T, Ziegler A, Cambien F, Goodall AH, Cupples LA, Quertermous T, Marz W, Hengstenberg C, Blankenberg S, Ouwehand WH, Hall AS, Deloukas P, Thompson JR, Stefansson K, Roberts R, Thorsteinsdottir U, O’Donnell CJ, McPherson R, Erdmann J, Samani NJ (2011) Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet 43:333–338
Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, Giannakopoulou O, Jiang T, Hamby SE, Di Angelantonio E, Assimes TL, Bottinger EP, Chambers JC, Clarke R, Palmer CNA, Cubbon RM, Ellinor P, Ermel R, Evangelou E, Franks PW, Grace C, Gu D, Hingorani AD, Howson JMM, Ingelsson E, Kastrati A, Kessler T, Kyriakou T, Lehtimaki T, Lu X, Lu Y, Marz W, McPherson R, Metspalu A, Pujades-Rodriguez M, Ruusalepp A, Schadt EE, Schmidt AF, Sweeting MJ, Zalloua PA, AlGhalayini K, Keavney BD, Kooner JS, Loos RJF, Patel RS, Rutter MK, Tomaszewski M, Tzoulaki I, Zeggini E, Erdmann J, Dedoussis G, Bjorkegren JLM,, Schunkert H, Farrall M, Danesh J, Samani NJ, Watkins H, Deloukas P, CardioGramplusC4D, UKBB (2017) Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 49:1385–1391
van der Harst P, Verweij N (2018) The identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 122:433–443
Erdmann J, Kessler T, Munoz Venegas L, Schunkert H (2018) A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. https://doi.org/10.1093/cvr/cvy084
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J (2017) 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 101(1):5–22
Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, van Iperen E, Kanoni S, Kruppa J, Mahajan A, Scott RA, Willenberg C, Braund PS, van Capelleveen JC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer CM, El-Mokhtari NE, Franke A, Gottesman O, Heilmann S, Hengstenberg C, Hoffman P, Holmen OL, Hveem K, Jansson JH, Jockel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alver M, Moebus S, Morris AD, Muller-Nurasyid M, Nikpay M, Olivieri O, Lemieux Perreault LP, AlQarawi A, Robertson NR, Akinsanya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Strauch K, Varga TV, Waldenberger M, Zeng L, Kraja AT, Liu C, Ehret GB, Newton-Cheh C, Chasman DI, Chowdhury R, Ferrario M, Ford I, Jukema JW, Kee F, Kuulasmaa K, Nordestgaard BG, Perola M, Saleheen D, Sattar N, Surendran P, Tregouet D, Young R, Howson JM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CN, Peters A, Rader D, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Kathiresan S, Deloukas P, Samani NJ, Schunkert H (2016) Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med 374:1134–1144
Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NG, Jansen H, Kanoni S, Nelson CP, Ferrario PG, König IR, Eicher JD, Johnson AD, Hamby SE, Betsholtz C, Ruusalepp A, Franzén O, Schadt EE, Björkegren JL, Weeke PE, Auer PL, Schick UM, Lu Y, Zhang H, Dube MP, Goel A, Farrall M, Peloso GM, Won HH, Do R, van Iperen E, Kruppa J, Mahajan A, Scott RA, Willenborg C, Braund PS, van Capelleveen JC, Doney AS, Donnelly LA, Asselta R, Merlini PA, Duga S, Marziliano N, Denny JC, Shaffer C, El-Mokhtari NE, Franke A, Heilmann S, Hengstenberg C, Hoffmann P, Holmen OL, Hveem K, Jansson JH, Jöckel KH, Kessler T, Kriebel J, Laugwitz KL, Marouli E, Martinelli N, McCarthy MI, Van Zuydam NR, Meisinger C, Esko T, Mihailov E, Escher SA, Alver M, Moebus S, Morris AD, Virtamo J, Nikpay M, Olivieri O, Provost S, AlQarawi A, Robertson NR, Akinsansya KO, Reilly DF, Vogt TF, Yin W, Asselbergs FW, Kooperberg C, Jackson RD, Stahl E, Müller-Nurasyid M, Strauch K, Varga TV, Waldenberger M, Zeng L, Chowdhury R, Salomaa V, Ford I, Jukema JW, Amouyel P, Kontto J, Nordestgaard BG, Ferrières J, Saleheen D, Sattar N, Surendran P, Wagner A, Young R, Howson JM, Butterworth AS, Danesh J, Ardissino D, Bottinger EP, Erbel R, Franks PW, Girelli D, Hall AS, Hovingh GK, Kastrati A, Lieb W, Meitinger T, Kraus WE, Shah SH, McPherson R, Orho-Melander M, Melander O, Metspalu A, Palmer CN, Peters A, Rader DJ, Reilly MP, Loos RJ, Reiner AP, Roden DM, Tardif JC, Thompson JR, Wareham NJ, Watkins H, Willer CJ, Samani NJ, Schunkert H, Deloukas P, Kathiresan S, MORGAM Investigators, Wellcome Trust Case Control Consortium, Myocardial infarction genetics and CARDIoGRAM exome consortia investigators (2017) Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol 69:823–836
Schunkert H (2016) Family or SNPs: what counts for hereditary risk of coronary artery disease? Eur Heart J 37:568–571
Braenne I, Kleinecke M, Reiz B, Graf E, Strom T, Wieland T, Fischer M, Kessler T, Hengstenberg C, Meitinger T, Erdmann J, Schunkert H (2016) Systematic analysis of variants related to familial hypercholesterolemia in families with premature myocardial infarction. Eur J Hum Genet 24:191–197
Stranneheim H, Wedell A (2016) Exome and genome sequencing: a revolution for the discovery and diagnosis of monogenic disorders. J Intern Med 279(1):3–15
Khera AV, Won HH, Peloso GM, O’Dushlaine C, Liu D, Stitziel NO, Natarajan P, Nomura A, Emdin CA, Gupta N, Borecki IB, Asselta R, Duga S, Merlini PA, Correa A, Kessler T, Wilson JG, Bown MJ, Hall AS, Braund PS, Carey DJ, Murray MF, Kirchner HL, Leader JB, Lavage DR, Manus JN, Hartzel DN, Samani NJ, Schunkert H, Marrugat J, Elosua R, McPherson R, Farrall M, Watkins H, Lander ES, Rader DJ, Danesh J, Ardissino D, Gabriel S, Willer C, Abecasis GR, Saleheen D, Dewey FE, Kathiresan S, Myocardial Infarction Genetics Consortium, DiscovEHR Study Group, CARDIoGRAM Exome Consortium, and Global Lipids Genetics Consortium (2017) Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA 7(9):937–946 317(
Ukraintseva S, Yashin A, Arbeev K, Kulminski A, Akushevich I, Wu D, Joshi G, Land KC, Stallard E (2016) Puzzling role of genetic risk factors in human longevity: “risk alleles” as pro-longevity variants. Biogerontology 17(1):109–127
Jansen H, Samani NJ, Schunkert H (2014) Mendelian randomization studies in coronary artery disease. Eur Heart J 35:1917–1924
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, Hindy G, Hólm H, Ding EL, Johnson T, Schunkert H, Samani NJ, Clarke R, Hopewell JC, Thompson JF, Li M, Thorleifsson G, Newton-Cheh C, Musunuru K, Pirruccello JP, Saleheen D, Chen L, Stewart A, Schillert A, Thorsteinsdottir U, Thorgeirsson G, Anand S, Engert JC, Morgan T, Spertus J, Stoll M, Berger K, Martinelli N, Girelli D, McKeown PP, Patterson CC, Epstein SE, Devaney J, Burnett MS, Mooser V, Ripatti S, Surakka I, Nieminen MS, Sinisalo J, Lokki ML, Perola M, Havulinna A, de Faire U, Gigante B, Ingelsson E, Zeller T, Wild P, de Bakker PI, Klungel OH, Maitland-van der Zee AH, Peters BJ, de Boer A, Grobbee DE, Kamphuisen PW, Deneer VH, Elbers CC, Onland-Moret NC, Hofker MH, Wijmenga C, Verschuren WM, Boer JM, van der Schouw YT, Rasheed A, Frossard P, Demissie S, Willer C, Do R, Ordovas JM, Abecasis GR, Boehnke M, Mohlke KL, Daly MJ, Guiducci C, Burtt NP, Surti A, Gonzalez E, Purcell S, Gabriel S, Marrugat J, Peden J, Erdmann J, Diemert P, Willenborg C, König IR, Fischer M, Hengstenberg C, Ziegler A, Buysschaert I, Lambrechts D, Van de Werf F, Fox KA, El Mokhtari NE, Rubin D, Schrezenmeir J, Schreiber S, Schäfer A, Danesh J, Blankenberg S, Roberts R, McPherson R, Watkins H, Hall AS, Overvad K, Rimm E, Boerwinkle E, Tybjaerg-Hansen A, Cupples LA, Reilly MP, Melander O, Mannucci PM, Ardissino D, Siscovick D, Elosua R, Stefansson K, O’Donnell CJ, Salomaa V, Rader DJ, Peltonen L, Schwartz SM, Altshuler D, Kathiresan S (2012) Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 380:572–580
Linsel-Nitschke P, Götz A, Erdmann J, Braenne I, Braund P, Hengstenberg C, Stark K, Fischer M, Schreiber S, El Mokhtari NE, Schaefer A, Schrezenmeir J, Rubin D, Hinney A, Reinehr T, Roth C, Ortlepp J, Hanrath P, Hall AS, Mangino M, Lieb W, Lamina C, Heid IM, Doering A, Gieger C, Peters A, Meitinger T, Wichmann HE, König IR, Ziegler A, Kronenberg F, Samani NJ (2008) Schunkert HLifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease—a Mendelian Randomisation study. PLoS One 3:e2986
Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J, Cardon LR, Whittaker JC, Sanseau P (2015) The support of human genetic evidence for approved drug indications. Nat Genet 47:856–860
Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L, Tikkanen E, Perola M, Schunkert H, Sijbrands EJ, Palotie A, Samani NJ, Salomaa V, Ripatti S, Inouye M (2016) Genomic prediction of coronary heart disease. Eur Heart J 37:3267–3278
International Consortium for Blood Pressure Genome-Wide Association Studies, Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, Pihur V, Vollenweider P, O’Reilly PF, Amin N, Bragg- Gresham JL, Teumer A, Glazer NL, Launer L, Zhao JH, Aulchenko Y, Heath S, Sõber S, Parsa A, Luan J, Arora P, Dehghan A, Zhang F, Lucas G, Hicks AA, Jackson AU, Peden JF, Tanaka T, Wild SH, Rudan I, Igl W, Milaneschi Y, Parker AN, Fava C, Chambers JC, Fox ER, Kumari M, Go MJ, van der Harst P, Kao WH, Sjögren M, Vinay DG, Alexander M, Tabara Y, Shaw-Hawkins S, Whincup PH, Liu Y, Shi G, Kuusisto J, Tayo B, Seielstad M, Sim X, Nguyen KD, Lehtimäki T, Matullo G, Wu Y, Gaunt TR, Onland-Moret NC, Cooper MN, Platou CG, Org E, Hardy R, Dahgam S, Palmen J, Vitart V, Braund PS, Kuznetsova T, Uiterwaal CS, Adeyemo A, Palmas W, Campbell H, Ludwig B, Tomaszewski M, Tzoulaki I, Palmer ND; CARDIoGRAM consortium;CKDGen Consortium;KidneyGen Consortium; EchoGen consortium; CHARGE-HF consortium, Aspelund T, Garcia M, Chang YP, O’Connell JR, Steinle NI, Grobbee DE, Arking DE, Kardia SL, Morrison AC, Hernandez D, Najjar S, McArdle WL, Hadley D, Brown MJ, Connell JM, Hingorani AD, Day IN, Lawlor DA, Beilby JP, Lawrence RW, Clarke R, Hopewell JC, Ongen H, Dreisbach AW, Li Y, Young JH, Bis JC, Kähönen M, Viikari J, Adair LS, Lee NR, Chen MH, Olden M, Pattaro C, Bolton JA, Köttgen A, Bergmann S, Mooser V, Chaturvedi N, Frayling TM, Islam M, Jafar TH, Erdmann J, Kulkarni SR, Bornstein SR, Grässler J, Groop L, Voight BF, Kettunen J, Howard P, Taylor A, Guarrera S, Ricceri F, Emilsson V, Plump A, Barroso I, Khaw KT, Weder AB, Hunt SC, Sun YV, Bergman RN, Collins FS, Bonnycastle LL, Scott LJ, Stringham HM, Peltonen L, Perola M, Vartiainen E, Brand SM, Staessen JA, Wang TJ, Burton PR, Soler Artigas M, Dong Y, Snieder H, Wang X, Zhu H, Lohman KK, Rudock ME, Heckbert SR, Smith NL, Wiggins KL, Doumatey A, Shriner D, Veldre G, Viigimaa M, Kinra S, Prabhakaran D, Tripathy V, Langefeld CD, Rosengren A, Thelle DS, Corsi AM, Singleton A, Forrester T, Hilton G, McKenzie CA, Salako T, Iwai N, Kita Y, Ogihara T, Ohkubo T, Okamura T, Ueshima H, Umemura S, Eyheramendy S, Meitinger T, Wichmann HE, Cho YS, Kim HL, Lee JY, Scott J, Sehmi JS, Zhang W, Hedblad B, Nilsson P, Smith GD, Wong A, Narisu N, Stančáková A, Raffel LJ, Yao J, Kathiresan S, O’Donnell CJ, Schwartz SM, Ikram MA, Longstreth WT Jr, Mosley TH, Seshadri S, Shrine NR, Wain LV, Morken MA, Swift AJ, Laitinen J, Prokopenko I, Zitting P, Cooper JA, Humphries SE, Danesh J, Rasheed A, Goel A, Hamsten A, Watkins H, Bakker SJ, van Gilst WH, Janipalli CS, Mani KR, Yajnik CS, Hofman A, Mattace-Raso FU, Oostra BA, Demirkan A, Isaacs A, Rivadeneira F, Lakatta EG, Orru M, Scuteri A, Ala-Korpela M, Kangas AJ, Lyytikäinen LP, Soininen P, Tukiainen T, Würtz P, Ong RT, Dörr M, Kroemer HK, Völker U, Völzke H, Galan P, Hercberg S, Lathrop M, Zelenika D, Deloukas P, Mangino M, Spector TD, Zhai G, Meschia JF, Nalls MA, Sharma P, Terzic J, Kumar MV, Denniff M, Zukowska-Szczechowska E, Wagenknecht LE, Fowkes FG, Charchar FJ, Schwarz PE, Hayward C, Guo X, Rotimi C, Bots ML, Brand E, Samani NJ, Polasek O, Talmud PJ, Nyberg F, Kuh D, Laan M, Hveem K, Palmer LJ, van der Schouw YT, Casas JP, Mohlke KL, Vineis P, Raitakari O, Ganesh SK, Wong TY, Tai ES, Cooper RS, Laakso M, Rao DC, Harris TB, Morris RW, Dominiczak AF, Kivimaki M, Marmot MG, Miki T, Saleheen D, Chandak GR, Coresh J, Navis G, Salomaa V, Han BG, Zhu X, Kooner JS, Melander O, Ridker PM, Bandinelli S, Gyllensten UB, Wright AF, Wilson JF, Ferrucci L, Farrall M, Tuomilehto J, C, Meneton P, Uitterlinden AG, Wareham NJ, Gudnason V, Rotter JI, Rettig R, Uda M, Strachan DP, Witteman JC, Hartikainen AL, Beckmann JS, Boerwinkle E, Vasan RS, Boehnke M, Larson MG, Järvelin MR, Psaty BM, Abecasis GR, Chakravarti A, Elliott P, van Duijn CM, Newton-Cheh C, Levy D, Caulfield MJ, Johnson T. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature 2011; 478(7367):103–109
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Müller- Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stančáková A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutškov K, Langford C, Leander K, Lindholm E, Lobbens S, MännistöS, Mirza G, Mühleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurðsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvänen AC, Eriksson JG, Peltonen L, Nöthen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L; Wellcome Trust Case Control Consortium; Meta-Analyses of Glucose and Insulin-related traits Consortium(MAGIC) Investigators; Genetic Investigation of ANthropometric Traits(GIANT) Consortium;Asian Genetic Epidemiology Network-Type 2 Diabetes (AGEN-T2D) Consortium; South Asian Type 2 Diabetes (SAT2D) Consortium, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njølstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyövälti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jöckel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI; DIAbetes Genetics Replication And Meta- analysis (DIAGRAM) Consortium. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44(9):981–990
Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, HeikkiläK, Hyppönen E, Isaacs A, Jackson AU, Johansson Å, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Nolte IM, O’Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Ingi Eyjolfsson G, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stančáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR; Global Lipids Genetics Consortium (2013) Discovery and refinement of loci associated with lipid levels. Nat Genet 45(11):1274–1283
Lempiäinen H, Brænne I, Michoel T, Tragante V, Vilne B, Webb TR, Kyriakou T, Eichner J, Zeng L, Willenborg C, Franzen O, Ruusalepp A, Goel A, van der Laan SW, Biegert C, Hamby S, Talukdar HA, Foroughi Asl H, Pasterkamp G, Watkins H, Samani NJ, Wittenberger T, Erdmann J, Schunkert H, Asselbergs FW, Björkegren JLM, CVgenes@target consortium (2018) Network analysis of coronary artery disease risk genes elucidates disease mechanisms and druggable targets. Sci Rep 8:3434
Braenne I, Civelek M, Vilne B, Di Narzo A, Johnson AD, Zhao Y, Reiz B, Codoni V, Webb TR, Foroughi Asl H, Hamby SE, Zeng L, Tregouet DA, Hao K, Topol EJ, Schadt EE, Yang X, Samani NJ, Bjorkegren JL, Erdmann J, Schunkert H, Lusis AJ, Leducq Consortium CADgenomics (2015) Prediction of causal candidate genes in coronary artery disease loci. Arterioscler Thromb Vasc Biol 35:2207–2217
Kessler T, Wobst J, Wolf B, Eckhold J, Vilne B, Hollstein R, von Ameln S, Dang TA, Sager HB, Moritz Rumpf P, Aherrahrou R, Kastrati A, Bjorkegren JLM, Erdmann J, Lusis AJ, Civelek M, Kaiser FJ, Schunkert H (2017) Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation 136:476–489
Chen CC1, Chen LL, Hsu YT, Liu KJ, Fan CS, Huang TS (2014) The endothelin-integrin axis is involved in macrophage-induced breast cancer cell chemotactic interactions with endothelial cells. J Biol Chem 289:10029–10044
Funding
This work was supported by grants from the Fondation Leducq [CADgenomics, 12CVD02], the German Federal Ministry of Education and Research (BMBF) within the framework of ERA-NET on Cardiovascular Disease, Joint Transnational Call 2017 [ERA-CVD: grant JTC2017_21-040], within the framework of target validation [BlockCAD: 16GW0198K], within the framework of the e:Med research and funding concept [AbCD-Net: grant 01ZX1706C and e:AtheroSysMed: grant 01ZX1313A-2014], and the European Union Seventh Framework Programme FP7/2007–2013, under grant agreement no. HEALTH-F2-2013-601456 (CVgenes-at-target). Further grants were received from the Deutsche Forschungsgemeinschaft (DFG) as part of the Sonderforschungsbereich CRC 1123 (B2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no competing interest.
Additional information
This article is part of the Spotlight Issue of Clinical Research in Cardiology with the title “German cardiac society welcomes ESC”.
Rights and permissions
About this article

Cite this article
Schunkert, H., von Scheidt, M., Kessler, T. et al. Genetics of coronary artery disease in the light of genome-wide association studies. Clin Res Cardiol 107 (Suppl 2), 2–9 (2018). https://doi.org/10.1007/s00392-018-1324-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00392-018-1324-1