
Abstract
A high-efficiency plant transgenic vector expressing multiple genes has been modified from expression vector pCAMBIA1302. Briefly, multiple cloning sites of a cloning vector pUCm-T were constructed into plant transgenic vector pCAMBIA1302 backbone using Bsp120 I and Spe I restriction digest site. Two Bt genes, cry1Ac and cry3A, were then constructed into the modified transgenic vector in different orders as p1870-35S::cry1Ac-Coy::cry3A and p1870-Coy::cry3A-35S::cry1Ac, respectively. Transgenic tobacco plants were generated using Agrobacterium-mediated transformation method. Polymerase chain reaction (PCR) results showed that both the exogenous genes were integrated into the genome of tobacco. Fluorescence qRT-PCR detected both transgenes and further ELISA assay validated the expressed Bt proteins. Transgenic lines showed enhanced resistance to larvae of Helicoverpa armigera Hubner in the further feeding assays. In conclusion, our modified transgenic vector was suitable for multiple gene expression and would greatly facilitate our future research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The innovations on transgenic techniques have greatly improved the plant transgenic research, from single-gene genetic transformation to multiple genes with various functions. Considerable researches have been conducted on transgenic plants using multiple gene complex (Rao et al. 2011; Sun et al. 2012; Azadi et al. 2010; Zhu et al. 2007; Qgawa et al. 2014). Various genes controlling insect resistance, drought resistance, and salt tolerance have been transformed into plants simultaneously, which greatly improve the comprehensive resistance of plants (Rao et al. 2011; Lian et al. 2008; Sun et al. 2012). All these transgenic plants with multiple transgenes mentioned above were obtained by either genetic crossing, co-transformation with multiple vectors or two-step transformation (Daley et al. 1991; Singla-Pareek et al. 2003; Rosati et al. 2003). However, all these experiments required long period of time for the combination of multiple transgenes. Moreover, the segregation of these transgenes in the offspring made it difficult to obtain homozygous transgenic lines with multiple transgenes. The ideal way for co-expression of multiple transgenes was constructing transgenic vectors with multiple genes, which would save a lot of time for transgenic operation and selection. With the development of vector constructing techniques, such as Gateway and Cre/LoxP recombination, a single gene or multiple genes can be easily constructed into plant transgenic vectors by one-step LR reaction (Chung et al. 2005; Chen et al. 2006; Earley et al. 2006; Tanaka et al. 2011).
This study, a high-efficiency plant transgenic vector expressing multiple genes has been modified from expression vector pCAMBIA1302. Two Bt genes, cry1Ac and cry3A, were then constructed into the modified transgenic vector. Transgenes in the transgenic lines were validated in three different layers, e.g., genomic DNA PCR, RNA expression qRT-PCR, and protein ELISA detection. The modified transgenic vector was suitable for multiple gene expression in plant and would greatly facilitate our future research.
Materials and methods
Strains, vectors, plant materials, and primary reagents
The strains Escherichia coli DH5α and Agrobacterium tumefaciens EHA105 were used in this study. Plasmids of PBtiA and pBCC3 carrying genes cry1Ac and cry3A, respectively, were derived from previous research. Plant transgenic vector pCAMBIA1302 was used as the backbone for modification and cloning vector p701, which modified from pUCm-T, provided the multiple cloning sites.
Tobacco (Nicotiana tabacum L.) variety Wisconsin35 was used for genetic transformation. T4 ligase was purchased from Promage. All the used restriction endonucleases, Taq DNA polymerase, RNase A, AB5000 DNA marker, DL2000 marker, and plasmid isolation kit were purchased from TaKaRa. Bt-Cry1Ab/1Ac and Bt-Cry3A ELISA kits were purchased from Agdia.
Modifications on expression vector pCAMBIA1302 and cloning vector pUCm-T
The pCAMBIA1302 were used as the backbone to construct the new transgenic vector p1870. Matrix attachment region (MAR) motif from tobacco genome was cloned and constructed to the both ends of multi-cloning site. Restriction digest sites Bsp120 I and Spe I were also introduced between these two MAR motifs. For cloning vector pUCm-T, AcADH 5′-UTR translocation enhancer was inserted to the downstream of the CaMV35S promoter with several restriction digest sites of Not I, Bsp120 I, Spe I, and Nhe I, etc. CoyMV or other promoter(s) could be institute easily by double digestion for the cut sites of Hind III and BamH I. We designated the modified pUCm-T as vector p701-T(35S) and p701-T(Coy).
Cloning of the target genes
Primers (Table 1) were designed to clone the ORF of two Bt genes cry1Ac (GenBank accession AF148644) and cry3A (M84650) from plasmid pBtiA and pBCC3, respectively. PCR reactions were carried out using Pfu DNA polymerase in MyCycle thermal cycler (BioRad) PCR machine. PCR products were further used for vector construction.
Construction and detection of complete ORF in the clone vector
Plasmids of p701-T(35S) and p701-T(Coy) were extracted and digested by restriction enzyme Xcm I. After separated on 1% TAE agarose gel, the appropriate fragment was then recycled. The amplified ORF fragments of cry1Ac and cry3A were ligated to p701-T(35S) and p701-T(Coy), respectively. The ligations were transformed to competence E. coli DH5α through thermal activation. Transformed E. coli DH5α was selected on LB solid medium with 50 mg L−1 Amp at 37 °C.
Several primers were used for PCR validations of the colonies, including 35SendF primer designed on CaMV35S promoter and Bt1fullR to detect cry1Ac gene, CoyendF on CoyMV promoter, and Bt3fullR to detect cry3A gene. Positive colonies were cultured and sequenced to validate correct insertion. The constructed cloning vector with target genes was named p701-35S::cry1Ac and p701-Coy::cry3A.
Construction of the plant transgenic vector expressing multiple genes
Transformation vector expressing multiple genes were constructed in a two-step way. Briefly, cloning vector p701-35S::cry1Ac was digested with Not I and Nhe I, whereas plant transgenic vector p1870 was digest with Bsp120 I and Spe I. The corresponding fragments were ligated as p1870-35S::cry1Ac. The p1870-35S::cry1Ac was further digested with Bsp120 I and Spe I and p701-Coy::cry3A using Not I and Nhe I. The corresponding fragments were ligated as p1870-35S::cry1Ac-Coy::cry3A. By a similar approach, vector of p1870-Coy::cry3A-35S::cry1Ac was constructed.
Tobacco transformation by Agrobacterium-mediated leaf disc method
Transformation vectors of p1870-35S::cry1Ac-Coy::cry3A and p1870-Coy::cry3A-35S::cry1Ac were transformed into Agrobacterium strain EHA105. Tobacco was transformed using the Agrobacterium-mediated leaf disc method (Horsch et al. 1985). Briefly, fresh and sterile tobacco leaves were cut into 1 cm2 leaf discs along the leaf veins. Leaf discs were incubated in Agrobacterium diluted with 5% sucrose (1:1) for 10 min. Leaf discs were dried using a sterile blotting paper and then incubated in the co-culture medium (MS + 2.0 mg·L−1 6-BA + 0.1 mg L−1 IBA) for 1 day in dark. Leaf discs were then transferred to the screening culture medium (MS + 1.0 mg L−1 6-BA + 0.1 mg L−1 IBA + 50 mg L−1 Kan + 400 mg L−1 Cef). The medium was refreshed every 2 weeks. After leaf discs grew to 2 cm, the positive buds were transferred to the rooting medium (MS + 75 mg L−1 Kan + 300 mg L−1 Cef). Samples were placed in a chamber at 25 ± 2 °C, white light with a light intensity of 30 μmol m−2 s−1, and a light/dark cycle of 14/10 h. The rooted plants were propagated and domesticated before transplanted into the field.
PCR detection of T0 transgenic plants
Genomic DNAs were extracted from the leaves of transgenic lines and wild-type tobacco plants using modified hexadecyl trimethyl ammonium bromide (CTAB) method (Wang and Fang 2002). Primers of cry1Ac and cry3A were designed (Table 1) for the PCR validation of integration of transgenes in the tobacco genome.
Fluorescence quantitative PCR detection of T0 plants
Total RNAs from transgenic and wild-type tobacco were extracted by ultrapure RNA kit (Beijing ComWin Biotech Co., Ltd.). The concentration of total RNA was measured by Nanophotometer p-class K5600. The first strand cDNA was synthesized by the TUREscript 1st Strand cDNA Synthesis Kit (Aidlab Biotechnologies Co., Ltd.) according to the manufacturer’s instructions by Mx3005P (Agilent). Three biological replicates were included. Experimental data were processed with the Microsoft Excel 2003 and IBM SPSS v21.0 software. The ANOVA among different indexes were calculated by Duncan’s test.
ELISA detection of Bt toxin in T0 plants
Leaves of tobacco seedlings for each transgenic line were collected to isolate the total protein. The Bt protein was detected using the Bt-Cry1Ac/Ab and Bt-Cry3A ELISA kits (Agdia, Inc) according to the manufacturer’s instructions. Positive control was provided with the kit, and wild-type tobacco served as the negative control. Data were detected using a BioRad 550 microplate reader. Concentrations of Bt protein (ng g−1 fresh weight) were evaluated by comparing with the standard curve. Three biological replicates were included. Experimental data were processed with the Microsoft Excel 2003 and IBM SPSS v21.0 software. The ANOVA among different indexes were calculated by Duncan’s test.
Insect-resistance test of T0 plants
Twenty newly hatched instar larvae of Helicoverpa armigera Hubner were fed on the seedling leaves of transgenic lines and wild type. Tobacco leaves were changed daily. The average corrected mortality was calculated as follows:
Results
Constructions of plant transgenic vector co-expressing multiple genes
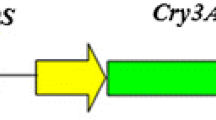
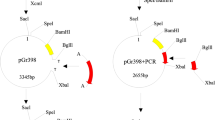
Two vectors, p701 and p1870 from pUCm-T and pCAMBIA1302, respectively, have been modified in this study to construct novel plant transgenic vector co-expressing multiple genes (Figs. 1, 2). Two Bt genes cry1Ac and cry3A were cloned and constructed to modified vector p701 under CaMV35S and CoyMV promoter, respectively (Fig. 3). The corresponding fragments from p701-35S::cry1Ac and p701-Coy::cry3A were constructed to p1870 in a two-step ligation way to prepare p1870-35S::cry1Ac-Coy::cry3A and p1870-Coy::cry3A-35S::cry1Ac (Fig. 2).

Structure of cloning vector p701 and plant transgenic vector p1870

Construction of plant transgenic vector co-expressing multiple genes. Fragments of transformation vector p1870 (digested by Bsp 120 I and Spe I) and mid-vector p701-35S::cry1Ac (digested by Not I and Nhe I) were ligated to construct transformation vector of p1870-35S::cry1Ac. The constructed p1870-35S::cry1Ac vector then further digested by Bsp 120 I and Spe I, and an other mid-vector p701-Coy::cry3A digested by Not I and Nhe I, the required fragments was then ligated to get p1870-35S::cry1Ac-Coy::cry3A. Similar approach was applied to prepare the p1870-Coy::cry3A-35S::cry1Ac

Validation of the multi-gene transformation vector. M AB5000 DNA Marker
Obtaining and detecting transgenic tobacco T0 plants
Leaf discs of tobacco plants were transformed using Agrobactium-mediated method. After kanamycin selection, a total of 20 transgenic lines from p1870-35S::cry1Ac-Coy::cry3A (named N4 lines) and 17 transgenic lines from p1870-Coy::cry3A-35S::cry1Ac (N5 lines) were obtained (Fig. 4).

Obtained transgenic lines. a Leaves grew on selective medium after Agrobacterium infection; b Kanamycin-resistant shoot grew; c Kanamycin-resistant shoot; d Kanamycin-resistant shoot took root; e Kanamycin-resistant plant; and f Kanamycin-rooting plant in the field
Genomic DNAs were isolated from five randomly picked N4 lines, N5 lines, and wild-type tobacco plants. Correct bands of cry1Ac (546 bp) and cry3A (667 bp) were detected in all the tested N4 and N5 lines, but not wild type, indicated that target genes were integrated into the tobacco genome.
Transcription and expression detection of Bt genes in T0 plants
Results of fluorescence quantitative PCR showed that the transcription abundance of cry1Ac and cry3A gene in selected N4 lines ranged from 1.14E+04 to 3.67E+04 (average 2.08E+04) and from 2.60E+06 to 6.67E+07 (average 1.78E+07), respectively (Table 2). The transcription abundance of these two genes in N5 lines ranged from 1.06E+05 to 5.24E+05 (average 2.24E+05) and from 6.09E+06 to 5.04E+07 (average 2.70E+07), respectively (Table 2). No transcription of Bt genes was detected in the wild-type control. These results indicated that the T-DNA insertion carrying cry1Ac and cry3A genes could transcript successfully in the transgenic lines. Among these two transgenic events, expression level and protein content of cry1Ac downstream transgene were 9.8 times and 7.9 times higher than of the upstream one, respectively. As for cry3A, there was no obviously change (about 1.5 times) when it arranged on upstream of cry1Ac (Fig. 5).

PCR validation of Bt transgenes in the transgenic plants. a cry1Ac; b cry3A; Lane 1 and 12 negative control of N4 and N5; lanes 2–6 samples of N4; lanes 7–11 samples of N5. M DL2000 DNA Marker
By ELISA analysis, the Bt products of cry1Ac and cry3A were detected (Table 2). Relative higher content of both Bt1 and Bt3 protein was observed in N5 lines compared with N4 lines, especially for the content of Bt1 protein (2828.28 ng g−1).
Insect-resistance test
Those transgenic lines used in qRT-PCR and ELISA assays were further tested with insect resistance. The results showed that each line had a different effect on the survival and growth of H. armigera Hübner larvae. When the average correction mortality of N5 lines reached 100%, the corresponding data for N4 lines were 51%. There are correlations between Bt toxins detected in the transgenic plants and correction mortality.
Discussion
Co-expressing multiple genes in the plants can be simplified by constructing plant transgenic vector expressing multiple genes. Modifications on the current transgenic vector are applicable in most of recent studies (Fitzgerald et al. 2006; Underhill et al. 2007; Zeevi et al. 2012). However, construction and assembly of vectors with multiple genes are hindered by several technical problems, such as duplicates of multiple cloning site, existence of digestion site in target gene and low digest efficiency, etc. One plant transgenic vector and one cloning vector were combined together in this study to generate the transformation vector co-expressing multiple genes. Isocaudamers, Not I/Bsp120 I and Spe I/Xba I/Nhe I, were used for the construction of this high-efficiency plant transgenic vector. In detail, Bsp120 I and Not I are isocaudamers that share a 3′ overhang CCGG cohesive terminus, whereas Spe I and Nhe I share GATC cohesive terminus. Since the original restriction digest site disappeared after the isocaudamer was connected, target genes could be transferred easily from cloning vector to expression vector. Using isocaudamers, digested vectors or fragments with different cohesive ends would not have intralooping problems during construction process. The modified vector does not require a dephosphorylation treatment after digestion, which simplifies the operating procedure. The applied digesting-ligating vector construction is reliable, simple operational, and costless. Plant transformation vectors co-expressing multiple genes were constructed, and transgenic tobacco was generated using Agrobacterium-mediated method. Transgenic tobacco was acquired screened by npt II resistance. Further amplification using specific primers designed on functional cry1Ac and cry3A genes showed the corresponding bands which indicate a successful integration of exogenous gene into the tobacco genome, and proves the T-DNA insertion ability of this modified vector.
Results of qRT-PCR indicated that the two Bt genes cry1Ac and cry3A could be transcripted in the transgenic tobacco lines. Further ELISA assay showed that the expression of Bt1 and Bt3 protein was different among each line. Such variation might be resulted from the insertion position of the exogenous gene in the tobacco genome. We did not observe any significant difference on the MAR motif, which was located on the borders of the exogenous gene protecting it from plant endogenic regulation. Moreover, Bt protein in N4 and N5 lines (318.51–2828.28 ng g−1) was significantly higher than transgenic lines expressing the same gene without MAR motifs (21.37–108.54 ng g−1, unpublished data), and no gene silencing was observed in the transgenic lines of the two transformation vectors. Which was consistent with the role of MAR motifs in avoiding of gene silencing of extrogenous gene and enhancing the expression level of transgenes (Zhou et al. 2012). Among these two transgenic events, expression level of cry1Ac downstream transgene was 8.9 times of the upstream one, and similar results were acquired when the gene was combined with other functional reporter gene (5.8 times). However, for cry3A, it was little higher (1.5 times) when the gene position on upstream, and this was coincident when there were two open reading frames downstream cry3A gene (unpublished data). Which requires further validation for other extrogenous gene or transformation on other plant. All these results proved that this transgenic vector is sufficient for co-expressing multiple transgenes.
As a practical value, the modified transgenic vector satisfied the demands of co-expressing multiple transgenes at the same time, even under different promoters. Upon the requirement of different studies in the future, various promoters could be constructed to the modified cloning vector p701 and subsequently introduced to the transgenic vector.
Author contribution statement
SY, YD, BG, and MY conceived and designed the experiments. SY, YD, and NZ contributed equally to this work. SY, YD, and NZ performed the experiments. SY, YD, and NZ analyzed the data. SY, YD, NZ, and YR contributed reagents/materials/analysis tools. SY and NZ wrote the paper.
References
Azadi P, Otang NV, Chin DP, Nakamura I, Fujisawa M, Harada H, Misawa N, Mii M (2010) Metabolic engineering of Lilium × formolongi using multiple genes of the carotenoid biosynthesis pathway. Plant Biotechnol Rep 4:269–280
Chen QJ, Ahou HM, Chen J, Wang XC (2006) A gateway-based platform for multigene plant transformation. Plant Mol Biol 62:927–936
Chung SM, Frankman EL, Tzfira T (2005) A versatile vector system for multiple gene expression in plants. Trends Plant Sci 10:357–361
Daley M, Knauf V, Summerfelt K, Turner J (1991) Co-transformation with one Agrobacterium tumefaciens strain containing two binary plasmids as a method for producing marker-free transgenic plants. Plant Cell Rep 17:489–496
Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song KM, Pikaard CS (2006) Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45:616–629
Fitzgerald DJ, Berger P, Schaffitzel C, Yamada K, Richmond TJ, Berger I (2006) Protein complex expression by using multigene baculoviral vectors. Nat Methods 3:1021–1032
Horsch RB, Fry JE, Hoffmann NL, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227:1229–1231
Li BJ, Zhu HC (2005) On the prospects of applying the multi-gene transformation strategy (MTS) to modify the inheritance of organisms. Acta Sci Nat Univ Sunyatseni 44:79–83
Lian Y, Jian ZW, He KL, Liu YJ, Song FP, Wang BW, Wang GY (2008) Transgenic tobacco plants expressing synthetic Cry1Ac and Cry1Ie genes are more toxic to cotton bollworm than those containing one gene. Chin Sci Bull 53:1381–1387
Qgawa Y, Shirakawa M, Koumoto Y, Honda M, Asami Y, Kondo Y, Nishimura IH (2014) A simple and reliable multi-gene transformation method for switchgrass. Plant Cell Rep 33:1161–1172
Rao MVR, Parameswari C, Sripriya R, Veluthambi K (2011) Transgene stacking and marker elimination in transgenic rice by sequential Agrobacterium-mediated co-transformation with the same selectable marker gene. Plant Cell Rep 30:1241–1252
Rosati C, Simoneau P, Treutter D, Poupard P, Cadot Y, Cadic A, Duron M (2003) Engineering of flower color in forsythia by expression of two independently-transformed dihydroflavonol 4-reductase and anthocyanidin synthase genes of flavonoid pathway. Mol Breeding 12:197–208
Singla-Pareek S, Reddy M, Sopory S (2003) Genetic engineering of the glyoxalase pathway in tobacco leads to enhanced salinity tolerance. Proc Natl Acad Sci USA 100:14672–14677
Sun H, Lang ZH, Zhu L, Huang DF (2012) Acquiring transgenic tobacco plants with insect resistance and glyphosate tolerance by fusion gene transformation. Plant Cell Rep 31:1877–1887
Tanaka Y, Nakamura S, Kawamukai M, Koizumi N, Nakagawa T (2011) Development of a series of gateway binary vectors possessing a tunicamycin resistance gene as a marker for the transformation of Arabidopsis thaliana. Biosci Biotechnol Biochem 75:80
Underhill MF, Smales CM, Naylor LH, Birch JR, James DC (2007) Transient gene expression levels from multigene expression vectors. Biotechnol Progr 23:435–443
Wang GL, Fang HJ (2002) Plant genetic engineering. Science Press, Beijing
Zeevi V, Liang Z, Arieli U, Tzfira T (2012) Zinc Finger nuclease and homing endonuclease-mediated assembly of multigene plant transformation vectors. Plant Physiol 158:132–144
Zhou Y, Yau YY, Ow DW, Wang Y (2012) Site-specific deletions in the tomato genome by the CinH-RS2 and ParA-MRS recombination systems. Plant Biotechnol Rep 6(3):225–232
Zhu HC, Xu XP, Xiao GY, Yuan LP, Li BJ (2007) Enhancing disease resistances of Super Hybrid Rice with four antifungal genes. Science China Ser C 50:31–39
Acknowledgements
This research was supported by the National High Technology Research and Development Program of China (863 Program) (No. 2013AA102703) and the National Natural Science Foundation of China (No. 31370663). Manuscript was polished by XD Wang, College of Plant Protection, Agricultural University of Hebei.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Y. Wang.
Rights and permissions
About this article

Cite this article
Yuan, S., Dong, Y., Zhang, N. et al. Construction of high-efficiency transformation vector with multiple insect-resistant genes and expression in tobacco. Acta Physiol Plant 39, 33 (2017). https://doi.org/10.1007/s11738-016-2338-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-016-2338-9