
Abstract
Key Message
A simple and reliable Agrobacterium -mediated transformation method was developed for switchgrass. Using this method, many transgenic plants carrying multiple genes-of-interest could be produced without untransformed escape.
Abstract
Switchgrass (Panicum virgatum L.) is a promising biomass crop for bioenergy. To obtain transgenic switchgrass plants carrying a multi-gene trait in a simple manner, an Agrobacterium-mediated transformation method was established by constructing a Gateway-based binary vector, optimizing transformation conditions and developing a novel selection method. A MultiRound Gateway-compatible destination binary vector carrying the bar selectable marker gene, pHKGB110, was constructed to introduce multiple genes of interest in a single transformation. Two reporter gene expression cassettes, GUSPlus and gfp, were constructed independently on two entry vectors and then introduced into a single T-DNA region of pHKGB110 via sequential LR reactions. Agrobacterium tumefaciens EHA101 carrying the resultant binary vector pHKGB112 and caryopsis-derived compact embryogenic calli were used for transformation experiments. Prolonged cocultivation for 7 days followed by cultivation on media containing meropenem improved transformation efficiency without overgrowth of Agrobacterium, which was, however, not inhibited by cefotaxime or Timentin. In addition, untransformed escape shoots were completely eliminated during the rooting stage by direct dipping the putatively transformed shoots into the herbicide Basta solution for a few seconds, designated as the ‘herbicide dipping method’. It was also demonstrated that more than 90 % of the bar-positive transformants carried both reporters delivered from pHKGB112. This simple and reliable transformation method, which incorporates a new selection technique and the use of a MultiRound Gateway-based binary vector, would be suitable for producing a large number of transgenic lines carrying multiple genes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Switchgrass (Panicum virgatum L.) is a warm season C4 perennial grass belonging to the subfamily Panicoideae within the family Poaceae (Gramineae). This species is one of the greatest potential bioenergy crops, along with miscanthus (Miscanthus spp.), reed canarygrass (Phalaris arundiacea L.) and the giant reed (Arundo donax L.) (Lewandowski et al. 2003; McLaughlin and Kszos 2005), and has the nature to combine high net energy efficiency with economic feasibility (Schmer et al. 2008). Because switchgrass is a highly self-incompatible out-crossing species, the use of conventional crossing-based breeding is limited. Therefore, molecular breeding techniques are desirable for this species (Bouton 2007, 2008).
Genetic transformation of switchgrass was first reported using a microprojectile bombardment method (Richards et al. 2001), and subsequently using an Agrobacterium-mediated method (Somleva et al. 2002). Recently, various modified Agrobacterium-mediated methods have also been reported (Burris et al. 2009; Li and Qu 2011; Ramamoorthy and Kumar 2012; Song et al. 2012; Xi et al. 2009). Although transgenic switchgrass plants overexpressing/suppressing a unique gene were produced by these transformation methods (Fu et al. 2011; Saathoff et al. 2011; Xu et al. 2011), these methods still had limitations due to transformation efficiency, availability of explants, albino formation or complexity of procedures.
In addition, because of recent accumulation of genetic and genomic information in this crop (Casler et al. 2011), introducing multi-gene traits, such as those in a metabolic pathway, into transgenic plants would be of more interest than introduction of single gene traits. Although several strategies (sexual crossing, re-transformation and cotransformation) have been developed to introduce multiple genes into plant genomes (Dafny-Yelin and Tzfira 2007; Halpin 2005), these methods have several drawbacks, such as complex transgene integration patterns, low coexpression and segregative separation of transgenes in the progeny. Because of the limited efficiency of biolistic transformation, delivery of multiple genes on a single T-DNA by a single Agrobacterium-mediated transformation is preferable. Several strategies have been employed for assembly of multiple gene expression cassettes on a binary vector, such as traditional type II restriction enzyme-mediated cloning, the Cre/loxP-mediated recombination system, rare-cutting enzymes and Gateway-based systems (Dafny-Yelin and Tzfira 2007). The MultiRound Gateway system is based on multiple rounds of two-component Gateway LR recombination (Chen et al. 2006). With this system, multiple transgenes can be delivered sequentially and indefinitely into a Gateway-compatible destination binary vector through alternate use of two series of entry vectors, pL12R34 and pL34R12.
Since caryopsis is the most readily available explant in switchgrass, and Type I callus is the most typical and prevalent type of callus induced from caryopsis (Denchev and Conger 1994; Vasil and Vasil 1994), we tried to establish a simple and reliable Agrobacterium-mediated transformation method for caryopsis-derived Type I calli of Alamo switchgrass. A MultiRound Gateway-compatible destination binary vector, pHKGB110, and a derivative expression binary vector carrying reporter genes, pHKGB112, were constructed and used for transformation experiments. To eliminate the production of untransformed escapes, a novel selection method at the rooting step was also developed.
Materials and methods
Vector construction
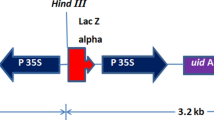
Two fragments derived from pB2GW7 (Karimi et al. 2002), a 35SΩ promoter (P35SΩ) (De Loose et al. 1995) and the selectable marker gene bar with a nopaline synthase terminator (bar-Tnos), were amplified and fused by the overlap extension polymerase chain reaction (OE-PCR) (Horton et al. 1989) to produce the expression cassette, P35SΩ-bar-Tnos. A primer set amplifying the whole fragment was designed for the In-Fusion PCR Cloning System (Clontech Laboratories, Mountain View, CA, USA) with an additional AsiSI recognition sequence. The bar expression cassette was cloned into the EcoRI site in the T-DNA region of the binary vector pPZP200 (Hajdukiewicz et al. 1994) using an In-Fusion Advantage PCR Cloning Kit (Clontech), and this binary vector was designated pHKB1. An AscI-PacI fragment of the entry vector pL34R12H-Cm, which contained attR1-Cmr-attR2 (Chen et al. 2006), was blunted and ligated into the BamHI site of pHKB1 using a Blunting-Convenience Kit (Nippon Gene, Tokyo, Japan), and the resulting destination binary vector was designated pHKGB110 (Fig. 1).

Physical map of the MultiRound Gateway-compatible destination binary vector, pHKGB110
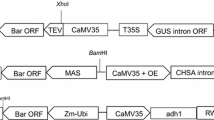
Two reporter genes, GUSPlus (Broothaerts et al. 2005) and gfp (sGFP-TYG) (Chiu et al. 1996), were introduced into pHKGB110 using the MultiRound Gateway system (Chen et al. 2006). A P35SΩ-GUSPlus-Tnos expression cassette was constructed by OE-PCR using pB2GW7 and pCAMBIA1105.1 (CAMBIA, Canberra, Australia) as templates, and was cloned into the HindIII/AscI sites of the entry vector pL12R34H-Ap (Chen et al. 2006) by In-Fusion PCR Cloning. A maize ubi1 gene promoter (Pubi1) (Christensen et al. 1992) -gfp-Tnos expression cassette was similarly constructed using maize genomic DNA (BioChain Institute, Hayward, CA, USA), pBI221/SP-GFP-HDEL (Mitsuhashi et al. 2000) and pB2GW7, and was cloned into the PacI site of the entry vector pL34R12H-Cm. The GUSPlus and gfp expression cassettes were sequentially introduced into pHKGB110 by two rounds of LR reactions (Chen et al. 2006), and the resulting binary vector was designated as pHKGB112 (Fig. 2). This binary vector was transformed into Agrobacterium tumefaciens EHA101 (Hood et al. 1986) using the freeze–thaw method (An et al. 1988) and was used for transformation experiments.

Schematic representation of the T-DNA region of pHKGB112. Double-headed arrows below the map indicate fragments amplified by PCR. The thin arrow below the map indicates the fragment detected by southern blot analysis using a bar-specific probe
Callus induction and maintenance
Mature seeds of switchgrass (Panicum virgatum L.), cultivar Alamo, were purchased from a local supplier. Embryogenic callus was induced from mature caryopses according to the methods of Denchev and Conger (1994). Mature caryopses were dehusked with 60 % (v/v) H2SO4 for 30 min, rinsed with distilled water and sterilized with 3.5 % (w/v) calcium hypochlorite for 30 min. After rinsing three times with sterilized distilled water, caryopses were placed on PVC medium [Murashige and Skoog (MS) salts and vitamins (Murashige and Skoog 1962), 30 g l−1 maltose, 22.6 μM 2,4-dichlorophenoxyacetic acid (2,4-D), 4.4 μM 6-benzylaminopurine (BAP) and solidified with 8 g l−1 agar] and cultured at 28 °C in the dark for 4 weeks. Because switchgrass is a self-incompatible open-pollinated species, calli formed on individual caryopses were maintained as independent genotypic lines. The regeneration ability of Type I (i.e., compact and embryogenic) calli was confirmed by growing the material on PVS medium [MS salts and vitamins, 30 g l−1 maltose, 1.4 μM gibberellic acid (GA3) and 8 g l−1 agar], and cell lines with a high regeneration ability were maintained by subculturing every 4 weeks. Seven independent lines of 6- to 18-month-old calli were used for the transformation experiments.
Agrobacterium-mediated transformation
Transformation experiments were performed according to Somleva et al. (2002) and Somleva (2007) with modifications. A glycerol stock of A. tumefaciens EHA101(pHKGB112) was streaked onto a YEP agar plate supplemented with 50 mg l−1 kanamycin, 100 mg l−1 spectinomycin and 35 mg l−1 chloramphenicol and cultured at 28 °C. After 2–3 days, bacterial cells were inoculated into liquid YEP medium containing the proper selective antibiotics and cultured overnight with shaking. Bacterial cells were collected by centrifugation and resuspended in liquid PVC-CO medium (PVC medium supplemented with 100 μM acetosyringone) at a density of A 600 = 0.5.
Type I calli were divided into 3–4 mm clumps and inoculated with the Agrobacterium suspension for 3 min with occasional agitation. After blotting on sterilized paper towels to remove excess Agrobacterium suspension, calli were transferred to PVC-CO agar plates. After 3–7 days of cocultivation at 28 °C in the dark, calli were transferred to PVC-S medium (PVC medium supplemented with 10 mg l−1 bialaphos and Agrobacterium-suppressing agents) without washing and cultured for 8 weeks at 28 °C in the dark, with bi-weekly subculturing. Bialaphos-resistant (BarR) and/or GFP-expressing (GFP+) calli were then transferred to PVS-S medium (PVS medium supplemented with 10 mg l−1 bialaphos and Agrobacterium-suppressing agents) and cultured for 6 weeks, with bi-weekly subculturing, at 28 °C under a 16/8-h light/dark cycle. The Agrobacterium-suppressing agents used in this study were 150 mg l−1 Timentin (PhytoTechnology Laboratories, Shawnee Mission, KS, USA), 250 mg l−1 cefotaxime sodium salt (Duchefa Biochemie, Haarlem, Netherlands) and 25 or 50 mg l−1 meropenem trihydrate (Wako Pure Chemical Industries, Osaka, Japan).
Herbicide selection of putatively transformed shoots
For rooting and selection, shoots regenerated on PVS-S medium were transferred onto PVR-S medium, comprising PVR medium (half-strength MS salts and vitamins, 15 g l−1 sucrose and 7 g l−1 agar) supplemented with the Agrobacterium-suppressing agents used in the PVS-S medium. Two herbicide selection methods were examined as follows: (1) the conventional method—shoots were placed onto PVR-S medium incorporated with 10 mg l−1 bialaphos; (2) the herbicide dipping method—shoots were dipped into a filter-sterilized 0.1 or 1 % (v/v) Basta (Bayer CropScience, Monheim am Rhein, Germany) solution for a few seconds, briefly blotted on sterilized paper towels, and then placed onto herbicide-free PVR-S medium. For both selection methods, shoots were cultured at 28 °C under a 16/8-h light/dark cycle, and herbicide resistance was evaluated after 2 weeks. Non-transformed control plantlets and putative transgenic plantlets were acclimatized and grown in a hydroponic solution (Mae and Ohira 1981) supplemented with 0.1 % (v/v) Plant Preservative Mixture (Plant Cell Technology, Washington DC, USA) to inhibit microbial growth. Thereafter plants were potted in soil and grown in the greenhouse.
Observation of GFP expression
GFP expression of inoculated calli at the macroscopic level was monitored and recorded using a ProDoc-LED470GFP macro imaging system (OptoCode, Tokyo, Japan). GFP expression of calli and shoots at the microscopic level was observed using a Leica MZ FLIII fluorescence stereomicroscope with filter sets of GFP Plus (excitation 480 ± 40 nm, emission ≥510 nm) and GFP Plant (excitation 470 ± 40 nm, emission 525 ± 50 nm) and recorded using a DFC310 FX digital color camera (Leica Microsystems, Wetzlar, Germany).
PCR analysis of putatively transformed plants
Small (ca. 3 mm2) segments of leaf blades sampled from 2- to 4-week-old putatively transformed plantlets were subjected to PCR using a Phire Plant Direct PCR Kit (Thermo Fisher Scientific, Waltham, MA, USA). PCR was initially performed using a primer set that amplified the virC gene (Sawada et al. 1995) to detect persistent Agrobacterium according to Cubero et al. (1999). Then, virC-negative plant samples were analyzed for transgene integration by PCR. The primer sequences, their annealing temperatures and the expected fragment lengths for each transgene were as follows: virC (VCF: 5′-ATCATTTGTAGCGACT-3′ and VCR: 5′-AGCTCAAACCTGCTTC-3′, 55 °C, 730 bp), bar (5′-CTGAAGTCCAGCTGCCAGAA-3′ and 5′-CATCGTCAACCACTACATCG-3′, 62 °C, 438 bp), GUSPlus (5′-CCACTGAACACGTATCTCTACCAG-3′ and 5′-GGTAGCCATCACAAACAGCA-3′, 62 °C, 621 bp) and gfp (5′-GTAAACGGCCACAAGTTCAG-3′ and 5′-AGATCTCCCTTGTACAGCTCG-3′, 62 °C, 659 bp). The amplification reaction was performed using a MultiGene Gradient thermal cycler (Labnet International, Edison, NJ, USA) using the following thermal cycling conditions: initial denaturation at 98 °C for 5 min followed by 35 cycles of denaturation at 98 °C for 5 s, annealing for 5 s at the temperatures described above, and extension at 72 °C for 20 s kb−1, with a final extension at 72 °C for 1 min. Amplified products were electrophoresed in a 1.5 % agarose gel and visualized by ethidium bromide staining. Because faint false-positive bands were occasionally detected in non-transformed control samples as reported by Xi et al. (2009), only clear, strong bands were determined to be positive.
Reverse-transcription (RT) PCR
Total RNA was extracted from leaves using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany), and first-strand cDNA was synthesized from total RNA using a PrimeScript II 1st Strand cDNA Synthesis Kit with an oligo-dT primer (Takara). RT-PCR analyses were performed using Premix Ex Taq Hot Start Version (Takara) and four primer sets for the elf1 gene as an internal control (Xi et al. 2009) and the three transgenes used for genomic PCR. All experiments were performed according to the manufacturer’s instructions.
Histochemical GUS (β-glucuronidase) assay
GUS assay was performed by 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (X-gluc) staining according to Kosugi et al. (1990). Tissue samples excised from transformed plants were stained with X-gluc solution at 37 °C overnight, and were fixed and cleared with ethanol before observation.
Southern blot hybridization of transgenic plants
Genomic DNA was extracted from leaf blades of PCR-positive plants using a Phytopure DNA Extraction Kit (GE Healthcare, Buckinghamshire, UK) and digested with Xma I. 15 μg of digested DNAs were electrophoresed in 0.8 % agarose gel, transferred onto a nylon membrane and hybridized with a bar gene-specific probe prepared using a PCR DIG Probe Synthesis Kit (Roche Applied Science, Mannheim, Germany) and the primer set described above. Hybridization and detection were performed with the DIG system according to the instruction manual (Roche Applied Science).
Experimental design and statistical analysis
All plant tissue culture media were buffered with 1 % (v/v) 2.5 mM 2-(N-morpholino)ethanesulfonic acid (pH 5.9) to adjust the pH to ca. 5.7 before and after autoclaving. Plates containing 40 ml of agar medium and cultures were sealed with Micropore surgical tape (3 M, St. Paul, MN, USA). Fifty calli and five shoots were used for transformation and herbicide selection experiments, respectively. The experiments were independently repeated three times and the data were represented as the mean ± standard deviation (SD) of the replicates. Analysis of variance was performed using single-factor ANOVA and followed by the Tukey–Kramer method using Excel 2003 software (Microsoft, Redmond, WA, USA) with the add-in software, Statcel3 (Yanai 2011).
Results
Construction of destination binary vector and expression binary vector
MultiRound Gateway (Chen et al. 2006) is one of the most useful systems for constructing a binary vector carrying multiple expression cassettes. To utilize this system for switchgrass transformation, we first constructed a MultiRound Gateway-compatible binary vector, pHKGB110 (Fig. 1), by introducing an attR1-Cmr-attR2 fragment for the LR recombination reaction as well as a selectable marker cassette, P35SΩ-bar-Tnos, into an empty binary vector, pPZP200. Subsequently, an expression binary vector, pHKGB112 (Fig. 2), was constructed for transformation experiments using a MultiRound Gateway. Two expression cassettes of reporter genes, GUSPlus and bar, could be easily constructed on two independent entry vectors, pL12R34H-Ap and pL34R12H-Cm, by general molecular techniques, and simply introduced into pHKGB110 by two sequential LR reactions. The resultant binary vector pHKGB112 was transformed into A. tumefaciens EHA101, and this strain was used for all switchgrass transformation experiments.
Effect of Agrobacterium-suppressing antibiotics on the transformation of Type I callus of switchgrass
For efficient recovery of transgenic plants, it is essential to select a suitable antibiotic for suppressing the overgrowth of Agrobacterium after cocultivation. In the first experiment, calli inoculated with EHA101 (pHKGB112) were cocultivated for 5 days as previously recommended by Somleva (2007) and were then cultured on medium supplemented with various antibiotics to suppress Agrobacterium overgrowth. Cefotaxime of 250 mg l−1, Timentin of 150 mg l−1 and meropenem of 25 and 50 mg l−1 were used in this study, and Agrobacterium overgrowth and transformation efficiency were evaluated (Table 1).
Cefotaxime of 250 mg l−1 failed to suppress overgrowth of Agrobacterium on almost all inoculated calli and also failed to produce any transgenic plants (Fig. 3a). Timentin of 150 mg l−1 allowed Agrobacterium overgrowth during both the callus selection and shoot regeneration stages at moderate frequency (18.7 %) (Fig. 3b). Meropenem of 25 mg l−1 also allowed Agrobacterium overgrowth at a similar frequency to Timentin. However, overgrowth of Agrobacterium was observed at the callus selection stage but not at the regeneration and rooting stages. Among the antibiotics tested, only 50 mg l−1 meropenem completely suppressed the overgrowth of Agrobacterium on inoculated calli (Fig. 3c).

Transformation of Type I calli of Alamo switchgrass. Calli inoculated with A. tumefaciens EHA101 (pHKGB112) and cultured on PVC-S medium supplemented with: a 250 mg l−1 cefotaxime; b 150 mg l−1 Timentin; c 50 mg l−1 meropenem. Overgrowth of Agrobacterium appears as white halos in (a) and (b). d Fluorescent image of c showing GFP expression. e Shoot regeneration of inoculated calli cultured on PVS-S medium supplemented with 50 mg l−1 meropenem. Transgenic plants cultivated in: f hydroponic solution in a growth chamber; g potted soil in a greenhouse. h X-gluc-stained internode section showing GUSPlus expression with weak blue staining in the stem (inner section), strong staining in an immature leaf (middle section) and no staining in the leaf sheath (outer section). i Shoot showing strong GFP expression fluorescence in the emerging leaf blade (white arrowhead), no fluorescence in the mature leaf blade (black arrowhead), and weak fluorescence in the leaf sheath (gray arrowhead)
Most calli survived and grew slightly on medium containing 10 mg l−1 bialaphos during the first 2–4 weeks of the callus selection stage. After the subsequent 6- to 8-week period, vigorously growing bialaphos-resistant (BarR) calli and severely browning bialaphos-sensitive (BarS) calli could be distinguished by visual inspection (Fig. 3c). Green fluorescent areas expressing the reporter GFP (GFP+) were observed on BarR calli (Fig. 3d), which eventually regenerated BarR shoots (Fig. 3e). GFP+ cells were also observed in bialaphos-sensitive/partially resistant calli as small foci (<1 mm diameter), but these calli generally failed to regenerate shoots. This result suggests that the expression level is different between the bar gene driven by a CaMV 35S-based promoter and the GUSPlus gene driven by a maize ubi1 promoter in switchgrass. The formation rates of BarR callus, GFP+ callus and BarR shoots did not vary among the antibiotics used except for cefotaxime (Table 1). Although treatment with 150 mg l−1 Timentin and 50 mg l−1 meropenem occasionally caused regeneration of albino shoots, these shoots formed with a frequency of less than 1.3 % (Table 1).
BarR shoots that were rooted on PVR-S medium containing 10 mg l−1 bialaphos and grown in hydroponic solution were subjected to PCR analysis to confirm Agrobacterium persistence and transgene integration (Fig. 4). Although the virC gene was detected in small plantlets at both the rooting and acclimatization stages, most of the plantlets grown in hydroponic culture became virC-negative 2–4 weeks after acclimatization. In subsequent PCR analyses using the bar-specific primer set, positive fragments could not be amplified from some of the BarR plantlets, which also did not show GFP fluorescence. Interestingly, these untransformed escapes were most frequently obtained with the 150 mg l−1 Timentin treatment (24 % of BarR events) (Table 1).

PCR analyses of putatively transformed BarR plantlets for virC, bar, GUSPlus and gfp genes. M marker, P A. tumefaciens EHA101 (pHKGB112), N non-transformed control plantlet, 1–6 putatively transformed plantlets treated with 50 mg l−1 meropenem, 7–12 putatively transformed plantlets treated with 150 mg l−1 Timentin
When 50 mg l−1 meropenem was used as an Agrobacterium-suppressing agent throughout the transformation, BarR plantlets were recovered at the highest efficiency (17.3 %) without overgrowth of Agrobacterium and the appearance of escape plantlets (Table 1). In addition, 25 mg l−1 meropenem successfully suppressed the overgrowth of Agrobacterium at the regeneration and rooting stages and produced BarR plantlets at a high frequency comparable to 50 mg l−1 meropenem. Therefore, 50 mg l−1 meropenem was used in the PVC-S medium for callus selection and 25 mg l−1 meropenem was used in the PVS-S and PVR-S media for regeneration and rooting, respectively, in subsequent experiments.
Cocultivation period for efficient recovery of transgenic switchgrass
Since we found that meropenem was effective for suppressing Agrobacterium overgrowth and producing transgenic switchgrass plants, we subsequently compared different cocultivation periods (3, 5, and 7 days) for further optimization of the transformation protocol. Transformation efficiencies (all BarR calli, shoots and plantlets) were positively correlated with the length of the cocultivation period (Table 2). The efficiency of BarR-plant recovery obtained with cocultivation for 7 days (17.3 %) was the highest among the tested conditions, and was 3.3- and 1.5-fold higher than with cocultivation for 3 and 5 days, respectively. The effectiveness of prolonged cocultivation was also confirmed in five other cell lines induced from independent caryopses that had different genotypes. All and four out of five lines failed to recover BarR plants with 3 and 5 days of cocultivation, respectively. In contrast, all lines produced BarR plants with 7 days of cocultivation at 2–12 % efficiency.
Novel selection of putatively transformed shoots
Selection of putatively transformed shoots was conventionally performed by placing shoots onto rooting medium supplemented with a selective agent. In this study, non-transformed switchgrass shoots partially survived and occasionally rooted on PVR-S medium supplemented with 10 mg l−1 bialaphos after 2 weeks of culture (Fig. 5). To develop a simpler and more reliable selection method, we tried a new selection technique as follows: shoots were dipped in herbicide solution (0.1 or 1 % Basta) for a few seconds, and then placed onto PVR-S medium without a selective agent. This was designated as the “herbicide dipping method”. Using this method, non-transformed shoots showed severe chlorosis within a week and were completely dead after 2 weeks. Conversely, transformed shoots rooted within 1 week after herbicide dipping even with 1 % Basta. After 2 weeks, they developed into healthy plantlets, which were similar to shoots grown without selection and were able to acclimatize. Integration of the bar gene into all of the resistant plantlets was confirmed by PCR.

Selection of regenerated shoots by the conventional and herbicide dipping methods
Multi-gene transformation with the MultiRound Gateway binary vector
Additional PCR analyses were performed on bar-positive plants (assessed by PCR) using specific primers for the GUSPlus and gfp genes (Table 3). Among the 128 bar-positive transformants examined, 119 (93.0 %) carried both reporter genes, and only 6 (4.7 %) and 3 (2.3 %) lacked either one or both reporters, respectively. These frequencies were similar to those of optimized transformation conditions (7 days of cocultivation, the use of meropenem for suppression of Agrobacterium, and selection by the herbicide dipping method).
These transgenic plants grew well in both hydroponic solution and soil (Fig. 3f, g). Expression of transgenes was examined by RT-PCR in transgenic plants carrying the three transgenes (bar, GUSPlus and gfp). Transcription products of all transgenes were detected from all plants tested (Fig. 6). GUSPlus expression in transgenic plants was confirmed by X-gluc staining, with blue staining clearly observed in immature tissues but absent or only slight in mature tissues (Fig. 3h). GFP expression fluorescence was also tissue dependent (Fig. 3i), but strong fluorescence was observed in all parts of the plant in some plants. These results indicated different promoter activities between the CaMV 35S-based promoter for GUSPlus and the ubi1 promoter for gfp in switchgrass, as was similarly observed in GFP-expressing calli showing bialaphos sensitivity/partial resistance. Southern blot hybridization confirmed integration of a T-DNA fragment containing the bar gene in all transgenic lines tested (Fig. 7). Six out of eight transgenic lines carried the bar gene in a single copy or low (2–3) copies. Signals smaller than the expected size (≥2.2 kb) were occasionally observed in lines carrying multiple copies of the transgenes, suggesting that the T-DNA was truncated at the left border sequence in these transgenic lines.

RT-PCR analyses of transgenic switchgrass plants for elf1, bar, GUSPlus and gfpgenes. M marker, N non-transformed control plant, 1–10 transgenic plants

Southern blot hybridization analysis of transgenic switchgrass plants for integration of bar gene. M marker, P pHKGB112, N non-transformed control plant, 1–8 transgenic plants
Discussion
Here, we established a simple and reliable method for multi-gene transformation of switchgrass that incorporates the construction and use of a MultiRound Gateway-compatible binary vector, the improvement of a transformation method for caryopsis-derived embryogenic Type I calli, and the establishment of a novel selection technique. The Gateway system is widely used for construction of plant transformation vectors (Karimi et al. 2002), and Gateway-compatible vectors for switchgrass and other monocots have been reported (Mann et al. 2012). However, most of these vectors were designed for construction of a single gene vector. The MultiRound Gateway is a simple, efficient and more flexible system for multi-gene assembly (Chen et al. 2006). Once the DNA fragments of interest are cloned into the two entry vectors, they can be introduced into a destination vector in a defined order and orientation through multiple rounds of Gateway reactions. In principle, the procedure may be repeated an infinite number of times. In this work, the binary vector pHKGB112 (Fig. 2) was constructed by introducing two reporter genes into the destination vector pHKGB110 (Fig. 1) using the MultiRound Gateway technique. By transformation with pHKGB112, transformants carrying both reporters as well as a selectable marker could be recovered at a high frequency (more than 90 % of Bar-resistant transformants). Although only three genes were stacked in a single T-DNA in the present proof-of principle experiments, it has been reported that eight genes were successfully introduced into a T-DNA by four rounds of LR recombination reactions (Chen et al. 2006). In addition, a series of binary vectors carrying various sets of genes-of-interest could be easily constructed by combining entry vectors harboring different genes (Ren et al. 2010). We have already been constructed various binary vectors carrying multiple genes-of-interest using this system and the work for switchgrass transformed with these binary vectors is in progress. In coupled with these findings, we demonstrated that the MultiRound Gateway would be a useful tool for introducing a series of genes, e.g., genes related to a unique metabolic pathway, in a single transformation for switchgrass.
We chose caryopsis-derived Type I calli for the explants of transformation experiments in this study. Embryogenic Type I callus is the most typical and prevalent type of callus induced from the most readily available explant type in switchgrass, the caryopsis (Denchev and Conger 1994; Vasil and Vasil 1994). Inflorescence-derived callus and friable Type II callus were used in other recent reports, but the former is limited in the time available for callus induction and the latter potentially induces albino shoots, particularly under prolonged cultivation (Li and Qu 2011; Ramamoorthy and Kumar 2012). In the present study, Type I callus lines were successfully used for transformation after at least 18 months of subculture without increasing albino shoot formation.
Cocultivation is an essential period for introducing transgenes from Agrobacterium cells to plant cells, and suppression of Agrobacterium overgrowth after cocultivation is important to recover transgenic plants. Somleva (2007) suggested that the optimal period for cocultivation is 3–5 days depending on the target explants, and the optimal cocultivation period is shorter for somatic embryos than for callus. Carbenicillin, cefotaxime and Timentin were employed alone or in combination for the transformation of switchgrass in previous reports. Xi et al. (2009) reported that the placement of inoculated calli directly onto agar-solidified medium during cocultivation led to overgrowth of Agrobacterium in subsequent cultivation with 250 mg l−1 cefotaxime. Thus, they cocultivated calli on filter papers wetted with medium for only 2 days. Other researchers also performed short cocultivation (2–4 days) and additional treatments after cocultivation such as washing with water/liquid medium/antibiotic solution (Burris et al. 2009; Ramamoorthy and Kumar 2012; Song et al. 2012) and resting on selective agent-free medium (Li and Qu 2011). Meropenem is a derivative of carbapenem and possesses high bactericidal activity against Agrobacterium and low phytotoxicity (Ogawa and Mii 2004, 2005, 2007). We found that 50 mg l−1 meropenem successfully suppressed the overgrowth of Agrobacterium throughout transformation, and that 25 mg l−1 meropenem was sufficient to suppress overgrowth at the regeneration and rooting stages, even after 5 days of cocultivation on agar medium (Table 1). Other antibiotics at elevated concentration may suppress overgrowth of Agrobacterium, while chemical cost also elevates and transformation efficiency would be affected negatively by their phytotoxicity (Ogawa and Mii 2007). Using meropenem, cocultivation could be prolonged to 7 days without overgrowth of Agrobacterium, and this simple modification achieved 3.3- and 1.5-fold higher efficiency than 3 and 5 days of cocultivation, respectively (Table 2). Compact Type I calli comprise almost entirely tightly-packed meristematic cells (Vasil and Vasil 1994) and somatic embryogenesis of mature caryopsis-derived Type I calli of switchgrass was observed to take place in embedded areas of callus tissue (Denchev and Conger 1994). Therefore, the prolonged cocultivation period may facilitate the invasion and attachment of Agrobacterium to transformation-competent plant embryogenic cells existing in embedded callus areas.
When 150 mg l−1 Timentin was used in the present study, untransformed escape plants were frequently recovered (Table 1) at levels similar to those reported by Somleva et al. (2002). Because Timentin and other penicillin derivatives have been reported to have auxin-like effects on various plant species/tissues (Holford and Newbury 1992; Nauerby et al. 1997; Robert et al. 1989), the formation of untransformed escapes on switchgrass calli might have been caused by unexpected growth of untransformed cells by supra-optimal concentrations of auxin. It was reported that untransformed escapes were also formed using the bar gene as a selectable marker to show herbicide resistance, which is a useful trait for field-grown transgenic plants and is preferred to omit horizontal transfer of antibiotic resistance gene to microbes. To identify escapes of regenerated shoots, Somleva et al. (2002) repeatedly applied a 0.1 % Basta solution to the leaf segments of in vitro- and greenhouse-grown putative transformants. To overcome the inconvenience of this method, we established a novel selection technique for complete elimination of untransformed escapes during the rooting stage (Fig. 5). The herbicide Basta contains glufosinate ammonium, which is a broad-spectrum, non-selective post-emergence herbicide that functions by inhibiting glutamine synthetase activity in plants, and is recommended for foliage application by spraying with a 100-fold dilution (final glufosinate concentration of 0.185 %) for perennial grass weeds (Bayer CropScience 2008). We succeeded in applying herbicide to foliage as follows: shoots of putative transformants were dipped in an herbicide solution and placed onto selective agent-free rooting medium. This technique, herbicide dipping selection, is simple and effective for the recovery of genuine transgenic switchgrass plants before acclimatization and cultivation, and could be used in the transformation of other plant species. Since Xi et al. (2009) used a hygromycin dipping test to detect hph expression in leaf blades of greenhouse-grown plants, we expect that the technique can be utilized in transformants carrying antibiotic resistance markers.
Using the present transformation method, independent transgenic switchgrass plants were recovered at a transformation efficiency of 2.0–17.4 % from all seven tested genotypes of the cultivar Alamo. These efficiencies were comparable to previous studies; 3.4 % by Somleva et al. (2002), 4.4 % by Burris et al. (2009) and 6 % by Ramamoorthy and Kumar (2012). Li and Qu (2011) have been reported a high-throughput transformation method achieving 56.6 % of transformation efficiency for Alamo, while this protocol is very laborious and tends to induce albino shoots. Because switchgrass is a self-incompatible, open-pollinated species, calli formed on an individual caryopsis can be of different genotypes. The present transformation method for Type I callus, which can be maintained long-term, would be suitable for obtaining a large set of transgenic plants carrying the same genetic background. In addition, a MultiRound Gateway-compatible vector would be useful to introduce multiple transgenes of interest in a single transformation. It would be interesting to determine whether the present transformation method could be applied to other cultivars and, moreover, could be improved to get the transformation efficiency to the maximum level reported previously.
Abbreviations
- MS:
-
Murashige and Skoog
- OE-PCR:
-
Overlap extension PCR
- PCR:
-
Polymerase chain reaction
- RT-PCR:
-
Reverse-transcription PCR
- SD:
-
Standard deviation
Referencess
An G, Ebert PR, Mitra A, Ha SB (1988) Binary vectors. In: Gelvin SB, Shilperoort RA (eds) Plant molecular biology manual. Kluwer Academic Publishers, Dordrecht, pp 1–19
Bayer CropScience (2008) Basta. http://www.basta.jp/index.html. Accessed 15 Jan 2014
Bouton JH (2007) Molecular breeding of switchgrass for use as a biofuel crop. Curr Opin Genet Dev 17:553–558
Bouton J (2008) Improvement of switchgrass as a bioenergy crop. In: Vermerris W (ed) Genetic improvement of bioenergy crops. Springer, New York, pp 295–308
Broothaerts W, Mitchell HJ, Weir B, Kaines S, Smith LMA, Yang W, Mayer JE, Roa-Rodríguez C, Jefferson RA (2005) Gene transfer to plants by diverse species of bacteria. Nature 433:629–633
Burris JN, Mann DGJ, Joyce BL, Stewart CN Jr (2009) An improved tissue culture system for embryogenic callus production and plant regeneration in switchgrass (Panicum virgatum L.). Bioenerg Res 2:267–274
Casler MD, Tobias CM, Kaeppler SM, Buell CR, Wang Z-Y, Cao P, Schmutz J, Ronald P (2011) The switchgrass genome: tools and strategies. Plant Genome 4:273–282
Chen Q-J, Zhou H-M, Chen J, Wang X-C (2006) A Gateway-based platform for multigene plant transformation. Plant Mol Biol 62:927–936
Chiu W, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6:325–330
Christensen AH, Sharrock RA, Quail PH (1992) Maize polyubiquitin genes-structure, thermal perturbation of expression and transcript splicing, and promoter activity following transfer to protoplasts by electroporation. Plant Mol Biol 18:675–689
Conger BV (2002) Development of in vitro systems for switchgrass (Panicum virgatum). Final Report for 1992–2002. ORNL/SUB-02-11XSY161/01. http://bioenergy.ornl.gov/pdfs/ornlsub_02_11xsy16101.pdf. Accessed 15 Jan 2014
Cubero J, Martínez MC, Llop P, López MM (1999) A simple and efficient PCR method for the detection of Agrobacterium tumefaciens in plant tumours. J Appl Microbiol 86:591–602
Dafny-Yelin M, Tzfira T (2007) Delivery of multiple transgenes to plant cells. Plant Physiol 145:1118–1128
De Loose M, Danthinne X, Van Bockstaele E, Van Montagu M, Depicker A (1995) Different 5’ leader sequences modulate β-glucuronidase accumulation levels in transgenic Nicotiana tabacum plants. Euphytica 85:209–216
Denchev PD, Conger BV (1994) Plant regeneration from callus cultures of switchgrass. Crop Sci 34:1623–1627
Fu C, Mielenz JR, Xiao X, Ge Y, Hamilton C, Rodriguez M, Chen F, Foston M, Ragauskas A, Bouton J, Dixon RA, Wang Z-Y (2011) Genetic manipulation of lignin reduces recalcitrance and improves ethanol production from switchgrass. Proc Natl Acad Sci USA 108:3803–3808
Hajdukiewicz P, Svab Z, Maliga P (1994) The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol Biol 25:989–994
Halpin C (2005) Gene stacking in transgenic plants—the challenge for 21st century plant biotechnology. Plant Biotechnol J 3:141–155
Holford P, Newbury HJ (1992) The effects of antibiotics and their breakdown products on the in vitro growth of Antirrhinum majus. Plant Cell Rep 11:93–96
Hood EE, Helmer GL, Fraley RT, Chilton MD (1986) The hypervirulence of Agrobacterium tumefaciens A281 is encoded in a region of pTiBo542 outside of T-DNA. J Bacteriol 168:1291–1301
Horton RM, Hunta HD, Ho SN, Pullen JK, Pease LR (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68
Karimi M, Inzé D, Depicker A (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7:193–195
Kosugi S, Ohashi Y, Nakajima K, Arai Y (1990) An improved assay for β-glucuronidase in transformed cells: methanol almost completely suppresses a putative endogenous β-glucuronidase activity. Plant Sci 70:133–140
Lewandowski I, Scurlock JMO, Lindvall E, Christou M (2003) The development and current status of perennial rhizomatous grasses as energy crops in the US and Europe. Biomass Bioenerg 25:335–361
Li R, Qu R (2011) High throughput Agrobacterium-mediated switchgrass transformation. Biomass Bioenerg 35:1046–1054
Mae T, Ohira K (1981) The remobilization of nitrogen related to leaf growth and senescence in rice plants (Oryza sativa L.). Plant Cell Physiol 22:1067–1074
Mann DG, Lafayette PR, Abercrombie LL, King ZR, Mazarei M, Halter MC, Poovaiah CR, Baxter H, Shen H, Dixon RA, Parrott WA, Stewart CN Jr. (2012) Gateway-compatible vectors for high-throughput gene functional analysis in switchgrass (Panicum virgatum L.) and other monocot species. Plant Biotechnol J 10:226–236
McLaughlin SB, Kszos LA (2005) Development of switchgrass (Panicum virgatum) as a bioenergy feedstock in the United States. Biomass Bioenerg 28:515–535
Mitsuhashi N, Shimada T, Mano S, Nishimura M, Hara-Nishimura I (2000) Characterization of organelles in the vacuolar-sorting pathway by visualization with GFP in tobacco BY-2 cells. Plant Cell Physiol 41:993–1001
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Nauerby B, Billing K, Wyndaele R (1997) Influence of the antibiotic timentin on plant regeneration compared to carbenicillin and cefotaxime in concentrations suitable for elimination of Agrobacterium tumefaciens. Plant Sci 123:169–177
Ogawa Y, Mii M (2004) Screening for highly active β-lactam antibiotics against Agrobacterium tumefaciens. Arch Microbiol 181:331–336
Ogawa Y, Mii M (2005) Evaluation of twelve β-lactam antibiotics for Agrobacterium-mediated transformation through in planta antibacterial activities and phytotoxicities. Plant Cell Rep 23:736–743
Ogawa Y, Mii M (2007) Meropenem and moxalactam: novel β-lactam antibiotics for efficient Agrobacterium-mediated transformation. Plant Sci 172:564–572
Ramamoorthy R, Kumar PP (2012) A simplified protocol for genetic transformation of switchgrass (Panicum virgatum L.). Plant Cell Rep 31:1923–1931
Ren F, Chen Q-J, Xie M, Li L-J, Wu W-H, Chen J, Wang X-C (2010) Engineering the K+ uptake regulatory pathway by MultiRound Gateway. J Plant Physiol 167:1412–1417
Richards HA, Rudas VA, Sun H, McDaniel JK, Tomaszewski Z, Conger BV (2001) Construction of a GFP-BAR plasmid and its use for switchgrass transformation. Plant Cell Rep 20:48–54
Robert ML, Flores MR, Loyola-Vargas VM (1989) Growth promoting activity of certain penicillins on cultivated cells of Bouvardia ternifolia. Phytochemistry 28:2659–2662
Saathoff AJ, Sarath G, Chow EK, Dien BS, Tobias CM (2011) Downregulation of cinnamyl-alcohol dehydrogenase in switchgrass by RNA silencing results in enhanced glucose release after cellulose treatment. PLoS One 6:e16416. doi:10.1371/journal.pone.0016416
Sawada H, Ieki H, Matsuda I (1995) PCR detection of Ti and Ri plasmids from phytopathogenic Agrobacterium strains. Appl Environ Microbiol 61:828–831
Schmer MR, Vogel KP, Mitchell RB, Perrin RK (2008) Net energy of cellulosic ethanol from switchgrass. Proc Natl Acad Sci USA 105:464–469
Somleva MN (2007) Switchgrass (Panicum virgatum L.). In: Wang K (ed) Agrobacterium protocols, methods in molecular biology 344, vol 2. Humana Press, Totowa, pp 65–74
Somleva MN, Tomaszewski Z, Conger BV (2002) Agrobacterium-mediated genetic transformation of switchgrass. Crop Sci 42:2080–2087
Song GQ, Walworth AE, Hancock JF (2012) Factors influencing Agrobacterium-mediated transformation of switchgrass cultivars. Plant Cell Tissue Org Cult 108:445–453
Vasil IK, Vasil V (1994) In vitro culture of cereals and grasses. In: Vasil IK, Thorpe TA (eds) Plant cell and tissue culture. Kluwer Academic Publishers, Dordrecht, pp 293–312
Xi Y, Fu C, Ge Y, Nandakumar R, Hisano H, Bouton J, Wang Z-Y (2009) Agrobacterium-mediated transformation of switchgrass and inheritance of the transgenes. Bioenerg Res 2:275–283
Xu B, Escamilla-Treviño LL, Sathitsuksanoh N, Shen Z, Shen H, Zhang YHP, Dixon RA, Zhao B (2011) Silencing of 4-coumarate:coenzyme A ligase in switchgrass leads to reduced lignin content and improved fermentable sugar yields for biofuel production. New Phytol 192:611–625
Yanai H (2011) Statcel—the useful addin forms on Excel, 3rd edn. OMS Publishing, Tokorozawa
Acknowledgments
We thank Dr. Xue-Chen Wang and Dr. Qi-Jun Chen (China Agricultural University, China) for their gift of pL12R34H-Ap and pL34R12H-Cm, Dr. Pal Maliga (Waksman Institute, USA) for his gift of pPZP200, the VIB Department of Plant Systems Biology (Belgium) and CAMBIA (Australia) for providing pB2GW7 and pCAMBIA1105.1, respectively. We are also grateful to Shuji Nitta and Miyoko Sakai (HRI-JP) for their management, support and encouragement of this research project. This work was funded by the New Energy and Industrial Technology Development Organization (NEDO), Japan, in the framework of “Development of Preparatory Basic Bioenergy Technology”.
Conflict of interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Ebinuma.
Rights and permissions
About this article
Cite this article
Ogawa, Y., Shirakawa, M., Koumoto, Y. et al. A simple and reliable multi-gene transformation method for switchgrass. Plant Cell Rep 33, 1161–1172 (2014). https://doi.org/10.1007/s00299-014-1605-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-014-1605-8