
Abstract
Fungal biodiversity significantly influences ecosystem dynamics through various interactions with plants, ranging from pathogenic to mutually beneficial associations. This study explores the fungal diversity associated with an ornamental orchid genus Epidendrum that is widely propagated globally but native to northern South America. Root samples were collected from Epidendrum in diverse geographic locations: Brazil, Colombia, Germany, Spain and six South African provinces. Fungal biodiversity was catalogued from the genomic DNA extracted from these roots using fungal-specific primers and Illumina MiSeq sequencing. Bioinformatic and statistical analyses revealed significant fungal diversity in the roots, with distinct dominant orders in each geographic region. Among the South African samples, significant differences were found in alpha diversity indices and species richness. Even though samples originating from different provinces overlapped in the PCoA plot, PERMANOVA indicated a significant difference in the fungal biodiversity, which was further supported by PERMDISP. In the global dataset, alpha diversity indices were insignificant, but species richness was. In the PCoA plot, data points clustered by sampling sites, indicating substantial differences in fungal biodiversity between the samples. This was validated by PERMANOVA and PERMDISP analyses. Outcomes from the core fungal analyses showed Epidendrum retained a conserved set of fungal orders from its native habitat when it transitioned to exotic regions, while it also formed new associations with local fungal communities in these introduced regions. These findings highlight the role of both core and region-specific fungal communities in the ecological adaptability and success of this widely planted orchid genus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungal biodiversity plays a crucial role in the functioning of ecosystems (Genevieve et al. 2019; Delgado-Baquerizo et al. 2016). This is because fungi can easily adapt and can form different interactions with plants, which can be either pathogenic, beneficial, or commensal (Zeilinger et al. 2016). For instance, beneficial fungi maintain important processes in all ecosystems such as litter decomposition, nutrient cycling and acquisition, improving plant vigour and resilience against pathogens, and facilitating or preventing habitat colonization by plants (Heilmann-Clausen et al. 2015; Trivedi et al. 2020).
Successful plant invasions and subsequent establishment often require plants to establish novel mutualistic interactions with microbes native to new environments (Nuñez and Dickie 2014). The availability of amenable mutualists can be an important limitation on the ability of invasive plants to successfully establish in an introduced habitat. Some plants can replace native mutualists with those present in their introduced ranges (Orlovich and Cairney 2004), while other plants are introduced together with mutualists from their native ranges (Nuñez and Dickie 2014). For example, mycorrhizal fungi were introduced with conifer (Pinaceae) hosts into the Southern Hemisphere (Dickie et al. 2010). Several exotic plants require fungal partners for successful invasions, such as Acacia longifolia and Robinia pseudoacacia which require co-invasion and novel associations respectively (Rodríguez-Echeverría 2010; Wei et al. 2009).
Some orchids can also be invasive, particularly due to their ability to adapt and thrive in new environments, such as Epipactis helleborine, Arundina graminifolia and Disa bracteata (Kolanowska 2013; Kolanowska and Konowalik 2014; Konowalik and Kolanowska 2018). It is common for orchids to co-invade with specialized fungi such as mycorrhizae and other non-mycorrhizal endophytes as they are introduced into new habitats (McCormick et al. 2018; Bonnardeaux et al. 2007; McCormick and Jacquemyn 2014). Orchids are dependent on these microorganisms for seed germination, growth, development and survival (Nuñez and Dickie 2014; Favre-Godal et al. 2020; Smith and Read 2008). Although it is widely understood that mycorrhizal fungi are necessary for the life cycle of orchids, little is known about how the overall fungal diversity and community composition differ within and between closely related orchid taxa (Jacquemyn et al. 2010).
High-throughput sequencing of fungal barcoding regions is used for resolving the broad taxonomic spectrum of fungi present in plants (Xu 2016). Recent studies have used high-throughput sequencing to unravel the microbial community that is associated with different orchids. For example, in a study by Esposito et al. (2016), the authors investigated the variance in the mycorrhizal communities of two closely related orchid species, Platanthera bifolia and P. chlorantha, as well as probable hybrids. Makwela et al. (2022b) used high-throughput sequencing to uncover the mycorrhizal community of two Habenaria orchids native to South Africa. Similar studies have been conducted to understand the composition of microbial communities that are associated with orchids (Makwela et al. 2022a; Böhmer et al. 2020; Huang et al. 2022; Park et al. 2018; Pecoraro et al. 2017).
From the few studies conducted, a majority have investigated the fungal diversity associated with orchids in a restricted geographic range. Only a few studies have investigated the distribution of microbial composition associated with specific orchids in a wider geographic area. For example, Davis et al. (2015) investigated the distribution of the mycorrhizal fungi of Pheladenia deforms across the Australian continent. Duffy et al. (2019) investigated the latitudinal variation in mycorrhizal diversity that is associated with Spiranthes spiralis. Nonetheless, there is no information on the fungal composition and its relationship with the distribution and establishment of any orchid globally.
The genus Epidendrum is one of the largest orchid genera in the Neotropics (Pinheiro and Cozzolino 2013) and includes many important ornamental plants. This allowed us to sample orchids from different geographic locations in this study and catalogue root-associated fungal diversity using a high-throughput sequencing platform. The present study aimed to (a) evaluate the fungal diversity associated with Epidendrum across various global locations, as well as within South Africa, and (b) identify the distinct and shared fungal taxa between these geographic locations and within South Africa. We hypothesized that Epidendrum would maintain a core group of conserved fungal taxa that overlaps among different geographic regions. However, we also anticipated that in each region there would be a unique fungal community.
Materials and methods
Sampling and preparation of root samples
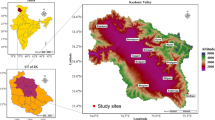
Root samples from Epidendrum were collected across various geographic locations, including Brazil, Colombia, Germany, Spain and South Africa (Table 1). In Brazil, Germany and Spain, three samples each were collected, whereas in Colombia, four samples were procured, with one from the field and three from a nursery. In South Africa, four root samples were collected from five nurseries located in the provinces of Gauteng, KwaZulu-Natal, Limpopo, Mpumalanga and North-West and three root samples from the Western Cape (Table 1; Fig. 1, A, B). Each sample was collected from the apical part of young roots, which had a thin velamen and a diameter of 0.2–0.35 cm and were penetrating the substrate surface (Fig. 1C).

Epidendrum root sampling sites at A a commercial nursery in Brits, North-West and B a private garden near Durban, Kwa-Zulu Natal. C All the root samples were 0.2–0.35 cm in diameter with a thin velamen
Irrespective of the collection sites, all root samples underwent a similar sample preparation protocol. The root samples were first rinsed with sterile deionized water to remove organic matter, followed by surface sterilization with 0.5% (v/v) sodium hypochlorite solution for 2 min. After sterilization, these root samples were rinsed thrice with sterile deionized water, freeze-dried and stored at − 20 °C until extraction of genomic DNA. All non-South African root specimens had their DNA extracted and shipped in a dehydrated form using the same DNA extraction kit listed below.
Extraction and high-throughput sequencing of genomic DNA
The freeze-dried root samples were pulverized using liquid nitrogen and a sterile mortar and pestle. In between pulverization of each root sample, the mortar and pestle were cleansed with detergent followed by 0.5% (v/v) hypochlorite solution and then rinsed thrice with sterile deionized water.
The total genomic DNA from each root sample was extracted individually using the Invisorb®Spin Plant Mini Kit (Invitek Molecular, Germany) following the manufacturer’s protocol and quantified using a NanoDrop™ OneC Microvolume Spectrophotometer (Thermo Scientific, USA). To confirm the presence of DNA, all the extracts were subjected to a polymerase chain reaction (PCR) amplification using fungal-specific primers ITS1F (5’-CTTGGTCATTTAGAGGAAGTAA-3’) and ITS4 (5’-TCCTCCGCTTATTGATATGC-3’) (Gardes and Bruns 1993; White et al. 1990) and visualized using agarose gel electrophoresis. If no band was observed following gel electrophoresis, the PCR was repeated. If the result remained negative, DNA was re-extracted from the root sample. All DNA samples were stored at − 20 °C.
Preparation of fungal amplicon libraries and Illumina MiSeq sequencing were outsourced to Macrogen, Inc., South Korea. Libraries were prepared using the Herculase II Fusion DNA Polymerase Nextera XT Index V2 Kit (2 × 300 bp paired) using the fungal-specific primers ITS1F (5’-TCCGTAGGTGAACCTGCGG-3’) and ITS2 (5’-GCTGCGTTCTTCATCGATGC-3’) (White et al. 1990). Following sequencing, the raw reads were trimmed and demultiplexed by the sequencing facility. These files were deposited in the NCBI Sequence Read Archive under the accession number PRJNA1068848.
Analysis of high-throughput sequence data
The quality of the raw reads was assessed using FastQC v0.12.1 (Andrews 2010). We used multiple approaches for preliminary analyses of the sequence data: merging forward and reverse reads, using forward reads only, and comparing Amplicon Sequencing Variants (ASV) versus Operational Taxonomic Units (OTUs) clustered using 100% sequence similarity. During the merging with BBMerge available through BBTools v39.05 (Bushnell et al. 2017), 85% of the reads were lost, with further losses during quality filtering with DADA2 (Callahan et al. 2016). Additionally, in this case, using ASVs instead of OTUs did not significantly improve the output. Therefore, all subsequent analyses were conducted using only the forward reads that were clustered as OTUs.
To investigate the fungal diversity at a ‘local’ and ‘global’ scale, the dataset was analysed as two distinct sets. The ‘local’ dataset included 23 samples from the six provinces of South Africa, with four replicates per province except for the Western Cape where only three samples were obtained. During the analysis of the ‘local’ dataset, samples from Mpumalanga were found to exhibit the greatest diversity among all provinces, and this diversity overlapped with the diversity observed in samples from other provinces in the PCoA plot. Thus, during the analysis of the ‘global’ dataset, three samples from Mpumalanga were included to represent South Africa along with three samples each from Brazil, Germany and Spain and four from Colombia. Both datasets were analysed using the same bioinformatics pipeline listed below.
The single-end forward reads were analysed using QIIME2 software v2023.7 (Bolyen et al. 2019). Denoising, chimera deletion and filtering of Amplicon Sequencing Variants (ASV) were done using the plugin ‘q2-dada2’. The resulting filtered non-chimeric reads were assembled into Operational Taxonomic Units (OTUs) using the plugin ‘vsearch’ (Rognes et al. 2016) with 100% sequence similarity. The ‘qiime feature classifier’ plugin (Bokulich et al. 2018) was then used for the taxonomic assignment of the OTUs using the UNITE fungal database version 9.0 (Abarenkov et al. 2024) as a reference.
Statistical analyses of biodiversity data
All statistical analyses were performed at the order level using MicrobiomeAnalyst v2.0 (Lu et al. 2023). Both the ‘local’ and ‘global’ datasets were analysed using an identical pipeline. To ensure the inclusivity of rare taxa, such as orchid mycorrhizal fungi, neither of the datasets was rarefied during any of the analyses (McMurdie and Holmes 2014). Low-count features were filtered based on a mean abundance threshold, with a minimum count of 4, while low-variance features were excluded using the interquartile range. The remaining features were normalized using total sum scaling (TSS). Alpha diversity was assessed by calculating Shannon and Simpson indices for each dataset, while species richness was evaluated using Chao1. Beta diversity was analysed through PCoA with the Bray–Curtis index, using PERMANOVA for statistical testing. In cases of significant results from PERMANOVA, PERMIDISP was used to determine if observed differences were due to dispersion. For PERMANOVA and PERMIDISP analyses, a p-value of 0.05 was considered significant.
To explore the core microbiome, the dataset was divided into three sets, Columbia (native habitat), South Africa and ‘other regions’ (Brazil, Germany and Spain). This allowed us to compare fungal biodiversity between the native region (Colombia), the location where the study was conducted (South Africa), and the remaining sampling sites. The analysis was independently conducted for each group, with a prevalence threshold of 20 and a relative abundance cutoff of 0.01. The results for each group were visualized using heat maps and a Venn diagram.
Results
Analysis of high-throughput sequence data
In total, 2,953,716 single-end reads were generated from Illumina MiSeq for the 36 root DNA samples. After quality filtering, 2,673,341 (90.5%) single reads were retained for subsequent downstream analysis.
Fungal biodiversity associated with Epidendrum roots
‘Local’ dataset
Out of 1,818,332 single-end raw reads in this dataset, 1,646,651 (91%) successfully passed the quality filtration process and were subsequently used for downstream analysis (Table S1). Following taxonomic assignment, 284,006 reads (17.25%) remained unclassified, while 1,362,645 reads (82.75%) were classified into 740 OTUs (Fig. 2A, Table S2). The majority of these OTUs were assigned to Ascomycota (~ 60%) and Basidiomycota (~ 34%). The remaining 6% represented Rozellomycota, Mortierellomycota, Mucoromycota, Glomeromycota, Kickxellomycota, Aphelidiomycota and Monoblepharomycota.

Hierarchy plot illustrating the abundance of fungal orders associated with Epidendrum roots from two datasets: A Local Dataset (covering six provinces in South Africa) and B Global Dataset (including data from Colombia, Brazil, Germany, Spain, and South Africa). Within each phylum, orders are listed in descending order of abundance. OTUs that cannot be assigned to a specific order are labeled as incertae sedis
The dominant orders within the Ascomycota were Pleosporales (93 OTUs), Hypocreales (46 OTUs) and Helotiales (33 OTUs), while in Basidiomycota, dominant orders included Tremellales (39 OTUs), Agaricales (33 OTUs), Polyporales (17 OTUs) and Cantharellales (11 OTUs). Among the six South African provinces, the highest number of OTUs were detected from Mpumalanga (382), followed by KwaZulu-Natal (373), Limpopo (286), North-West (208), Gauteng (194) and Western Cape (159).
‘Global’ dataset
The ‘global’ dataset included 1,429,194 single-end raw reads. Following quality filtration, 1,293,606 reads (91%) of the reads were retained for downstream analysis (Table S3). Following taxonomic assignment, 109,011 reads (8%) remained unclassified, while 1,184,595 reads (92%) were classified into 731 OTUs (Fig. 2B, Table S4).
Ascomycota constituted 76% of the classified reads, Basidiomycota 23%, and the remaining 1% encompassed various phyla including Rozellomycota, Chytridiomycota, Mortierellomycota, Mucoromycota, Aphelidiomycota, Glomeromycota and Kickxellomycota. The dominant orders within the Ascomycota were Pleosporales (99 OTUs), Hypocreales (49 OTUs), Helotiales (28 OTUs) and Xylariales (28 OTUs). For the Basidiomycota, dominant orders included Agaricales (41 OTUs), Polyporales (40 OTUs) and Tremellales (21 OTUs). The highest number of OTUs was detected in Colombia (431), followed by South Africa (347), Brazil (264), Spain (206) and Germany (170).
Community composition of fungi associated with Epidendrum roots
‘Local’ dataset
Significant differences were observed in the Shannon and Simpson diversity indices as well as richness among the samples collected from six provinces of South Africa (p < 0.05; Fig. 3A, B, C). Samples from Mpumalanga and KwaZulu-Natal exhibited high richness, with the latter having less variability. In the PCoA plot, samples from different provinces overlapped. However, PERMANOVA indicated that there were significant differences between the samples (p-value < 0.05; Fig. 3D), which was further supported by PERMDISP (p-value < 0.05). The samples from all provinces clustered closely except for those from North-West province indicating a higher fungal diversity within these samples.

Box plots representing the diversity indices in the fungal community associated with Epidendrum roots in South Africa: A Shannon index, B Simpson index and C species richness. D Principal coordinate analysis (PCoA) of fungal community composition associated with the roots of Epidendrum across different South African provinces. The provinces are labelled as follows: GP = Gauteng, NW = North-West, KZN = KwaZulu-Natal, LP = Limpopo, WC = Western Cape and MP = Mpumalanga
‘Global’ dataset
There were no significant differences observed in the Shannon and Simpson diversity indices (p-value > 0.05; Fig. 4A, B) of the ‘global’ dataset, suggesting no notable changes in the overall fungal diversity. However, a significant difference in richness was observed (p-value < 0.05; Fig. 4C), indicating a variation in the number of taxa present. The PCoA plot revealed a distinct separation between the Colombian samples and those from other introduced countries (Fig. 4D). This suggests a significant difference (p-value < 0.05) in fungal biodiversity between native and introduced regions, which was further confirmed by the PERMANOVA (p-value < 0.05) and PERMDISP analysis (p-value > 0.05).

Box plots representing the diversity indices in the fungal community associated with Epidendrum roots globally: A Shannon index, B Simpson index and C species richness. D Principal coordinate analysis (PCoA) of fungal community composition associated with the roots of Epidendrum across different sampling countries. The countries are labelled as follows: ZA = South Africa, BR = Brazil, DE = Germany, ES = Spain and CO = Colombia
Core fungal taxa associated with Epidendrum roots
This analysis focused on the 13 most prevalent fungal orders across three datasets, Colombia, South Africa and ‘other regions’. Chaetothyriales, Hypocreales, Pleosporales and Tremellales were detected in all three datasets, with varying relative abundances (Fig. 5A, B). Each region also exhibited unique prevalent fungal taxa (Fig. 5A). Amphisphaeriales, Chaetosphaeriales and Trechisporales were detected from Columbia, whereas South Africa had Coniochaetales and Sebacinales, with the latter known to form symbiotic relationships with orchids in South Africa. Cystobasidiales, Diaporthales, Diothiadeales, Laconorales, Mycosphaerellales, Myrmecridiales and Sordariales were exclusive to ‘other regions’ dataset (Fig. 5A). There were no shared fungal orders between Columbia and ‘other regions’. Cantharellales, which includes orchid symbionts, was among the most abundant orders detected in South Africa and ‘other regions’ but not in Colombia (Table S4). Six fungal orders were shared between Colombia and South Africa, including Agaricales, which also includes orchid mycorrhizal fungi. These observations suggest that a core set of fungal orders is acquired regardless of where Epidendrum are cultivated.

Core fungal orders associated with Epidendrum roots. A Venn diagram shows the intersection and commonalities among the top 13 fungal taxa, and B their respective percentage abundance across three groups: ‘native habitat’ (Colombia), ‘introduced habitat 1’ (Brazil, Germany and Spain) and ‘introduced habitat 2’ (South Africa)
Discussion
In this study, we catalogued the fungal biodiversity associated with roots of Epidendrum from Brazil, Colombia, Germany, South Africa and Spain using a next-generation sequencing approach. The Illumina high-throughput sequence data from the root DNA samples were separated into two datasets. The ‘local’ dataset comprised samples collected from six provinces within South Africa. The ‘global’ dataset included samples from Brazil, Colombia, Germany, Spain and samples from one location in South Africa. The analyses of these datasets showed that Epidendrum harbours a diverse group of fungi in its roots. This orchid also maintains a core group of fungal taxa from its native habitat when moving to exotic regions, while also establishing new associations with local fungal communities in these introduced areas globally.
Total fungal biodiversity associated with roots of Epidendrum
In this study, fungal orders from Ascomycota that include dark septate endophytes were abundant. These fungi from Pleosporales, Xylariales and Helotiales are recognized for promoting plant growth through mutualistic associations (Newsham 2011; Usuki and Narisawa 2007). These fungi facilitate carbon transfer to the host by employing extrametrical mycelia for accessing additional resources (Jumpponen 2001). Consequently, these fungi could potentially confer nutritional benefits or other advantages to Epidendrum, although specific benefits are yet to be fully understood.
While orchids are usually known for their association with Rhizoctonia-like fungi from families such as Ceratobasidiaceae, Tulasnellaceae and Sebacinales (Dearnaley 2007; Rasmussen and Rasmussen 2009), these fungi were not the predominant fungal OTUs identified in this study. Although numerous OTUs of Rhizoctonia-like fungi were found in South Africa, they were absent in root samples from other geographic locations. This observation prompts the hypothesis that Epidendrum may not consistently engage with Rhizoctonia-like fungi as symbionts. Instead, they appear to form diverse partnerships, including associations with saprophytes and plant pathogens, such as Fusarium oxysporum (Jiang et al. 2019) and Leptodontidium orchidicola (Currah et al. 1988) as indicated in previous studies (Martos et al. 2009). Saprobes and plant pathogens consistently emerge as abundant components within the endophytic communities of various orchids in different orchid diversity studies (Kartzinel et al. 2013; Makwela et al. 2022b; de los Angeles Beltrán-Nambo et al. 2018; Kohout et al. 2013). Additionally, in this study, the detection of OTUs belonging to undescribed Fusarium and various saprobic fungi suggest that Epidendrum may engage in symbiotic associations with these fungi in the absence of Rhizoctonia-like fungi (Jiang et al. 2019; Currah et al. 1988).
OTUs from Tulasnellaceae were not commonly detected in this study. However, fungi from this family form symbiotic associations with orchids (Xing et al. 2019; Dearnaley et al. 2012; Martos et al. 2012). This is consistent with previous studies by Hernández-Ramírez et al. (2023) and Kohout et al. (2013) where Tulasnella species were not the most commonly identified fungi from orchid roots. Nonetheless, Tulasnella species have been isolated from various Epidendrum species, such as E. secundum (Pereira et al. 2014), E. dendrobioides (Nogueira et al. 2005) and E. rigidum (Pereira et al. 2005), with reported mycorrhizal associations. The limited number of OTUs from Tulasnellaceae detected in this study may be attributed to the constraints of the universal fungal primers used for metabarcoding, which have been documented to have limitations in amplifying Tulasnella and other fungi (Usyk et al. 2017; Stielow et al. 2015). To overcome this limitation, future studies on fungal diversity in orchids should consider using Tulasnella-specific primers, as proposed by Taylor and McCormick (2008), in conjunction with universal fungal primers. This dual-primer approach holds the promise of providing more comprehensive insights into the root-associated fungal communities of orchids, contributing to a better understanding of their symbiotic relationships.
In the present study, Atractiellomycetes were identified in the roots of Epidendrum. The initial hypothesis by Kottke et al. (2010) proposed a mycorrhizal association between fungi from the rust lineage Atractiellomycetes and orchids. Subsequent studies have also confirmed the presence of Atractiellomycetes in the roots of various orchids (Ávila-Díaz et al. 2013; Suárez et al. 2016; Makwela et al. 2022b; Fernández et al. 2023; Herrera et al. 2019). However, none of these studies conducted infection trials to validate the hypothesis. This step is crucial because the mere presence of these rust fungi does not inherently reveal their specific role within orchids. To illustrate, Hoang et al. (2017) identified different Ceratobasidium species in Dendrophylax lindenii; among them, Ceratobasidium sp. 394 facilitated seed germination, while Ceratobasidium sp. 379 did not. It is also important to note that conducting plant trials with Atractiellomycetes presents challenges, as these fungi, like all rust fungi, are likely obligate organisms (Spirin et al. 2018). Despite these challenges, infection trials are essential for a comprehensive understanding of the functional dynamics of Atractiellomycetes in orchids.
Yeast species within Tremellomycetes and Microbotryomycetes emerged as prominent fungi in this study. Despite not being previously documented in orchids, yeasts are hypothesized to play a partial role in a tripartite interaction involving mycorrhizal fungi and plants (Mirabal Alonso et al. 2008; Fracchia et al. 2003). For example, Rhodotorula mucilaginosa (Microbotryomycetes) actively recruits arbuscular mycorrhizal colonization in soybeans and red clover (Fracchia et al. 2003). Consequently, the yeast species identified in this study may potentially serve as a mediator for recruiting saprophytes and plant pathogenic fungi as symbionts of Epidendrum. However, substantiating this hypothesis requires dedicated future studies.
Community composition and diversity of fungi differed between the South African provinces
In this study, root samples collected from Gauteng, Limpopo, North-West and Western Cape originated from nurseries. Thus, these samples had lower fungal richness and diversity compared to samples from Mpumalanga and KwaZulu-Natal, which were sourced from soil-grown plants. This disparity between nursery and non-nursery samples emphasizes the need to consider the propagation environment when studying plant-associated microbial diversity, including orchids. Henry et al. (2017) noted a significant shift in fungal composition when Asteropeia mcphersonii seedlings were transplanted from the wild to the nursery. This negatively impacted the survival of ectomycorrhizal fungi associated with A. mcphersonii wildlings. Conversely, Marčiulynienė et al. (2021) detected a diverse range of fungal taxa, including beneficial mycorrhizal fungi in forest nurseries in Lithuania. However, it is important to note that these were bare-root forest nurseries, where the plants were not propagated in pots. While these studies are not orchid-specific, still they underscore the potential influence of the nursery environment on fungal community richness and diversity.
In all South African provinces, fungi from the Sebacinales were the only Rhizoctonia-like fungi detected, aligning with a study by Herrera et al. (2019), where these fungi were also common in Epidendrum marsupiale. The family Ceratobasidiaceae was detected from samples collected from all provinces in South Africa except Gauteng and Western Cape. Previously, Makwela et al. (2022a) and Makwela et al. (2022b) did not detect Ceratobasidiaceae in Gauteng. However, in earlier studies, Waterman et al. (2011), consistently detected Ceratobasidiaceae associated with orchids from the tribe Coryciinae along the eastern coastline of South Africa extending into the Western Cape province. This disparity could emerge from the difference in sampling and source between this study and Waterman et al. (2011). The roots samples from the Western Cape province originated from nurseries whereas Waterman et al. (2011) worked on wild orchids. This observation further reinforces the above hypothesis that fungal biodiversity associated with orchids differs depending on cultivation strategies (in soil vs. nurseries). It also underscores the flexibility of orchids in selecting their symbiotic partners based on geographic location, aligning with the proposition made by Jacquemyn et al. (2016).
Community composition and diversity of fungi differed between the sampled countries
Despite variations in the richness of fungal species across the global samples, the overall diversity, as measured by Shannon and Simpson indices, remained consistent. Nonetheless, the presence of distinct clusters in the PCoA plot indicated significant differences in fungal communities among the samples, emphasizing the potential influence of geographic factors on fungal diversity (Tedersoo et al. 2013; Poulin et al. 2011). However, as seen in the core fungal taxa analysis, there was substantial overlap between taxa between these regions.
In the core fungal taxa analysis, the most prevalent fungal orders detected from all three datasets were Chaetothyriales, Hypocreales, Pleosporales and Tremellales. None of these orders include fungi that are recognized as orchid mycorrhizae. Nevertheless, Pleosporales are frequently detected in orchid roots, although their mycorrhizal status remains unconfirmed (Makwela et al. 2022a). Even though many fungi detected in this study may not be recognized as symbionts of orchids, they may contribute to mobilizing soil nutrients in the rhizosphere, thereby promoting orchid growth, or they may inhabit the roots as endophytes (Zhao et al. 2014).
In this study, core fungal communities associated with roots of Epidendrum included both overlapping and distinct fungal orders. Thus, we hypothesize that Epidendrum likely acquires a core set of fungal orders irrespective of where it is propagated, while also associating with region-specific fungi. To validate this, further research on the fungal biodiversity associated with the roots of various orchids and Epidendrum across more diverse regions is needed. Nevertheless, this flexibility of Epidendrum in recruiting diverse fungi was previously reported for E. firmum populations from Costa Rica (Kartzinel et al. 2013). It was shown that E. firmum from this region exhibited a broad specificity and the potential for opportunistic associations with diverse fungi. Similar trends have also been observed in studies exploring fungal biodiversity associated with roots of woody and herbaceous plants. For example, Klironomos (2003), Porter et al. (2011) and Policelli et al. (2019) demonstrated that arbuscular mycorrhizal plants often establish relationships with widely distributed fungal species or form new partnerships when introduced to new areas, potentially replacing some of those from their native habitats (Nuñez and Dickie 2014; Orlovich and Cairney 2004).
Current challenges and way forward in orchid mycorrhizal research
Orchid mycorrhizal research has made significant progress in recent decades, but there are still several limitations and areas for future exploration. One key area is gaining an understanding of how the distribution and abundance of appropriate mycorrhizal fungi influence the population dynamics of introduced orchids (Li et al. 2021; McCormick et al. 2018). As seen in this study, while mycorrhizal specificity did not necessarily limit the distribution range of Epidendrum, it is not known how the complex interactions between the orchids, their fungal partners and environmental conditions are affecting their biogeography (Jacquemyn et al. 2017; Li et al. 2021; McCormick et al. 2018). For this, understanding the biodiversity of orchid-associated fungi from different regions is an essential step forward.
Next-generation sequencing has become an effective technology for studying microbial diversity from various environments. This platform has been used for identifying the fungal diversity associated with various orchid species. Still, our knowledge of the specificity, dynamics and functional significance of fungi that associate with orchids as symbionts or endophytes is limited (Li et al. 2021). For example, in this study, we detected various fungi from the roots of Epidendrum across three continents. However, our identification of the fungi up to the species level was limited by the choice of a short-read sequencing platform and reference database. Currently, commonly used references such as those from UNITE and NCBI lack representative sequences for orchid-associated fungi. We also could not in most cases identify the specific functional guilds and trophic modes of these fungi using popularly used databases like such and FUNGuild or FungalTraits (Nguyen et al. 2016; Põlme et al. 2020). Previous studies conducted by our team also faced similar challenges (Makwela et al. 2022a, b) using earlier versions of these databases. This illustrates that the rate of discovering new orchid-associated fungi is slow, resulting in almost no new reference sequences in the updated versions of the databases. This is primarily because isolating and identifying orchid mycorrhizae is challenging (Zhu et al. 2008).
In another section of this study, we made numerous attempts to isolate orchid mycorrhizae from the root samples collected from South Africa using the various previously published protocols (Athipunyakom et al. 2004; Chen et al. 2012; Tian et al. 2022; Xi et al. 2020; Yamato et al. 2005; Zettler and Corey 2018; Zhu et al. 2008). However, we realized that there is no single user-friendly protocol for isolating orchid mycorrhizae; instead, it requires continuous effort. Through persistent attempts, we successfully isolated Tulasnella epidendrea from the roots of Epidendrum × obrienianum (Crous et al. 2023). Like other species of Tulasnella, we predicted that this species could be an orchid mycorrhizal fungus. However, we have not conducted infection trials to confirm this due to technical challenges. However, such trials are essential to increase our current knowledge of the infection biology of orchid mycorrhizal fungi and their function (Li et al. 2021). Applying techniques such as stable isotope labelling, enzyme assays and transcriptomics could help us to elucidate the functional diversity and ecological roles of orchid mycorrhizal fungi, and how these functions vary across different orchid species and environments.
The knowledge gained from the abovementioned studies and the fungal isolates can be used for seed germination, growth promotion and conservation of orchids (Dearnaley 2007; Li et al. 2021). Many orchid species are threatened by habitat loss, overexploitation and climate change, such as Brachycorythis conica subsp. transvaalensis from South Africa (Makwela et al. 2022a). Knowledge of the use of mycorrhizal fungi in orchid seed germination and seedling establishment can have important implications for ex situ conservation and reintroduction efforts (Dearnaley 2007; Li et al. 2021). Additionally, exploring the potential of mycorrhizal fungi for growth promotion and stress tolerance in orchids could lead to the development of more efficient cultivation techniques and improved orchid production (Li et al. 2021).
Conclusion
This study showed that the roots of Epidendrum orchids harbour a wide range of fungi with various ecological roles. We also showed that Epidendrum maintained core fungal orders from their native habitats when moved to new regions, while also forming new associations with local fungi. We believe that orchids of the same genus with a broad distribution range likely associate with different mycorrhizal partners based on their geographical locations, although further studies are needed to confirm this.
Using widely propagated horticultural orchids, such as Epidendrum, for fungal biodiversity studies presents several limitations. All these limitations could have affected the observed fungal biodiversity associated with the roots of Epidendrum. One significant challenge is determining the parentage of these orchids, as Epidendrums are frequently crossed with various other species within the genus to achieve desired floral morphologies. Many of these hybrids are not necessarily registered (see The Royal Horticultural Society, The International Orchid Register). Our sample size was greatly affected by our criterion to use plants not treated with fungicides or biocontrol agents like Trichoderma species. While we can confirm that our immediate sources did not use these antifungal agents, we cannot rule out the possibility of prior treatments before the plants reached our sampling sources. In this study, we also collected samples from both private (Colombia) and public (Germany) orchidariums, each housing a diverse orchid collection from various parts of the world. This exposure likely subjected the Epidendrum to a higher diversity of fungi than usual in the regions. Nevertheless, future studies involving other orchid genera or species, while considering the limitations mentioned above, could further validate if the strategy implemented by Epidendrum for recruiting fungal partners is consistent across the Orchidaceae or exclusive to this genus.
Data availability
Raw Illumina Miseq data is available through NCBI Sequence Read Archive under the accession number PRJNA1068848.
References
Abarenkov K, Nilsson RH, Larsson K-H, Andy T, Frøslev TG, Pawlowska J, Lindahl B, Põldmaa K, Truong C, Vu D, Hosoya T, Niskanen T, Piirmann T, Ivanov F, Zirk A, Peterson M, Tanya IY, Arnold T, Kristiansson E, Mikryukov V, Joseph OR, Francisco PJ, Saar I, Schigel D, Suija A, Tedersoo L, Kõljalg U (2024) The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: sequences, taxa and classifications reconsidered. Nucleic Acids Res 52(D1):D791–D797. https://doi.org/10.1093/nar/gkad1039
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Athipunyakom P, Manoch L, Piluek C (2004) Isolation and identification of mycorrhizal fungi from eleven terrestrial orchids. Agric Nat Resour 38(2):216–228
Ávila-Díaz I, Garibay-Orijel R, Magaña-Lemus RE, Oyama K (2013) Molecular evidence reveals fungi associated within the epiphytic orchid Laelia speciosa (HBK) Schltr. Bot Sci 91(4):523–529. https://doi.org/10.17129/botsci.429
Böhmer M, Ozdín D, Račko M, Lichvár M, Budiš J, Szemes T (2020) Identification of bacterial and fungal communities in the roots of orchids and surrounding soil in heavy metal contaminated area of mining heaps. Appl Sci 10(20):7367. https://doi.org/10.3390/app10207367
Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J (2018) Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:1–17. https://doi.org/10.1186/s40168-018-0470-z
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu Y-X, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, Van Der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, Von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37(8):852–857. https://doi.org/10.1038/s41587-019-0209-9
Bonnardeaux Y, Brundrett M, Batty A, Dixon K, Koch J, Sivasithamparam K (2007) Diversity of mycorrhizal fungi of terrestrial orchids: compatibility webs, brief encounters, lasting relationships and alien invasions. Mycol Res 111(1):51–61. https://doi.org/10.1016/j.mycres.2006.11.006
Bushnell B, Rood J, Singer E (2017) BBMerge – accurate paired shotgun read merging via overlap. PLoS ONE 12(10):e0185056. https://doi.org/10.1371/journal.pone.0185056
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13(7):581–583. https://doi.org/10.1038/nmeth.3869
Chen J, Wang H, Guo S-X (2012) Isolation and identification of endophytic and mycorrhizal fungi from seeds and roots of Dendrobium (Orchidaceae). Mycorrhiza 22(4):297–307. https://doi.org/10.1007/s00572-011-0404-0
Crous PW, Osieck ER, Shivas RG, Tan YP, Bishop-Hurley SL, Esteve-Raventós F, Larsson E, Luangsa-ard JJ, Pancorbo F, Balashov S, Baseia IG, Boekhout T, Chandranayaka S, Cowan DA, Cruz RHSF, Czachura P, De la Peña-Lastra S, Dovana F, Drury B, Fell J, Flakus A, Fotedar R, Jurjević Ž, Kolecka A, Mack J, Maggs-Kölling G, Mahadevakumar S, Mateos A, Mongkolsamrit S, Noisripoom W, Plaza M, Overy DP, Pitek M, Sandoval-Denis M, Vauras J, Wingfield MJ, Abell SE, Ahmadpour A, Akulov A, Alavi F, Alavi Z, Altés A, Alvarado P, Anand G, Ashtekar N, Assyov B, Banc-Prandi G, Barbosa KD, Barreto GG, Bellanger JM, Bezerra JL, Bhat DJ, Bilański P, Bose T, Bozok F, Chaves J, Costa-Rezende DH, Danteswari C, Darmostuk V, Delgado G, Denman S, Eichmeier A, Etayo J, Eyssartier G, Faulwetter S, Ganga KGG, Ghosta Y, Goh J, Góis JS, Gramaje D, Granit L, Groenewald M, Gulden G, Gusmão LFP, Hammerbacher A, Heidarian Z, Hywel-Jones N, Jankowiak R, Kaliyaperumal M, Kaygusuz O, Kezo K, Khonsanit A, Kumar S, Kuo CH, Laessøe T, Latha KPD, Loizides M, Luo SM, Maciá-Vicente JG, Manimohan P, Marbach PAS, Marinho P, Marney TS, Marques G, Martín MP, Miller AN, Mondello F, Moreno G, Mufeeda KT, Mun HY, Nau T, Nkomo T, Okrasińska A, Oliveira JPAF, Oliveira RL, Ortiz DA, Pawłowska J, Pérez-De-Gregorio MA, Podile AR, Portugal A, Privitera N, Rajeshkumar KC, Rauf I, Rian B, Rigueiro-Rodríguez A, Rivas-Torres GF, Rodriguez-Flakus P, Romero-Gordillo M, Saar I, Saba M, Santos CD, Sarma PVSRN, Siquier JL, Sleiman S, Spetik M, Sridhar KR, Stryjak-Bogacka M, Szczepańska K, Taşikn H, Tennakoon DS, Thanakitpipattana D, Trovão J, Türkekul A, van Iperen AL, van ‘t Hof P, Vasquez G, Visagie CM, PTW BD, WingfieldWong, Yang WX, Yarar M, Yarden O, Yilmaz N, Zhang N, Zhu YN, Groenewald JZ (2023) Fungal Planet description sheets 1478–1549. Persoonia 50:158–310. https://doi.org/10.3767/persoonia.2023.50.05
Currah R, Hambleton S, Smreciu A (1988) Mycorrhizae and mycorrhizal fungi of Calypso bulbosa. Am J Bot 75(5):739–752. https://doi.org/10.1002/j.1537-2197.1988.tb13495.x
Davis BJ, Phillips RD, Wright M, Linde CC, Dixon KW (2015) Continent-wide distribution in mycorrhizal fungi: implications for the biogeography of specialized orchids. Ann Bot 116(3):413–421. https://doi.org/10.1093/aob/mcv084
de los AngelesBeltrán-NamboMartínez-TrujilloMontero-CastroSalgado-GarcigliaOtero-OspinaCarreón-Abud MMJCRJTY (2018) Fungal diversity in the roots of four epiphytic orchids endemic to Southwest Mexico is related to the breadth of plant distribution. Rhizosphere 7:49–56. https://doi.org/10.1016/j.rhisph.2018.07.001
Dearnaley JD (2007) Further advances in orchid mycorrhizal research. Mycorrhiza 17(6):475–486. https://doi.org/10.1007/s00572-007-0138-1
Dearnaley, J.D.W., Martos, F., Selosse, MA. (2012). 12 Orchid Mycorrhizas: molecular ecology, physiology, evolution and conservation aspects. In: Hock, B. (eds) Fungal Associations. The Mycota, vol 9. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30826-0_12
Delgado-Baquerizo M, Maestre FT, Reich PB, Jeffries TC, Gaitan JJ, Encinar D, Berdugo M, Campbell CD, Singh BK (2016) Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat Commun 7(1):10541. https://doi.org/10.1038/ncomms10541
Dickie IA, Bolstridge N, Cooper JA, Peltzer DA (2010) Co-invasion by Pinus and its mycorrhizal fungi. New Phytol 187(2):475–484. https://doi.org/10.1111/j.1469-8137.2010.03277.x
Duffy KJ, Waud M, Schatz B, Petanidou T, Jacquemyn H (2019) Latitudinal variation in mycorrhizal diversity associated with a European orchid. J Biogeogr 46(5):968–980. https://doi.org/10.1111/jbi.13548
Esposito F, Jacquemyn H, Waud M, Tyteca D (2016) Mycorrhizal fungal diversity and community composition in two closely related Platanthera (Orchidaceae) species. PLoS ONE 11(10):e0164108. https://doi.org/10.1371/journal.pone.0164108
Favre-Godal Q, Gourguillon L, Lordel-Madeleine S, Gindro K, Choisy P (2020) Orchids and their mycorrhizal fungi: an insufficiently explored relationship. Mycorrhiza 30(1):5–22. https://doi.org/10.1007/s00572-020-00934-2
Fernández M, Kaur J, Sharma J (2023) Co-occurring epiphytic orchids have specialized mycorrhizal fungal niches that are also linked to ontogeny. Mycorrhiza 33(1):87–105. https://doi.org/10.1007/s00572-022-01099-w
Fracchia S, Godeas A, Scervino JM, Sampedro I, OcampoGarcı́A-Romera JAI (2003) Interaction between the soil yeast Rhodotorula mucilaginosa and the arbuscular mycorrhizal fungi Glomus mosseae and Gigaspora rosea. Soil Biol Biochem 35(5):701–707. https://doi.org/10.1016/s0038-0717(03)00086-5
Gardes M, Bruns TD (1993) ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Molecular Ecology 2(2):113–118. https://doi.org/10.1111/j.1365-294X.1993.tb00005.x
Genevieve L, Pierre-Luc C, Roxanne G-T, Amélie M, Danny B, Vincent M, Hugo G (2019) Estimation of fungal diversity and identification of major abiotic drivers influencing fungal richness and communities in northern temperate and boreal Quebec forests. Forests 10(12):1096. https://doi.org/10.3390/f10121096
Heilmann-Clausen J, Barron ES, Boddy L, Dahlberg A, Griffith GW, Nordén J, Ovaskainen O, Perini C, Senn-Irlet B, Halme P (2015) A fungal perspective on conservation biology. Biol Conserv 29(1):61–68. https://doi.org/10.1111/cobi.12388
Henry C, Raivoarisoa J-F, Razafimamonjy A, Ramanankierana H, Andrianaivomahefa P, Ducousso M, Selosse M-A (2017) Transfer to forest nurseries significantly affects mycorrhizal community composition of Asteropeia mcphersonii wildings. Mycorrhiza 27(4):321–330. https://doi.org/10.1007/s00572-016-0750-z
Hernández-Ramírez F, Damon A, Fernández Pavía SP, Guillén-Navarro K, Iracheta-Donjuan L, Zarza E, Castro-Chan RA (2023) Community richness and diversity of endophytic fungi associated with the orchid Guarianthe skinneri infested with “Black Blotch” in the Soconusco region, Chiapas. Mexico Diversity 15(7):807. https://doi.org/10.3390/d15070807
Herrera P, Suárez JP, Sánchez-Rodríguez A, Molina MC, Prieto M, Méndez M (2019) Many broadly-shared mycobionts characterize mycorrhizal interactions of two coexisting epiphytic orchids in a high elevation tropical forest. Fungal Ecol 39:26–36. https://doi.org/10.1016/j.funeco.2018.11.003
Hoang NH, Kane ME, Radcliffe EN, Zettler LW, Richardson LW (2017) Comparative seed germination and seedling development of the ghost orchid, Dendrophylax lindenii (Orchidaceae), and molecular identification of its mycorrhizal fungus from South Florida. Ann Bot 119(3):379–393. https://doi.org/10.1093/aob/mcw220
Huang M, Gao D, Lin L, Wang S, Xing S (2022) Spatiotemporal dynamics and functional characteristics of the composition of the main fungal taxa in the root microhabitat of Calanthe sieboldii (Orchidaceae). BMC Plant Biol 22(1):556. https://doi.org/10.1186/s12870-022-03940-y
Jacquemyn H, Honnay O, Cammue BPA, Brys R, Lievens B (2010) Low specificity and nested subset structure characterize mycorrhizal associations in five closely related species of the genus Orchis. Mol Ecol 19(18):4086–4095. https://doi.org/10.1111/j.1365-294X.2010.04785.x
Jacquemyn H, Waud M, Lievens B, Brys R (2016) Differences in mycorrhizal communities between Epipactis palustris, E. helleborine and its presumed sister species E. neerlandica. Ann Bot 118(1):105–114. https://doi.org/10.1093/aob/mcw015
Jacquemyn H, Duffy KJ, Selosse M-A (2017) Biogeography of orchid mycorrhizas. In:Tedersoo L (ed) Biogeography of mycorrhizal symbiosis. Springer International Publishing, Cham, pp 159–177. https://doi.org/10.1007/978-3-319-56363-3_8
Jiang J, Zhang K, Cheng S, Nie Q, Zhou S-X, Chen Q, Zhou J, Zhen X, Li XT, Zhen TW, Xu M, Hsiang T, Sun Z, Zhou Y (2019) Fusarium oxysporum KB-3 from Bletilla striata: an orchid mycorrhizal fungus. Mycorrhiza 29(5):531–540. https://doi.org/10.1007/s00572-019-00904-3
Jumpponen A (2001) Dark septate endophytes - are they mycorrhizal? Mycorrhiza 11(4):207–211. https://doi.org/10.1007/s005720100112
Kartzinel TR, Trapnell DW, Shefferson RP (2013) Highly diverse and spatially heterogeneous mycorrhizal symbiosis in a rare epiphyte is unrelated to broad biogeographic or environmental features. Mol Ecol 22(23):5949–5961. https://doi.org/10.1111/mec.12536
Klironomos JN (2003) Variation in plant response to native and exotic arbuscular mycorrhizal fungi. Ecology 84(9):2292–2301. https://doi.org/10.1890/02-0413
Kohout P, Těšitelová T, Roy M, Vohnik M, Jersakova J (2013) A diverse fungal community associated with Pseudorchis albida (Orchidaceae) roots. Fun Ecol 6(1):50–64. https://doi.org/10.1016/j.funeco.2012.08.005
Kolanowska M (2013) Niche conservatism and the future potential range of Epipactis helleborine (Orchidaceae). PLoS ONE 8:e77352. https://doi.org/10.1371/journal.pone.0077352
Kolanowska M, Konowalik K (2014) Niche conservatism and future changes in the potential area coverage of Arundina graminifolia, an invasive orchid species from Southeast Asia. Biotropica 46:157–165. https://doi.org/10.1111/btp.12089
Konowalik K, Kolanowska M (2018) Climatic niche shift and possible future spread of the invasive South African orchid Disa bracteata in Australia and adjacent areas. PeerJ 6:e6107. https://doi.org/10.7717/peerj.6107
Kottke I, Suárez JP, Herrera P, Cruz D, Bauer R, Haug I, Garnica S (2010) Atractiellomycetes belonging to the ‘rust’ lineage (Pucciniomycotina) form mycorrhizae with terrestrial and epiphytic neotropical orchids. Proc R Soc B 277(1685):1289–1298. https://doi.org/10.1098/rspb.2009.1884
Li T, Wu S, Yang W, Selosse M-A, Gao J (2021) How mycorrhizal associations influence orchid distribution and population dynamics. Front Plant Sci 12:647114. https://doi.org/10.3389/fpls.2021.647114
Lu Y, Zhou G, Ewald J, Pang Z, Shiri T, Xia J (2023) MicrobiomeAnalyst 2.0: comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res 51(W1):W310–W318. https://doi.org/10.1093/nar/gkad407
Makwela MC, Hammerbacher A, Coetzee MP, Wingfield BD, van Ede G, Bose T (2022a) Fungal diversity associated with the mycorrhizosphere soil of Brachycorythis conica subsp. transvaalensis, a critically endangered and endemic terrestrial orchid from South Africa. S Afr J Bot 146:807–814. https://doi.org/10.1016/j.sajb.2022.01.019
Makwela MC, Hammerbacher A, Vivas M, Coetzee MP, Wingfield BD, van Ede G, Bose T (2022b) Uncovering the mycorrhizal community of two Habenaria orchids in South Africa. S Afr J Bot 146:856–863. https://doi.org/10.1016/j.sajb.2022.02.020
Marčiulynienė D, Marčiulynas A, Lynikienė J, Vaičiukynė M, Gedminas A, Menkis A (2021) DNA-metabarcoding of belowground fungal communities in bare-root forest nurseries: focus on different tree species. Microorganisms 9(1):150. https://doi.org/10.3390/microorganisms9010150
Martos F, Dulormne M, Pailler T, Bonfante P, Faccio A, Fournel J, Dubois MP, Selosse MA (2009) Independent recruitment of saprotrophic fungi as mycorrhizal partners by tropical achlorophyllous orchids. New Phytol 184(3):668–681. https://doi.org/10.1111/j.1469-8137.2009.02987.x
Martos F, Munoz F, Pailler T, Kottke I, Gonneau C, Selosse MA (2012) The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Mol Ecol 21(20):5098–5109. https://doi.org/10.1111/j.1365-294x.2012.05692.x
McCormick MK, Jacquemyn H (2014) What constrains the distribution of orchid populations? New Phytol 202(2):392–400. https://doi.org/10.1111/nph.12639
McCormick MK, Whigham DF, Canchani-Viruet A (2018) Mycorrhizal fungi affect orchid distribution and population dynamics. New Phytol 219(4):1207–1215. https://doi.org/10.1111/nph.15223
McMurdie PJ, Holmes S (2014) Waste Not, Want Not: why rarefying microbiome data is inadmissible. PLoS Comput Biol 10(4):e1003531. https://doi.org/10.1371/journal.pcbi.1003531
Mirabal Alonso L, Kleiner D, Ortega E (2008) Spores of the mycorrhizal fungus Glomus mosseae host yeasts that solubilize phosphate and accumulate polyphosphates. Mycorrhiza 18(4):197–204. https://doi.org/10.1007/s00572-008-0172-7
Newsham KK (2011) A meta-analysis of plant responses to dark septate root endophytes. New Phytol 190(3):783–793. https://doi.org/10.1111/j.1469-8137.2010.03611.x
Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG (2016) FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248. https://doi.org/10.1016/j.funeco.2015.06.006
Nogueira RE, Pereira OL, Kasuya MCM, Lanna MCDS, Mendonça MP (2005) Fungos micorrízicos associados a orquídeas em campos rupestres na região do Quadrilátero Ferrífero, MG. Brasil Acta Bot Bras 19(3):417–424. https://doi.org/10.1590/s0102-33062005000300001
Nuñez MA, Dickie IA (2014) Invasive belowground mutualists of woody plants. Biol Invasions 16:645–661. https://doi.org/10.1007/s10530-013-0612-y
Orlovich DA, Cairney JG (2004) Ectomycorrhizal fungi in New Zealand: current perspectives and future directions. N Z J Bot 42(5):721–738. https://doi.org/10.1080/0028825X.2004.9512926
Park MS, Eimes JA, Oh SH, Suh HJ, Oh S-Y, Lee S, Park KH, Kwon HJ, Kim S-Y, Lim YW (2018) Diversity of fungi associated with roots of Calanthe orchid species in Korea. J Microbiol 56(1):49–55. https://doi.org/10.1007/s12275-018-7319-9
Pecoraro L, Huang L, Caruso T, Perotto S, Girlanda M, Cai L, Liu ZJ (2017) Fungal diversity and specificity in Cephalanthera damasonium and C. longifolia (Orchidaceae) mycorrhizas. J Syst Evol 55(2):158–169. https://doi.org/10.1111/jse.12238
Pereira OL, Kasuya MCM, Borges AC, Araújo EFD (2005) Morphological and molecular characterization of mycorrhizal fungi isolated from neotropical orchids in Brazil. Canad J Bot 83(1):54–65. https://doi.org/10.1139/b04-151
Pereira MC, Da Silva Coelho I, Da Silva Valadares RB, Oliveira SF, Bocayuva M, Pereira OL, Araújo EF, Kasuya MCM (2014) Morphological and molecular characterization of Tulasnella spp. fungi isolated from the roots of Epidendrum secundum, a widespread Brazilian orchid. Symbiosis 62(2):111–121. https://doi.org/10.1007/s13199-014-0276-0
Pinheiro F, Cozzolino S (2013) Epidendrum (Orchidaceae) as a model system for ecological and evolutionary studies in the Neotropics. Taxon 62(1):77–88. https://doi.org/10.1002/tax.621007
Policelli N, Bruns TD, Vilgalys R, Nuñez MA (2019) Suilloid fungi as global drivers of pine invasions. New Phytol 222(2):714–725. https://doi.org/10.1111/nph.15660
Põlme S, Abarenkov K, Henrik Nilsson R, Lindahl BD, Clemmensen KE, Kauserud H, Nguyen N, Kjøller R, Bates ST, Baldrian P, Frøslev TG, Adojaan K, Vizzini A, Suija A, Pfister D, Baral H-O, Järv H, Madrid H, Nordén J, Liu J-K, Pawlowska J, Põldmaa K, Pärtel K, Runnel K, Hansen K, Larsson K-H, Hyde KD, Sandoval-Denis M, Smith ME, Toome-Heller M, Wijayawardene NN, Menolli N, Reynolds NK, Drenkhan R, Maharachchikumbura SSN, Gibertoni TB, Læssøe T, Davis W, Tokarev Y, Corrales A, Soares AM, Agan A, Machado AR, Argüelles-Moyao A, Detheridge A, de Meiras-Ottoni A, Verbeken A, Dutta AK, Cui B-K, Pradeep CK, Marín C, Stanton D, Gohar D, Wanasinghe DN, Otsing E, Aslani F, Griffith GW, Lumbsch TH, Grossart H-P, Masigol H, Timling I, Hiiesalu I, Oja J, Kupagme JY, Geml J, Alvarez-Manjarrez J, Ilves K, Loit K, Adamson K, Nara K, Küngas K, Rojas-Jimenez K, Bitenieks K, Irinyi L, Nagy LG, Soonvald L, Zhou L-W, Wagner L, Aime MC, Öpik M, Mujica MI, Metsoja M, Ryberg M, Vasar M, Murata M, Nelsen MP, Cleary M, Samarakoon MC, lomBahram MM, Hagh-Doust N, Dulya O, Johnston P, Kohout P, Chen Q, Tian Q, Nandi R, Amiri R, Perera RH, dos SantosChikowski R, Mendes-Alvarenga RL, Garibay-Orijel R, Gielen R, Phookamsak R, Jayawardena RS, Rahimlou S, Karunarathna SC, Tibpromma S, Brown SP, Sepp S-K, Mundra S, Luo Z-H, Bose T, Vahter T, Netherway T, Yang T, May T, Varga T, Li W, Coimbra VRM, de Oliveira VRT, de Lima VX, Mikryukov VS, Lu Y, Matsuda Y, Miyamoto Y, Kõljalg U, Tedersoo L (2020) FungalTraits: a user-friendly traits database of fungi and fungus-like stramenopiles. Fungal Divers 105(1):1–16. https://doi.org/10.1007/s13225-020-00466-2
Porter SS, Stanton ML, Rice KJ (2011) Mutualism and adaptive divergence: co-invasion of a heterogeneous grassland by an exotic legume-rhizobium symbiosis. PLoS ONE 6(12):e27935. https://doi.org/10.1371/journal.pone.0027935
Poulin R, Krasnov BR, Mouillot D (2011) Host specificity in phylogenetic and geographic space. Trends in Parasitol 27(8):355–361. https://doi.org/10.1016/j.pt.2011.05.003
Rasmussen HN, Rasmussen FN (2009) Orchid mycorrhiza: implications of a mycophagous lifestyle. Oikos 118(3):334–345. https://doi.org/10.1111/j.1600-0706.2008.17116.x
Rodríguez-Echeverría S (2010) Rhizobial hitchhikers from Down Under: invasional meltdown in a plant–bacteria mutualism? J Biogeogr 37(8):1611–1622. https://doi.org/10.1111/j.1365-2699.2010.02284.x
Rognes T, Flouri T, Nichols B, Quince C, Mahé F (2016) VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. https://doi.org/10.7717/peerj.2584
Smith SE, Read DJ (2008) Mycorrhizal symbiosis. Academic Press, New York, London, Burlington, San Diego
Spirin V, Malysheva V, Trichies G, Savchenko A, Põldmaa K, Nordén J, Miettinen O, Larsson KH (2018) A preliminary overview of the corticioid Atractiellomycetes (Pucciniomycotina, Basidiomycetes). Fung Syst Evol 2:311–340. https://doi.org/10.3114/fuse.2018.02.09
Stielow JB, Lévesque CA, Seifert KA, Meyer W, Irinyi L, Smits D, Renfurm R, Verkley GJM, Groenewald M, Chaduli D, Lomascolo A, Welti S, Lesage-Meessen L, Favel A, Al-Hatmi AMS, Damm U, Yilmaz N, Houbraken J, Lombard L, Quaedvlieg W, Binder M, Vaas LAI, Vu D, Yurkov A, Begerow D, Roehl O, Guerreiro M, Fonseca A, Samerpitak K, Van Diepeningen AD, Dolatabadi S, Moreno LF, Casaregola S, Mallet S, Jacques N, Roscini L, Egidi E, Bizet C, Garcia-Hermoso D, Martín MP, Deng S, Groenewald JZ, Boekhout T, De Beer ZW, Barnes I, Duong TA, Wingfield MJ, De Hoog GS, Crous PW, Lewis CT, Hambleton S, Moussa TAA, Al-Zahrani HS, Almaghrabi OA, Louis-Seize G, Assabgui R, McCormick W, Omer G, Dukik K, Cardinali G, Eberhardt U, De Vries M, Robert V (2015) One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35(1):242–263. https://doi.org/10.3767/003158515x689135
Suárez JP, Eguiguren JS, Herrera P, Jost L (2016) Do mycorrhizal fungi drive speciation in Teagueia (Orchidaceae) in the upper Pastaza watershed of Ecuador? Symbiosis 69(3):161–168. https://doi.org/10.1007/s13199-016-0399-6
Taylor DL, McCormick MK (2008) Internal transcribed spacer primers and sequences for improved characterization of basidiomycetous orchid mycorrhizas. New Phytol 177(4):1020–1033. https://doi.org/10.1111/j.1469-8137.2007.02320.x
Tedersoo L, Mett M, Ishida TA, Bahram M (2013) Phylogenetic relationships among host plants explain differences in fungal species richness and community composition in ectomycorrhizal symbiosis. New Phytol 199(3):822–831. https://doi.org/10.1111/nph.12328
Tian F, Liao X-F, Wang L-H, Bai X-X, Yang Y-B, Luo Z-Q, Yan F-X (2022) Isolation and identification of beneficial orchid mycorrhizal fungi in Paphiopedilum barbigerum (Orchidaceae). Plant Signal Behav 17(1):2005882. https://doi.org/10.1080/15592324.2021.2005882
Trivedi P, Leach JE, Tringe SG, Sa T, Singh BK (2020) Plant–microbiome interactions: from community assembly to plant health. Nat Rev Microbiol 18(11):607–621. https://doi.org/10.1038/s41579-020-0412-1
Usuki F, Narisawa K (2007) A mutualistic symbiosis between a dark septate endophytic fungus, Heteroconium chaetospira, and a nonmycorrhizal plant. Chinese Cabbage Mycologia 99(2):175–184. https://doi.org/10.1080/15572536.2007.11832577
Usyk M, Zolnik CP, Patel H, Levi MH, Burk RD (2017) Novel ITS1 fungal primers for characterization of the mycobiome. mSphere 2(6):10–1128. https://doi.org/10.1128/msphere.00488-17
Waterman RJ, Bidartondo MI, Stofberg J, Combs JK, Gebauer G, Savolainen V, Barraclough TG, Pauw A (2011) The effects of above-and belowground mutualisms on orchid speciation and coexistence. Am Nat 177(2):E54–E68. https://doi.org/10.1086/657955
Wei G, Chen W, Zhu W, Chen C, Young JPW, Bontemps C (2009) Invasive Robinia pseudoacacia in China is nodulated by Mesorhizobium and Sinorhizobium species that share similar nodulation genes with native American symbionts. FEMS Microbiol Ecol 68(3):320–328. https://doi.org/10.1111/j.1574-6941.2009.00673.x
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc 18(1):315–322
Xi G, Shi J, Li J, Han Z (2020) Isolation and identification of beneficial orchid mycorrhizal fungi in Bletilla striata (Thunb.) Rchb.f.(Orchidaceae). Plant Signal Behav 15(12):1816644. https://doi.org/10.1080/15592324.2020.1816644
Xing X, Jacquemyn H, Gai X, Gao Y, Liu Q, Zhao Z, Guo S (2019) The impact of life form on the architecture of orchid mycorrhizal networks in tropical forest. Oikos 128(9):1254–1264. https://doi.org/10.1111/oik.06363
Xu J (2016) Fungal DNA barcoding. The 6th International Barcode of Life Conference 01(01):913–932. https://doi.org/10.1139/gen-2016-0046
Yamato M, Iwase K, Yagame T, Suzuki A (2005) Isolation and identification of mycorrhizal fungi associating with an achlorophyllous plant, Epipogium roseum (Orchidaceae). Mycoscience 46(2):73–77. https://doi.org/10.1007/S10267-004-0218-4
Zeilinger S, Gupta VK, Dahms TES, Silva RN, Singh HB, Upadhyay RS, Gomes EV, Tsui CK-M, Nayak SC (2016) Friends or foes? Emerging insights from fungal interactions with plants. FEMS Microbiol Rev 40(2):182–207. https://doi.org/10.1093/femsre/fuv045
Zettler LW, Corey LL (2018) Orchid mycorrhizal fungi: isolation and identification techniques. In:Lee Y-I, Yeung EC-T (eds) Orchid propagation: from laboratories to greenhouses—methods and protocols. Springer New York, New York, NY, pp 27–59. https://doi.org/10.1007/978-1-4939-7771-0_2
Zhao X, Zhang J, Chen C, Yang J, Zhu H, Liu M, Lv F (2014) Deep sequencing–based comparative transcriptional profiles of Cymbidium hybridum roots in response to mycorrhizal and non-mycorrhizal beneficial fungi. BMC Genom 15(1):1–22. https://doi.org/10.1186/1471-2164-15-747
Zhu G, Yu Z, Gui Y, Liu Z (2008) A novel technique for isolating orchid mycorrhizal fungi. Fungal Divers 33(12):123–137
Acknowledgements
We thank the University of Pretoria and several members of the Forestry and Agricultural Biotechnology Institute (FABI) for their cooperation during this study. Thanks to Thomas Bopp and Burkhard Witt from the Botanical Gardens of the Friedrich Schiller University, Jena, Germany, for their help in collecting Epidendrum root samples.
Funding
Open access funding provided by University of Pretoria. This research was funded by the South African National Research Foundation (NRF) through Grant No. SRUG210318590365 (Almuth Hammerbacher), Grant No. MND210621613813 (Tiphany Nkomo) and the Department of Science and Innovation (DSI)-NRF SARChI chair in Fungal Genomics (Brenda Wingfield). The University of Pretoria and Forestry and Agricultural Biotechnology Institute (FABI) provided additional financial support.
Author information
Authors and Affiliations
Contributions
AH, TB and TN conceived the study. TN performed all laboratory work and conducted the bioinformatics and statistical analyses, with assistance from RK and OM. NMG, CAR, CP, CTC and MAF contributed to sample collection from Spain, Colombia, Germany, South Africa and Brazil. TN wrote the initial draft, which was revised by TB, AH and BDW, followed by contributions from RK, OM, NMG, CAR, CP, CTC and MAF. The study was supervised by TB, AH and BDW.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed with the submission of the manuscript.
Conflict of interest
Tanay Bose is the Section Editor of Mycological Progress but was not involved in the editorial processes associated with handling this manuscript. All other authors declare no competing interests.
Additional information
Section Editor: Marco Thines
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary material can also be found at https://doi.org/10.17632/tg3fyzgttz.1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nkomo, T., Bose, T., Wingfield, B.D. et al. Geographic location shapes fungal communities associated with Epidendrum roots. Mycol Progress 23, 54 (2024). https://doi.org/10.1007/s11557-024-01990-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11557-024-01990-0