
Abstract
Dissolved sulfide, iron (Fe), and phosphorus (P) in a mudflat (Jiaozhou Bay, China) impacted by shellfish aquaculture were measured in situ by the diffusive gradients in thin films (DGT) technique. A combination of porewater and solid-phase chemistry was used to characterize the interplays of Fe and S, and their control on P mobilization. Below the subsurface layer, two times higher fluxes (FDGT) of dissolved Fe2+ from porewater to the DGT device than those of dissolved sulfide indicate that dissimilatory iron reduction (DIR) dominates over sulfate reduction (SR). Spatial coupling of dissolved Fe2+ and P points to P release driven mainly by reductive dissolution of Fe. Much higher FDGT values of dissolved Fe2+ relative to dissolved P imply that oxidative regeneration of Fe oxides at the sediment–water interfaces (SWIs) of the transitional mudflat serves as an effective “iron curtain” of upward diffusing P. In the mudflat sediments of DIR prevalence, the accumulation of total reduced inorganic sulfur (TRIS) is dampened, which can largely ascribed to enhanced oxidative loss of sulfide and/or limited availability of degradable organic carbon in the dynamic regimes. Low dissolved sulfide concentrations in the sediments leave the majority of reactive Fe unsulfidized and thus abundantly available to buffer newly produced sulfide.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Intertidal mudflats are a transitional zone of the land–sea continuum and play an important role in trapping nutrients and pollutants from the land (Taillefert et al. 2007). Physical and chemical properties of intertidal mudflats such as temperature, light, salinity, and concentrations of dissolved oxygen and nutrients strongly vary during a tidal cycle (Taillefert et al. 2007; Al-Raei et al. 2009; Jansen et al. 2009; Rigaud et al. 2018). Therefore, dynamic regimes of the intertidal mudflats exert profound impacts on the geochemical processes of oxygen, carbon, sulfur (S), and iron (Fe), which are the four key elements dominating the behavior and fates of numerous other elements including phosphorus (P) (Rozan et al. 2002; Burdige 2006; Raiswell and Canfield 2012).
Intertidal mudflats are important locations for shellfish aquaculture worldwide. Aquaculture usually produces a great amount of biological deposits enriched in organic matter (OM) (Carlsson et al. 2009). For example, labile OM concentrations could be up to 25 times higher in aquaculture areas than in similar areas not impacted by aquaculture (Otero et al. 2006). The enrichment of OM usually stimulates benthic mineralization and quick depletion of oxygen and consequently promotes sulfate reduction (SR). For example, integrated SR rates within the upper 10 cm of sediments in an aquaculture area (Hardangerfjord, Denmark) are 39 times as high as those found at a reference site (Valdemarsen et al. 2012). Under these circumstances, abundant sulfide generated by SR may not be quickly buffered by reaction with reactive Fe oxides, leading to an accumulation of highly toxic sulfide in porewater and even in the overlying water to the levels detrimental to the benthic organisms (Valdemarsen et al. 2009). In addition, abundant dissolved sulfide in anoxic condition can result in depletion of reactive Fe oxides and, consequently, an increase in P flux from sediments towards the overlying water (Rozan et al. 2002; Lehtoranta et al. 2009).
In intertidal mudflats, however, the impacts of aquaculture on the benthic biogeochemistry may be mitigated due to (i) strong sediment resuspension and rapid transport away of dissolved components by dynamic tidal currents during flooding and ebbing (Taillefert et al. 2007; Rigaud et al. 2018), (ii) periodic exposure of the sediments to the atmosphere (Al-Raei et al. 2009; Jansen et al. 2009), and (iii) an increase in bioturbation and bioirrigation by shellfish (Aller 1994). Therefore, we hypothesize that aquaculture in intertidal mudflats exerts different influences on diagenetic cycling of S, Fe, and P, and thus their spatial heterogeneity than what is generally expected in typical coastal marine sediments impacted by aquaculture, due to highly redox-sensitive nature of dissolved Fe2+ and sulfide (Robertson et al. 2008; Pagès et al. 2014).
Traditional geochemical methods, including porewater extraction techniques, have been used to investigate S, Fe, and P biogeochemistry in coastal sediments impacted by aquaculture (Otero et al. 2006, 2009; Hyun et al. 2013). However, these techniques are incapable of in situ characterizing porewater chemistry in high resolution to unravel a nuanced picture of the distributions and interactions of S, Fe, and P. Up to date, only limited efforts have been devoted to simultaneous high-resolution characterization of the three constituents in sediment porewater (Pagès et al. 2011, 2012; Cesbron et al. 2014; Ma et al. 2017). Because of the gap, a detailed picture of S, Fe, and P interactions in dynamic intertidal mudflats has not been well depicted. Simultaneous measurements of porewater Fe2+ (dissolved ferrous Fe is denoted as Fe2+ and solid-phase ferrous Fe as Fe(II) hereafter), sulfide, and P using an in situ high-resolution diffusive gradients in thin films (DGT) technique, by combining solid-phase chemistry, promise to reveal a more nuanced picture of their spatial heterogeneity and offer an integrated understanding of their interplays.
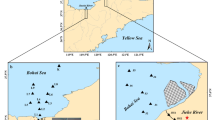
A shellfish aquaculture area in an intertidal mudflat of Jiaozhou Bay, China, was selected for this study (Fig. 1). In situ simultaneous measurements of porewater Fe2+, sulfide, and P were implemented using the DGT technique to provide detailed distributions of the three constituents in the mudflat. In addition, solid-phase S and Fe were speciated for further insights into the linkages between solid-phase and porewater chemistry. This study aims to (i) elucidate diagenetic interactions of S and Fe, sulfide accumulation, and its potential ecotoxicity in the mudflat sediments; (ii) reveal the mechanisms of P mobilization and potential role of the intertidal mudflat as a source/sink of P; (iii) quantitatively assess buffering capacity of reactive Fe towards dissolved sulfide.

Map of triplicate sampling sites (J1, J2, J3) in mudflat sediments of Jiaozhou Bay
Sampling and analytical methods
Study area and sampling
Jiaozhou Bay is a semi-enclosed water body linked by a narrow channel to the Yellow Sea (Fig. 1). The bay has an area of 390 km2, and one fifth of which is an intertidal zone with an average tidal range of 2.7–3.0 m. The intertidal mudflat selected for this study is within a clam (Ruditapes philippinarum) aquaculture area, which has been utilized for aquaculture since the 1980s (Han et al. 2017). Sampling sites (Fig. 1) were located at the center of the aquaculture area, which is in the mid-tidal zone with an average water depth of 2.6 m at high tide and 6-h exposure time to the atmosphere in a tidal cycle. Thus, the sampling localities represent the typical hydrogeochemical regimes of the aquaculture area.
Sample collection was carried out on June 9 to 11, 2017. Shellfish aquaculture in this season is generally characterized by rapid shellfish growth and high bioturbation intensity (Soudant et al. 2004). Thus, the sampling was expected to capture the typical geochemical features of the mariculture-impacted mudflat. After 3-h exposure of the mudflat to the air, sediments in triplicate (J1, J2, and J3) were collected manually using PVC tubes (inner diameter 110 mm). The three sites were linearly aligned and 35 m apart away from the neighboring site(s) (Fig. 1). Upon retrieval, the three cores were immediately sealed, leaving almost no headspace (the sediment surfaces were not disturbed), and then transported to the laboratory within 1.5 h. DGT probes were vertically deployed in sediments beside the three sampling sites and left for 24 h before retrieval (see the section “Application of the DGT technique” for detail). Upon in the laboratory, the cores were immediately sectioned at 2–3-cm intervals in N2, and subsamples were sealed in ziplock plastic bags and then frozen at − 20 °C for later handling.
Analysis of total organic carbon
Pre-weighed wet sediment subsamples in duplicate were dried at 105 °C until constant weight for gravimetric analysis of water contents. For total organic carbon (TOC) analysis, pre-weighed dry sediments (ca. 0.5 g) were treated with 1 M HCl overnight for decarbonation. After rinsing and redrying, the samples were ground to ~ 100 meshes for TOC analysis using a vario MACRO cube CHNS elemental analyzer (Elementar, Germany). Relative deviations between duplicates were 1.6–5.4%, except for an outlier of 10%.
Analyses of acid volatile sulfides and chromium(II) reducible sulfides
Acid volatile sulfides (AVSs) and chromium(II) reducible sulfides (CRSs) were quantified in this study. Procedures for the extractions and analyses have been detailed elsewhere (Cornwell and Morse 1987; Burton et al. 2008), and thus, only brief information was provided here. AVS in pre-weighed (~ 1 g), frozen wet subsamples was released under N2 by the addition of 6 M HCl together with 1 mL ascorbic acid (0.1 M). Ascorbic acid was used to inhibit oxidation of released sulfide by concomitantly released Fe3+. Evolved H2S gas was trapped as ZnS precipitate by alkaline ZnAc solution. After 24 h, trapped sulfide was measured by colorimetry (Cline 1969).
For CRS quantification, residual sediment pellets after AVS extraction above were rinsed twice using deoxygenated deionized water and then immediately treated with a mixed solution of acidic chromium(II) solution plus acetone under N2 at room temperature to reduce elemental S and pyrite (FeS2) to H2S (Burton et al. 2008). After 48 h, evolved H2S was measured by the Cline method (Cline 1969). Relative deviations (RDs) of duplicates for AVS were 0.34–10.4%, and the RDs for CRS were 0.5–8.9%, except for an outlier of 31%.
Speciation of solid-phase Fe
The methods of Raiswell et al. (1994) and Poulton and Canfield (2005) were used to determine two reactive Fe pools: (i) buffered acetate-extractable Fe(II) [acetate-Fe(II)], which consists mainly of Fe(II) sulfides (pyrite excluded) and Fe(II) carbonates (siderite and ankerite), and (ii) bicarbonate–citrate-buffered sodium dithionite (BCD)-extractable Fe(III) oxides [BCD-Fe(III)]. Briefly, buffered acetate solution (pH 4.5) was used to extract diagenetic Fe(II) (pyrite excluded) (Poulton and Canfield 2005). After the extraction, residual sediment pellets were rinsed twice with deionized water and then immediately subject to BCD extraction at pH 4.8 to target all amorphous and well-crystalline Fe(III) phases such as ferrihydrite, lepidocrocite goethite, and hematite (Raiswell et al. 1994; Poulton and Canfield 2005).
All extracted Fe was measured by phenanthroline colorimetry with hydroxylamine reductant (Stookey 1970). RDs of duplicates were 0.04–9.5% for the acetate extraction and 0.02–12% for the BCD extraction.
Sediment subsamples were also subject to 1-h extractions with 1 M HCl to estimate labile Fe that is capable of quickly buffering dissolved sulfide in the mudflat (Burton et al. 2011). The extraction targets Fe(II) sulfides (pyrite-Fe excluded), the most labile amorphous and poorly crystalline Fe(III) oxides (ferrihydrite and lepidocrocite) with half-lives from hours to days (Canfield et al. 1992; Raiswell and Canfield 1998; Poulton et al. 2004), and Fe in authigenic clays without extended aging (collectively termed LFe hereafter) (Aller et al. 2004). Wet sediments of known weight (~ 5 g) were extracted by 1 M HCl in N2 for 1 h. After centrifuge and filtration (0.22 μm), resulting supernatants were used for analyses of Fe(II) [i.e., LFe(II)] and Fe(III) [i.e., LFe(III)]. Total extracted Fe was measured using the phenanthroline colorimetry with hydroxylamine reductant (Stookey 1970), and Fe(II) was measured using the same method but omitting hydroxylamine reductant. LFe(III) was calculated by the difference between total extracted LFe and LFe(II). RDs of duplicates for LFe and LFe(II) were 0.15–15% and 0.25–14%, respectively.
Application of the DGT technique
A combination of ZrO-Chelex DGT probes (ZrO for P binding and Chelex for Fe binding) and AgI DGT probes (AgI for sulfide binding) was applied for the in situ measurements of dissolved sulfide, Fe, and P concentrations in the sediments.
At J1 and J2, an AgI DGT probe and two ZrO-Chelex probes with different diffusion layer thicknesses (0.1 mm and 0.9 mm) were deployed as a triangle. At J3, only a ZrO-Chelex probe (diffusion layer thickness 0.9 mm) and an AgI DGT probe were deployed back to back. Results from the 0.9-mm diffusion layer were used to calculate time-averaged fluxes (FDGT) of dissolved Fe2+ and P from porewater to the DGT devices, while results from the 0.1-mm diffusion layer were used only for characterization of resupply kinetics from solid phases to porewater (see the section “Phosphor mobilization and its relevance to Fe reduction” for detail). After 24 h, the probes were retrieved from the sediments and immediately rinsed thoroughly with deionized water and then protected in black plastic bags for transport to the laboratory (within 1.5 h). Upon in the laboratory, the ZrO-Chelex binding gels were immediately sliced into 5-mm intervals for the elution of Fe in 1.0 M HNO3 solutions (1 mL). It was assumed that dissolved Fe measured by the DGT technique was Fe2+ because Fe(III) is almost insoluble at pH of seawater. After 24 h, eluates were collected for Fe analysis, and then the binding gels were subject to 24-h elution of P in 1.0 M NaOH solutions (1 mL). Eluted P and Fe were analyzed by the molybdenum blue method and phenanthroline colorimetric method (with hydroxylamine reductant), respectively, on an Epoch Microplate Spectrophotometer. Accumulated masses (M, μg) of Fe and P in binding gels at given times were calculated according to Eq. (1) (Ding et al. 2012):
where Ce is the concentrations of Fe and P in the eluates; Vg and Ve are the volumes of the gel and eluates, respectively; and fe is the elution efficiency (0.88 for Fe and 0.98 for P) (Xu et al. 2013). Time-averaged FDGT values of dissolved P and Fe2+ from porewater to the DGT devices were calculated by Eq. (2), and DGT concentrations (CDGT) were calculated via Eq. (3). CDGT values were only used to estimate equilibrium concentrations of porewater (Cpw) (see the “Discussion” section for detail).
where Δg (cm) is the thickness of the diffusion layer, t (s) is the deployment time, A (cm2) is the exposure area of the gel, and D (cm2/s) is the diffusion coefficient of the analyte in the diffusion layer at in situ sediment temperature. Temperature-dependent D data for Fe2+, P, and sulfide were measured by Wang et al. (2016) (available at www.easysensor.net).
For sulfide measurement, AgI binding gels were immediately scanned using a flat-bed scanner (600 dpi), and the images were then converted to grayscale density using a computer-imaging densitometry technique (Ding et al. 2012). Sulfide mass accumulation (x) (μg/cm2) was calculated via the calibration curve (Eq. (4)) of grayscale density (y) versus sulfide accumulation (x) (Ding et al. 2012), and then two-dimensional (2-D) FDGT and CDGT of dissolved sulfide were obtained via Eqs. (2) and (3), respectively.
Results
TOC
All data reported hereafter are averages of triplicate samplings at J1, J2, and J3, unless otherwise stated.
Range of TOC contents of the sediments was 0.69–0.94% (Fig. 2). The TOC contents displayed a gradual downcore increase, which is opposite to a commonly observed downcore decrease with prolonged burial in coastal marine sediments (Burdige 2006). The atypical TOC profile in the mudflat may have been resulted from unsteady-state sedimentation caused by several possible factors, including increased dilution by terrestrial inputs due to an increase in anthropogenic perturbations (Yang et al. 2016), decreased density of aquaculture in recent years (Liu et al. 2010), and more stringent environmental measures for wastewater discharge to the bay since the 2000s (Dai et al. 2007; Yang et al. 2011). Because of the complexity, we will not quantitatively link OM mineralization and diagenesis of S, Fe, and P to the TOC profile in later discussion.

Vertical profiles of total organic carbon (TOC) in mudflat sediments of Jiaozhou Bay. Error bars indicate standard deviations of triplicate samplings (J1, J2, J3)
AVS and CRS
AVS contents in the mudflat were 2.70–19.7 μmol/g (Fig. 3a). The contents were very low (< 5.0 μmol/g) in the uppermost layer (1–2 cm) but increased gradually with depth to a maximum of 19.7 μmol/g at 19 cm depth and then decreased downcore. CRS contents were 20.1–42.8 μmol/g (Fig. 3b) and displayed a downcore increase.

Vertical profiles of acid volatile sulfide (AVS) (a), chromium(II) reducible sulfide (CRS) (b), and degree of sulfidization (DOS) (c) in mudflat sediments of Jiaozhou Bay. Error bars indicate standard deviations of triplicate samplings (J1, J2, J3)
Solid-phase Fe speciation
Acetate-Fe(II) contents were 23.5–43.3 μmol/g (Fig. 4a). The contents increased from the surface to a maximum of 43.3 μmol/g at 15 cm depth and then decreased gradually to the bottom of the cores. The depth pattern is similar to that of the AVS. BCD-Fe(III) contents were almost invariable over all the depths, with an average of 75.1 ± 4.07 μmol/g (Fig. 4b).

Vertical profiles acetate-extractable Fe(II) [acetate-Fe(II)] (a) and bicarbonate–citrate-buffered sodium dithionite (BCD)-extractable Fe(III) oxides [BCD-Fe(III)] (b) in mudflat sediments of Jiaozhou Bay. Error bars indicate standard deviations of triplicate samplings (J1, J2, J3)
LFe(II) contents were in the range of 24.8–35.5 μmol/g (Fig. 5a) and displayed a slight downcore increase; LFe(III) contents were in the range of 19.2–28.8 μmol/g with no clear depth variability (Fig. 5b). LFe(II) contents were somewhat lower than those of LFe(III) at similar depths, indicating that a large portion of reactive Fe(III) remained unreduced. LFe(II) contents (24.8–35.5 μmol/g) were roughly comparable to those of acetate-Fe(II) (23.5–43.3 μmol/g) at corresponding depths.

Vertical profiles of ferrous LFe [LFe(II)] (a), ferric LFe [LFe(III)] (b), and quick sulfide’s buffering capacity (βL) (c) in mudflat sediments of Jiaozhou Bay. Error bars indicate standard deviations of triplicate samplings (J1, J2, J3)
Two-dimensional images of dissolved sulfide F DGT and vertical F DGT of dissolved Fe2+ and P
Two-dimensional FDGT values of dissolved sulfide at the three sites (J1, J2, J3) were in the range of 0.37–0.87 μmol/cm2/s (Fig. 6a–c). Note that FDGT results in the uppermost 1 cm were excluded because of possible coloration of the sulfide binding gel during the ebbing period of the deployment. The 2-D FDGT images indicated that dissolved sulfide was omnipresent but spatially heterogeneous.

Two-dimensional images of dissolved sulfide FDGT measured by DGT at sites J1 (a), J2 (b), and J3 (c) and vertical distributions of dissolved Fe2+ (c), P (d), and sulfide (e) FDGT measured by DGT. Dissolved sulfide FDGT data in the upper 1 cm were excluded due to possible coloration of the sulfide binding gel during the period of the deployment. Error bars for FDGT-Fe and FDGT-P and gray area for FDGT-S indicate standard deviations of triplicate samplings (J1, J2, J3)
FDGT profiles of dissolved Fe2+ and P are shown in Fig. 6d–f, and FDGT profiles of dissolved sulfide obtained by lateral averaging of the 2-D FDGT for the triplicate are also shown for the purpose of later comparison. FDGT values of dissolved Fe2+ were 0.011–2.04 μmol/cm2/s and showed a quick increase from the surface to a depth of 5 cm. Deeper than this depth, FDGT values of dissolved Fe2+ remained roughly invariable. FDGT values of dissolved P were 0.012–0.505 μmol/cm2/s and showed a gradual downcore increase. FDGT values of both dissolved sulfide and P were much lower than those of dissolved Fe2+ at similar depths.
Discussion
Sulfate and Fe reductions recorded by in situ DGT measurement
As shown in Fig. 6a–c, the omnipresence of dissolved sulfide from the subsurface to the bottom of the cores, despite high spatial heterogeneity, indicates that SR was not restricted to the deep zone, but it also occurred close to the surface layer, though the uppermost layer is subject to bioturbation/bioirrigation and periodical exposure to the atmosphere, which tend to dampen sulfate reduction and sulfide accumulation.
A quick downcore increase in FDGT of Fe2+ from the surface to a depth of 5 cm (Fig. 6d) indicates a rapid increase in Fe(III) reduction rates. We note that FDGT values of dissolved Fe2+ are more than two factors higher than those of dissolved sulfide below a depth of 3 cm. This difference implies that dissimilatory iron reduction (DIR)–coupled OM oxidation prevails in the mudflat, and as a result, the DIR may have competitively dampened SR to some extent. Fe reduction by dissolved sulfide (i.e., chemical reduction) as the dominant process could be excluded because precipitation of dissolved sulfide with dissolved Fe2+ is kinetically more favored than reaction of dissolved sulfide with solid-phase Fe(III) oxides, and thus, porewater Fe2+ is not allowed to outnumber dissolved sulfide as long as Fe chemical reduction prevails in sulfidic porewaters (Canfield 1989; Thamdrup et al. 1994). This DIR-dominated feature in the mudflat sediments is contrast to SR-dominated feature usually observed in many other aquaculture-impacted marine sediments (Otero et al. 2006, 2009; Álvarez-Iglesias and Rubio 2012; Hyun et al. 2013). The omnipresence of dissolved sulfide, despite much lower FDGT values relative to dissolve Fe2+, testifies to a widespread concurrence of SR in the DIR-dominated mudflat sediments, as inferred in other sediments (Postma and Jakobsen 1996).
Phosphor mobilization and its relevance to Fe reduction
Either reductive dissolution of Fe (Slomp et al. 1996; Ding et al. 2016) or OM decomposition (Ruttenberg and Berner 1993; Mucci et al. 2000) is the source of P mobilization in marine sediments. In the mudflat sediments, the FDGT values of dissolved P and Fe2+ were highly coupled to each other over the upper 8 cm (Figs. 6 and 7). This suggests that P release is closely associated with reductive dissolution of Fe oxides. Below the depth of 8 cm, decoupling of dissolved P from Fe2+ could not invalidate the statement above because the decoupling is not caused by their release processes but by different processes of their removal from the porewaters. Specifically, a small downcore decrease in FDGT of Fe2+, while a gradual increase in P FDGT (Fig. 6d), could be ascribed to the titration of dissolved Fe2+ by dissolved sulfide, while the process has no a significant influence on P in porewaters because Fe(II) sulfide phases have low sorptive affinity for P (Bebie et al. 1998; Rozan et al. 2002).

Correlations between dissolved Fe2+FDGT (FDGT-Fe) and dissolved P FDGT (FDGT-P) measured by DGT in mudflat sediments of Jiaozhou Bay. Data over the upper 8 cm of sediments are indicated by solid circles, and data below the depth of 8 cm are indicated by empty circles
The mechanism of Fe reduction-driven P release is further confirmed by resupply dynamics of dissolved Fe2+ and P from sediment solid phases to the porewaters. The dynamics can be quantitatively characterized by parameter R (Eq. (5)) (Harper et al. 1998), which reflects the extent of solid-phase resupply to sustain equilibrium concentrations of the porewater during DGT uptake.
CDGT of dissolved Fe2+ and P measured by a thin diffusive layer (0.1 mm) can be used to characterize dynamic resupply from solid phases to the porewater due to a quick depletion of porewater solutes through the thin diffusive layer (Harper et al. 1998; Ding et al. 2016). High-resolution measurement of Cpw was not made in this study. Instead, here, CDGT obtained from the thick diffusive layer (0.9 mm) was used as an estimate of Cpw, by assuming that equilibrium porewater concentrations have been sustained by resupply from solid phases given that a long time is needed to deplete the porewater through the thick diffusive layer. With this assumption, an apparent ratio (R′) (Eq. (6)) can be calculated to characterize the resupply dynamics (Ding et al. 2016).
where FDGT(0.1) and FDGT(0.9) are the FDGT of P or Fe2+ obtained from the measurements using 0.1-mm and 0.9-mm diffusive layers, respectively. The calculated averages of R′ for J1 and J2 (Fig. 8) indicate that the resupply dynamics of dissolved Fe2+ and P is broadly similar to each other over the sampling depths, with the exception of two points in the uppermost layer. This confirms a synchronous release of dissolved Fe2+ and P from solid phases, and thus the mechanism of P mobilization driven by reductive dissolution of Fe oxides.

Vertical distributions of R′ averages for sites J1 and J2. R′ values are the ratio of FDGT(0.1) of P or Fe2+ measured by the thin diffusive layer (0.1 mm) to FDGT(0.9) measured by the thick one (0.9 mm)
Given that Fe(III) reduction is mainly driven by DIR coupled to OM mineralization, P release from OM mineralization is logically dependent of Fe oxide reduction. Its importance can be assessed via Eq. (7) by assuming that OM composition follows the Redfield ratio, and Fe oxides occur as FeOOH.
The stoichiometric ratio of Fe2+/P (424) in Eq. (7) is much higher than the FDGT ratios of dissolve Fe2+ to dissolved P (11.8 ± 4.30) in the upper 8 cm of the sediments. This means that P release from OM mineralization coupled to DIR is only a minor contribution to the total P release, which further lends credence to the importance of Fe reduction-driven P release.
At the sediment–water interface (SWI), Fe2+ can be rapidly oxidized to Fe(III) oxides. Regenerated Fe oxides serve as an effective adsorbent of P (Lehtoranta et al. 2008, 2009). The effective sequestration of porewater P by the Fe(III) oxides is largely dependent on the number of adsorption sites available on the oxides, and thus on Fe2+ concentration. Thus, the concentration ratios of dissolved Fe2+ to dissolved P in porewaters are a good proxy to infer the sequestration efficiency of upward diffusing P by newly regenerated Fe(III) oxides at the SWI. It has been demonstrated that the concentration ratios of at least larger than 2 are required for an efficient sequestration of porewater P by newly formed Fe oxides from Fe2+ oxidation at the SWI (Sundby et al. 1986; Gunnars et al. 2002; Blomqvist et al. 2004; Lehtoranta et al. 2008). Actually, ratios of in situ FDGT values of the two constituents are more straightforward than their concentration ratios for this assessment. FDGT ratios of dissolved Fe2+ to P (11.8 ± 4.30) over the upper 8 cm of the mudflat sediments are much larger than 2. This suggests that upon oxidation of upward diffusing Fe2+, newly formed Fe oxides at the SWI can function as an efficient “iron curtain” of upward diffusing P, and thus the mudflat sediments are unlikely to be an important P source of the bay water. We note that FDGT ratios in the mudflat sediments are much higher than those at the center of the bay (2.44 ± 1.62, unpublished) and in the inner shelf of the East China Sea (5.31 ± 3.65) (Ma et al. 2017). This implies that, relative to typical marine sediments, the SWI of the intertidal mudflat has higher capacity to sequestrate upward diffusing P and thus has larger resilience to pulsed nutrient supplies. The transitional mudflat zone may also serve as an effective sink of terrestrially delivered P as well as seawater P due to the presence of iron curtain at the SWI.
Sulfide accumulation
The net accumulation of AVS at any given depth in a sediment represents the balance of the relative rates of AVS formation and its loss (conversion to pyrite, oxidation to sulfate by O2 at the uppermost layer or to intermediate S by manganese and Fe oxides at depth) taking place at the depth. AVS profile in this study (Fig. 3a) is similar to the classic maximum distribution pattern (i.e., a maximum appears at a subsurface layer), as observed in typical marine sediments (Rickard and Morse 2005). Very low but detectable contents of AVS (average 2.7 μmol/g) in the uppermost layer (0–2 cm) of the sediments are a combined result of rapid oxidation of sulfide, conversion to pyrite through polysulfides, and more importantly, low activity of sulfate-reducing bacteria in the suboxic conditions (Jørgensen 1977; Rickard and Morse 2005). The downcore buildup of AVS could be attributed to a combined result of the decreased rate of AVS oxidation below the oxic zone and the downward migration of previously accumulated AVS with prolonged burial, though SR rates usually decrease with depth (Jørgensen et al. 1990; Burdige 2006). Below the depth of the AVS maximum, a downcore decline of AVS contents is ascribable to two factors: (i) low rate of sulfide production due to further decreased degradability of OM with prolonged burial and (ii) AVS consumption through pyrite formation. AVS-associated Fe (AVS-Fe) contents (roughly equal to AVS contents) in the mudflat account for only 32.4 ± 9.6% (11.5–52.9%) of acetate-Fe(II). This indicates that, on average, most of the acetate-Fe(II) remains unsulfidized, which confirms the DIR-dominated regime of the sediments inferred from the DGT measurements. The results are much different from the observations in many other aquaculture-impacted marine sediments, in which reactive Fe(III) oxides have usually been highly sulfidized and thus become depleted (Otero et al. 2006, 2009; Álvarez-Iglesias and Rubio 2012).
Here, we assume that pyrite is the dominant constituent of CRS in the mudflat because usually elemental S is only a minor faction of CRS (Rickard and Morse 2005). The presence of CRS (20.1–20.8 μmol/g) in the uppermost layer (1–3 cm) (Fig. 3b) indicates that AVS is capable of rapid conversion to pyrite, and that periodic exposure of pyrite to oxygen has not resulted in its substantial oxidative loss, consistent with the observation that pyrite is oxidized slowly in oxic seawater (Morse 1991). Actually, AVS oxidation and elemental S formation may facilitate pyrite formation via the polysulfide pathways (Rickard 1974; Luther 1991). A gradual increase in CRS with increasing depth suggests continuous pyrite formation at the expense of AVS with prolonged burial, as indicated by a rapid AVS decrease below the depth of AVS maximum (Fig. 3a vs. b).
A study of Yokoyama (2003) indicates that a substantial decrease of macro benthic biota occurs only when AVS concentration is up to 53 μmol/g. AVS concentrations in the mudflat sediments (2.70–19.7 μmol/g) are far below the critical value. In addition, a study shows that a 24-h exposure of the Manila clam (Ruditapes philippinarum) juveniles to 1000 μmol/L of dissolved sulfide has no impact on their survival (Kodama et al. 2018). As a first approximation, here, we used CDGT values of dissolved sulfide from the 0.9-mm diffusive layer (Eq. (3)) as estimates of its Cpw. Calculated CDGT concentrations of dissolved sulfide are in the range of 1.53–9.53 μmol/L, more than 2 orders of magnitude lower than the concentration of 1000 μmol/L. Actually, shellfishes are not always exposed to dissolved sulfide in sediments because they frequently introduce oxygen around their borrows via bioirrigation. Thus, shellfish aquaculture in the mudflat will not have the risk of sulfide ecotoxicity.
In typical marine sediments, the availability of degradable OC is usually the limiting factor of TRIS (TRIS = AVS + CRS) formation/accumulation, which renders TRIS linearly dependent of TOC with a TOC/TRIS weight ratio around 2.8 after early diagenesis (Berner and Raiswell 1984). As a first approximation, here, we assume that sediments below 20 cm depth represent the zone of eventual burial of TOC and TRIS, and thus, only the TOC/TRIS ratios below this depth are included in the following discussion. The average TOC/TRIS in the mudflat is 6.3 ± 2.1 (Fig. 9), much higher than the value in typical marine sediments, but close to the average value of 7 for marine sediments with intense physical reworking (Aller 2014). This suggests that TRIS accumulation in the mudflat sediments has been greatly dampened relative to typical marine sediments, which is starkly in contrast to TRIS enrichment often observed in aquaculture-impacted marine sediments (Otero et al. 2006, 2009; Álvarez-Iglesias and Rubio 2012). The lower TRIS burial in the mudflat can be ascribed to enhanced oxidative loss of sulfide and low availability of OC due to strong sediment resuspension and bioturbation, which favors aerobic respiration and metal reduction (e.g., iron oxide reduction) at the expense of SR (Aller et al. 2004).

Plot of TOC (wt%) versus TRIS (wt%) in mudflat sediments of Jiaozhou Bay. Note that only the data below 20 cm depth are included (see the text for detail). Also shown are averages of TOC/TRIS ratios for normal marine shelf sediments (2.8) (Berner and Raiswell 1984) and for mobile mud belts in the Amazon shelf (7) (Aller 2014)
Fe oxide sulfidization and its quick buffering towards dissolves sulfide
In anoxic marine sediments, reaction of Fe oxides with dissolved sulfide and formation of Fe(II) sulfides (i.e., sulfidization of Fe) are the key processes of sulfide buffering. The degree of sulfidization (DOS) of Fe estimated by Eq. (8) (Boesen and Postma 1988) can be used to quantitatively assess the availability of reactive Fe for further buffering of dissolved sulfide.
where AVS-Fe is the AVS-associated Fe estimated by AVS according to the 1:1 stoichiometry of FeS, Fepy is the pyrite-Fe estimated by CRS/2 according to the 1:2 stoichiometry of FeS2, and FeHR is the total highly reactive Fe estimated by Σ[acetate-Fe(II) + BCD-Fe(III) + Fepy] (Morse et al. 2007). Calculated DOS values range from 0.12 to 0.30 (Fig. 3c). This suggests that most of reactive Fe in the sediments remains unsulfidized over the whole depths of the cores and thus is poised to buffer newly formed sulfide.
In the case of rapid production of dissolved sulfide induced by pulsed inputs of OC, quick buffering of dissolved sulfide is essential to prevent the accumulation of the toxic compound to high levels. Quick buffering capacity (βL), as defined by Eq. (9), can be quantitatively assessed on the basis of sulfide buffering reaction (Eq. (10)) (Azzoni et al. 2005; Giordani et al. 2008).
where [LFe(II) − AVS] is the buffering capacity of Fe(II) that has not yet been sulfidized and 1.5 LFe(III) is the buffering capacity of reactive Fe(III) oxides that are capable of quick reaction with dissolved sulfide. Here, LFe(III) is operationally defined as 1 M HCl-extractable Fe(III) in 1 h (Burton et al. 2011). Calculated βL values for the mudflat sediments are 48.2–67.1 μmol/g, with a mean of 57.4 ± 5.6 μmol/g (Fig. 5c). The buffering capacities together with very low AVS/LFe ratios (0.21 ± 0.10) in the mudflat are characteristic of effective buffering of episodic production of sulfide, according to our previous studies (Zhu et al. 2012; Liu et al. 2014) and others (Azzoni et al. 2005; Giordani et al. 2008). With the βL, we can further estimate the duration for effective buffering of labile Fe towards episodically increased dissolved sulfide due to high SR rates. Here, we use a maximum SR rate of 2000 nmol/cm3/day reported for a eutrophic estuary (the Peel-Harvey Estuary, Australia) (Kraal et al. 2013), together with an average density of dry marine sediment (2.6 g/cm3) and measured porosity in this study for the estimation. Estimated durations are 19.8–45.6 days with a mean of 33.1 ± 5.3 days. It is worth noting that these represent only conservative estimates because an unusually high SR rate is used in the estimation, and more importantly, oxidative regeneration of Fe oxides caused by bioirrigation in the upper layer of the sediments is not taken into account. The conservative estimates still indicate that the available βL can secure quick buffering of dissolved sulfide induced by pulsed OC enrichment at least on 1 month timescale.
Summary and conclusion
Much higher FDGT values of dissolved Fe2+ than those of dissolved sulfide below the subsurface layer (3 cm) suggest that DIR prevails in the mudflat, which is further confirmed by the abundant presence of unsulfidized Fe(II). Due to enhanced oxidative loss and/or low SR rates, TRIS contents in the mudflat sediments are obviously lower than those in typical marine sediment. Low dissolved sulfide concentrations in the sediments render most of reactive Fe unsulfidized and thus abundantly available to secure quick buffering of newly formed sulfide.
Strong coupling between dissolved P and Fe2+ over the upper 8 cm suggests Fe reduction-driven P release. High FDGT ratios of dissolved Fe2+ to P (11.8 ± 4.30) over the upper 8 cm of the mudflat suggest that Fe oxide regeneration by oxidation at the SWI can serve as an iron curtain of upward diffusing P. Given the high FDGT ratios of dissolved Fe2+ to P and effective oxidative regeneration of Fe oxides at the SWI of the mudflat, it is inferred that the transitional zone may also serve as an effective sink of seawater and terrestrially delivered P.
In comparison with many other marine sediments impacted by aquaculture, the aquaculture-impacted mudflat has distinctly different features: (i) DIR, rather than SR, is the dominant pathway of anaerobic diagenesis; (ii) TRIS accumulation is dampened rather than enhanced; and (iii) the sediments serve as a sink of P rather than a source of P.
References
Aller RC (1994) Bioturbation and remineralization of sedimentary organic matter: effects of redox oscillation. Chem Geol 114:331–345
Aller RC (2014) Sedimentary diagenesis, depositional environments, and benthic fluxes. In: Holland HD, Turekian KK (eds) Treatise on geochemistry, 2nd edn. Elsevier, Oxford, pp 293–334
Aller RC, Heilbrun C, Panzeca C, Zhu ZB, Baltzer F (2004) Coupling between sedimentary dynamics, early diagenetic processes, and biogeochemical cycling in the Amazon–Guianas mobile mud belt: coastal French Guiana. Mar Geol 208:331–360
Al-Raei AM, Bosselmann K, Böttcher ME, Hespenheide B, Tauber F (2009) Seasonal dynamics of microbial sulfate reduction in temperate intertidal surface sediments: controls by temperature and organic matter. Ocean Dynam 59:351–370
Álvarez-Iglesias P, Rubio B (2012) Early diagenesis of organic-matter-rich sediments in a ría environment: organic matter sources, pyrites morphology and limitation of pyritization at depth. Estuar Coast Shelf Sci 100:113–123
Azzoni R, Giordani G, Viaroli P (2005) Iron-sulphur-phosphorus interactions: implications for sediment buffering capacity in a Mediterranean eutrophic lagoon (Sacca di Goro, Italy). Hydrobiologia 550:131–148
Bebie J, Schoonemn MAA, Uhrmanannd F, Strongin DR (1998) Surface charge development on transition meal sulfides: an electrokinetic study. Geochim Cosmochim Acta 62:633–642
Berner RA, Raiswell R (1984) C/S method for distinguishing fresh-water from marine sedimentary rocks. Geology 12:365–368
Blomqvist S, Gunnars A, Elmgren R (2004) Why the limiting nutrient differs between temperate coastal seas and freshwater lakes: a matter of salt. Limnol Oceanogr 49:2236–2241
Boesen C, Postma D (1988) Pyrite formation in anoxic environments of the Baltic. Am J Sci 288:575–603
Burdige DJ (2006) Geochemistry of marine sediments. Princeton University Press, Princeton
Burton ED, Sullivan LA, Bush RT, Johnston SG, Keene AF (2008) A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils. Appl Geochem 23:2759–2766
Burton ED, Bush RT, Johnston SG, Sullivan LA, Keene AF (2011) Sulfur biogeochemical cycling and novel Fe-S mineralization pathways in a tidally re-flooded wetland. Geochim Cosmochim Acta 75:3434–3451
Canfield DE (1989) Reactive iron in marine sediments. Geochim Cosmochim Acta 53:619–632
Canfield DE, Raiswell R, Bottrell S (1992) The reactivity of sedimentary iron minerals toward sulfide. Am J Sci 292:659–683
Carlsson MS, Holmer M, Petersen JK (2009) Seasonal and spatial variations of benthic impacts of mussel longline farming in a eutrophic Danish Fjord, Limfjorden. J Shellfish Res 28:791–801
Cesbron F, Metzger E, Launeau P, Deflandre B, Delgard M-L, Thibault de Chanvalon A, Geslin E, Anschutz P, Jézéquel D (2014) Simultaneous 2D imaging of dissolved iron and reactive phosphorus in sediment porewaters by thin-film and hyperspectral methods. Environ Sci Technol 48:2816–2826
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Cornwell JC, Morse JW (1987) The characterization of iron sulfide minerals in anoxic sediments. Mar Chem 22:193–206
Dai J, Song J, Li X, Yuan H, Li N, Zheng G (2007) Environmental changes reflected by sedimentary geochemistry in recent hundred years of Jiaozhou Bay, North China. Environ Pollut 145:656–667
Ding S, Sun Q, Xu D, Jia F, He X, Zhang C (2012) High-resolution simultaneous measurements of dissolved reactive phosphorus and dissolved sulfide: the first observation of their simultaneous release in sediments. Environ Sci Technol 46:8297–8304
Ding S, Wang Y, Wang D, Li YY, Gong M, Zhang C (2016) In situ, high-resolution evidence for iron-coupled mobilization of phosphorus in sediments. Sci Rep 6:24341 https://doi.org/10.1038/srep24341
Giordani G, Azzoni R, Viaroli P (2008) A rapid assessment of the sedimentary buffering capacity towards free sulphides. Hydrobiologia 611:55–66
Gunnars A, Blomqvist S, Johansson P, Andersson C (2002) Formation of Fe(III) oxyhydroxide colloids in freshwater and brackish seawater, with incorporation of phosphate and calcium. Geochim Cosmochim Acta 66:745–758
Han DY, Chen Y, Zhang CL, Ren Y, Xue Y, Wan R (2017) Evaluating impacts of intensive shellfish aquaculture on a semi-closed marine ecosystem. Ecol Model 359:193–200
Harper MP, Davison W, Zhang H, Tych W (1998) Kinetics of metal exchange between solids and solutions in sediments and soils interpreted from DGT measured fluxes. Geochim Cosmochim Acta 62:2757–2770
Hyun J-H, Kim S-H, Mok J-S, Lee J-S, An S-U, Lee W-C, Jung R-H (2013) Impacts of long-line aquaculture of Pacific oysters (Crassostrea gigas) on sulfate reduction and diffusive nutrient flux in the coastal sediments of Jinhae-Tongyeong, Korea. Mar Pollut Bull 74:187–198
Jansen S, Walpersdorf E, Werner U, Billerbeck M, Böttcher ME, de Beer D (2009) Functioning of intertidal flats inferred from temporal and spatial dynamics of O2, H2S and PH in their surface sediment. Ocean Dynam 59:317–332
Jørgensen BB (1977) The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark). Limnol Oceanol 22:814–832
Jørgensen BB, Bang M, Blackburn TH (1990) Anaerobic mineralization in marine sediments from the Baltic Sea-North Sea transition. Mar Ecol Prog Ser 59:39–54
Kodama K, Waku M, Sone R, Miyawaki D, Ishida T, Akatsuka T, Horiguchi T (2018) Ontogenetic and temperature-dependent changes in tolerance to hypoxia and hydrogen sulfide during the early life stages of the Manila clam Ruditapes philippinarum. Mar Environ Res 137:177–187
Kraal P, Burton ED, Rose AL, Cheetham MD, Bush RT, Sullivan LA (2013) Decoupling between water column oxygenation and benthic phosphate dynamics in a shallow eutrophic estuary. Environ Sci Technol 47:3114–3121
Lehtoranta J, Ekholm P, Heikki P (2008) Eutrophication-driven sediment microbial processes can explain the regional variation in phosphorus concentrations between Baltic Sea sub-basins. J Mar Syst 74:495–504
Lehtoranta J, Ekholm P, Heikki P (2009) Coastal eutrophication thresholds: a matter of sediment microbial processes. Ambio 38:303–308
Liu SM, Zhu BD, Zhang J, Wu Y, Liu GS, Deng B, Zhao MX, Liu GQ, Du JZ, Ren JL, Zhang GL (2010) Environmental change in Jiaozhou bay recorded by nutrient components in sediments. Mar Pollut Bull 60:1591–1599
Liu J, Zhu M-X, Yang G-P, Shi X-N, Yang R-J (2014) Quick sulfide buffering in inner shelf sediments of the East China Sea impacted by eutrophication. Environ Earth Sci 71:465–473
Luther GW (1991) Pyrite synthesis via polysulfide compounds. Geochim Cosmochim Acta 55:2839–2849
Ma W-W, Zhu M-X, Yang G-P, Li T (2017) In situ, high-resolution DGT measurements of dissolved sulfide, iron and phosphorus in sediments of the East China Sea: insights into phosphorus mobilization and microbial iron reduction. Mar Pollut Bull 124:400–410
Morse JW (1991) Oxidation kinetics of sedimentary pyrite in seawater. Geochim Cosmochim Acta 55:3665–3667
Morse JW, Thomson H, Finneran DW (2007) Factors controlling sulfide geochemistry in sub-tropical estuarine and bay sediments. Aquat Geochem 13:143–156
Mucci A, Richard L, Lucotte M, Guignard C (2000) The differential geochemical behavior of arsenic and phosphorus in the water column and sediments of the Saguenay Fjord Estuary, Canada. Aquat Geochem 6:293–324
Otero XL, Calvo de Anta RM, Macías F (2006) Sulphur partitioning in sediments and biodeposits below mussel rafts in the Ria de Arousa (Galicia, NW Spain). Mar Environ Res 61:305–325
Otero XL, Calvo de Anta RM, Macías F (2009) Iron geochemistry under mussel rafts in the Galician ria system (Galicia-NW Spain). Estuar Coast Shelf Sci 81:83–93
Pagès A, Teasdale PR, Robertson D, Bennett WW, Schäfer J, Welsh DT (2011) Representative measurement of two-dimensional reactive phosphate distributions and co-distributed iron(II) and sulfide in seagrass sediment porewaters. Chemosphere 85:1256–1261
Pagès A, Welsh DT, Robertson D, Panther JG, Schäfer J, Tomlinson RB, Teasdale PR (2012) Diurnal shifts in co-distributions of sulfide and iron(II) and profiles of phosphate and ammonium in the rhizosphere of Zostera capricorni. Estuar Coast Shelf Sci 115:282–290
Pagès A, Welsh DT, Peter R, Teasdale PR, Grice K, Vacher M, Bennett WW, Visscher PT (2014) Diel fluctuations in solute distributions and biogeochemical cycling in a hypersaline microbial mat from Shark Bay, WA. Mar Chem 137:102–112
Postma D, Jakobsen R (1996) Redox zonation: equilibrium constraints on the Fe(III)/SO4-reduction interface. Geochim Cosmochim Acta 60:3169–3175
Poulton SW, Krom MD, Raiswell R (2004) A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim Cosmochim Acta 68:3703–3715
Poulton SW, Canfield DE (2005) Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem Geol 214:209–221
Raiswell R, Canfield DE (2012) The iron biogeochemical cycle past and present. Geochem Perspect 1:1–220
Raiswell R, Canfield DE (1998) Sources of iron for pyrite formation in marine sediments. Am J Sci 298: 219–245
Raiswell R, Canfield DE, Berner RA (1994) A comparison of iron extraction methods for the determination of degree of pyritisation and the recognition of iron-limited pyrite formation. Chem Geol 111:101–110
Rickard DT (1974) Kinetics and mechanism of pyrite formation at low temperatures. Am J Sci 275:636–652
Rickard D, Morse JW (2005) Acid volatile sulfide (AVS). Mar Chem 97:141–197
Rigaud S, Deflandre B, Maire O, Bernard G, Duchêne JC, Poirier D, Anschutz P (2018) Transient biogeochemistry in intertidal sediments: new insights from tidal pools in Zostera noltei meadows of Arcachon bay (France). Mar Chem 200:1–13
Robertson D, Teasdale PR, Welsh DT (2008) A novel gel-based technique for the high resolution, two-dimensional determination of iron (II) and sulfide in sediment. Limnol Oceanogr Methods 6:502–512
Rozan TF, Taillefert M, Trouwborst RE, Glazer BT, Ma S, Herszage J, Valdes LM, Price KS, Luther GW (2002) Iron-sulfur-phosphorus cycling in the sediments of a shallow coastal bay: implications for sediment nutrient release and benthic macroalgal blooms. Limnol Oceanol 47:1346–1354
Ruttenberg KC, Berner RA (1993) Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochim Cosmochim Acta 57:991–1007
Slomp CP, van der Gaast SJ, van Raaphorst W (1996) Phosphorus binding by poorly crystalline iron oxides in North Sea sediments. Mar Chem 52:55–73
Soudant P, Paillard C, Choquet G, Lambert C, Reid HI, Marhic A, Donaghy L, Birkbeck TH (2004) Impact of season and rearing site on the physiological and immunological parameters of the Manila clam Venerupis (=Tapes, =Ruditapes) philippinarum. Aquaculture 229:401–418
Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42:779–781
Sundby B, Anderson LG, Hall POJ, Iverfeldt A, van der Loeff MMR, Westerlund SFG (1986) The effect of oxygen on release of and uptake of cobalt, manganese, iron and phosphate at the sediment-water interface. Geochim Cosmochim Acta 50:1281–1288
Taillefert M, Neuhuber S, Bristow G (2007) The effect of tidal forcing on biogeochemical processes in intertidal salt marsh sediments. Geochem Trans 8:1–15
Thamdrup B, Fossing H, Jørgensen BB (1994) Manganese, iron, and sulfur cycling in a coastal marine sediment, Aarhus Bay, Denmark. Geochim Cosmochim Acta 58:5115–5129
Valdemarsen T, Kristensen E, Holmer M (2009) Metabolic threshold and sulfide-buffering in diffusion controlled marine sediments impacted by continuous organic enrichment. Biogeochemistry 95:335–353
Valdemarsen T, Bannister RJ, Hansen PK, Holmer M, Ervik A (2012) Biogeochemical malfunctioning in sediments beneath a deep-water fish farm. Environ Pollut 170:15–25
Wang Y, Ding S, Gong M, Xu S, Xu W, Zhang C (2016) Diffusion characteristics of agarose hydrogel used in diffusive gradients in thin film for measurements of cations and anions. Anal Chim Acta 945:47–56
Xu D, Chen Y, Ding S, Sun Q, Wang Y, Zhang C (2013) Diffusive gradients in thin films technique equipped with a mixed binding gel for simultaneous measurements of dissolved reactive phosphorus and dissolved iron. Environ Sci Technol 47:10477–10484
Yang L, Wu Y, Zhang J, Liu S, Deng B (2011) Burial of terrestrial and marine organic carbon in Jiaozhou Bay: different responses to urbanization. Reg Environ Chang 11:707–714
Yang W, Chen M, Zhang F, Zhao X, Fang Z, Ma H (2016) Anthropogenic impacts on sedimentation in Jiaozhou Bay, China. J Coast Conserv 20:501–506
Yokoyama H (2003) Environmental quality criteria for fish farms in Japan. Aquaculture 226:45–56
Zhu M-X, Liu J, Yang G-P, Li T, Yang R-J (2012) Reactive iron and its buffering capacity towards dissolved sulfide in sediments of Jiaozhou Bay, China. Mar Environ Res 80:46–55
Funding
The research was jointly supported by the National Key Research and Development Program of China (grant 2016YFA0601301), the National Natural Science Foundation of China (grants 41576078 and 41776085), and the Shandong Province Natural Science Foundation, China (grant ZR2015DM006).
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Meng, T., Zhu, MX., Ma, WW. et al. Sulfur, iron, and phosphorus geochemistry in an intertidal mudflat impacted by shellfish aquaculture. Environ Sci Pollut Res 26, 6460–6471 (2019). https://doi.org/10.1007/s11356-018-04114-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-018-04114-w