
Abstract
The effects of organic enrichment on sediment biogeochemistry was studied in diffusion controlled sediment mesocosms, where labile organic matter (OM) (fish feed) pulses were added once a week to the sediment surface. Two types of sediments, differing mainly in content of reactive Fe, were used. The aim of this experiment was two-fold, (1) to evaluate the importance of Fe-driven sulfide buffering for sulfide accumulation in surface enriched sediments, and (2) to estimate the diagenetic capacity for degradation of labile OM near the sediment surface. The simulated OM loading rate of 375 mmol C m−2 day−1 led to a 5–6 times increase in CO2-production and a 4–5 times increase in O2-uptake. Sulfate reduction estimated by radiotracer experiments and CO2-release was 105–131 mmol m−2 day−1, but accumulation of porewater sulfide was low in both sediment types. Instead 99% of sulfide was oxidized with O2 at the sediment water interface in the low Fe treatment, whereas 46% of produced sulfide precipitated as Fe-S compound in the high Fe treatment resulting in significantly lower O2-uptake. Furthermore, the accumulation of up to 30% of added OM by the end of the experiment indicated a saturation of the heterotrophic microbial communities in the upper enriched surface layer. These results suggest a maximum diagenetic capacity for OM degradation in the range of ~25 μmol C cm−3 day−1 or 260 mmol m−2 day−1 for the present sediment types.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Enhanced sedimentation of organic matter (OM) occurs in marine areas affected by eutrophication (Vaalgamaa 2004), marine fin-fish farming (Holmer et al. 2007; Kutti et al. 2008) or shell-fish farming (Callier et al. 2006; Giles et al. 2006), and the up to 25 times increased sedimentation of labile OM markedly alters sediment biogeochemical processes. OM enrichment leads to a stimulation of benthic metabolism as evidenced by increased sediment oxygen uptake (SOU) and release of TCO2 (CO2 + H2CO3 + HCO3 − + CO3 2−). SOU is primarily due to O2-requirements for reoxidation of metabolites while TCO2 is released in proportion to total carbon (C) oxidation. Both SOU and TCO2-release in organic impacted areas may reach >400 mmol m−2 day−1 as compared to 20–40 mmol m−2 day−1 in reference areas (Holmer and Kristensen 1992; Holmer et al. 2003; Nickell et al. 2003). O2 penetration into organically enriched sediments is greatly reduced and C-oxidation is mediated almost exclusively by anaerobic bacteria. Sulfate reduction (SR) rapidly becomes the dominant respiration pathway, since other respiration pathways are hampered by depletion of their electron acceptors, e.g. NO3 − and oxidized Fe (Sloth et al. 1995; Thamdrup 2000; Christensen et al. 2000).
The OM induced stimulation of SR is usually accompanied by accumulation of mM-levels of sulfide (TH2S = H2S + HS− + S2−) in sediment pore waters (Holmer and Kristensen 1992; Macleod et al. 2004; Porrello et al. 2005). This may have negative ecological consequences since TH2S is toxic at μM-concentrations and has been linked to both decreased fauna diversity and increased mortality of sea grasses (Hargrave et al. 2008 and references therein). Thus the coupling between OM input and stimulation of SR as well as the controls of TH2S in porewater has been widely studied in the past (e.g. Heijs et al. 1999; Kristensen and Holmer 2001). Initial diagenesis of OM is performed by hydrolysing and fermenting bacteria. They split organic macromolecules into smaller moieties and release nutrients (NH4 + and PO4 3−) and low molecular dissolved organic C (LM-DOC, e.g. acetate) to the surrounding microenvironment (Canfield et al. 2005). The generated LM-DOC is oxidized by sulfate reducing bacteria to TCO2 with concurrent reduction of SO4 2− to TH2S. The initial hydrolysis and fermentation of OM is usually considered the rate limiting step for SR and is controlled by both OM quality and quantity (Arnosti and Holmer 1999; Kristensen et al. 1995; Kristensen and Holmer 2001). SR varies from <3 mmol m−2 d−1 (or 10% of total C-oxidation) in continental margin sediments receiving low OM input (Canfield et al. 1993) to >400 mmol m−2 d−1 (or 100% of total C-oxidation) in near shore sediments receiving high amounts of labile OM (Holmer et al. 2003). Under extreme conditions, a shift towards methanogenesis has been observed (Hall et al. 1990; Hoehler et al. 1998; Morrisey et al. 2000). High OM loading may facilitate LM-DOC and H2 accumulation and coexistence of SR and methanogenesis (e.g. Holmer and Kristensen 1994b), whereas excessive OM loading and SO4 2− depletion may lead to domination of methanogenesis or hampered sediment metabolism due to LM-DOC accumulation (Canfield 1994; Kristensen and Hansen 1995; Canfield et al. 2005).
Several processes mitigate the mM-levels of TH2S observed in organic enriched sediments. TH2S is removed from solution either through reoxidation by chemical or microbial processes or by precipitation as iron-sulfur compounds. Reoxidation of TH2S to products like S0 or SO4 2− occurs typically along oxidized interfaces near the sediment surface, infaunal burrows or seagrass rhizospheres (Kristensen 2000; Mascaro et al. 2009). The process occurs as a chemical reaction with O2 or Fe- and Mn-oxides (Schippers and Jorgensen 2002) or is catalyzed by sulfide oxidizing bacteria (Fossing et al. 1995; Preisler et al. 2007). Reoxidation of TH2S leads to increased SOU, and positive linear relationships between area specific C-sedimentation and SOU are often observed beneath fish farms (Findlay and Watling 1997). This is interpreted as a tight coupling between sedimentation, SR and reoxidation of TH2S with O2. However, above a certain threshold SOU becomes limited by factors such as O2-concentration in the water column and water flow (Jorgensen and Marais 1990) and TH2S may escape to the overlying water column (Morrisey et al. 2000). Fe-containing sediments may have significant TH2S buffering capacity (Heijs and van Gemerden 2000; Giordani et al. 2008), where TH2S precipitates with dissolved Fe2+ to form FexSy (e.g. mackinawite, greigite or pyrite) (Rickard and Morse 2005). The generated FexSy compounds may be permanently buried or transported to oxidized interfaces by bioturbation where they are reoxidized to Fe(III)-oxides and oxidized sulfur compounds.
The effects of OM-enrichment on sediment biogeochemistry have mostly been studied in closed sediment incubations (Holmer and Kristensen 1994a, b; Kristensen and Hansen 1995) or in plug incubations open to an overlying water reservoir (Kristensen and Hansen 1995; Sloth et al. 1995; Banta et al. 1999; Papaspyrou et al. 2004; Karle et al. 2007). These batch studies have provided valuable basic knowledge on the impact of organic enrichment on sediment metabolism. However, they cannot simulate the behavior of e.g. SR, FexSy-buffering and metabolic capacity under in situ conditions, because OM-enrichment often occurs as a gradual process where labile OM is continuously supplied to the sediment surface from above. There are to our knowledge no available studies which have simulated such continuous OM-enrichment.
In this study we identify the effects of organic matter pulses on sediment metabolism, sulfide behavior and iron dynamics in diffusion controlled sediment mesocosms. Two types of organic-poor sediments, differing mainly in Fe-content, were exposed to weekly pulses of labile OM for 5 weeks. Benthic metabolism was determined as the sediment-water exchange of TCO2, NH4 +, O2, Fe2+ and TH2S. Temporal changes in vertical profiles of solid phase and dissolved sulfide and iron provided evidence of key oxidation, reduction, precipitation and dissolution processes. Fe- and S-budgets provided the basis for estimates of iron-driven TH2S-buffering, and rates of TH2S-oxidation and FeS-formation in marine sediments. Furthermore, from C- and N-budgets the metabolic capacity of the examined sediments was estimated under the present experimental conditions and the increased OM-burial in enriched sediments was explored by diagenetic modeling. In this article we discuss continuous OM enrichment at the sediment-water interface in diffusion controlled situation only. For a more detailed discussion of how fauna bioturbation influences the biogeochemical cycling of particulate and dissolved phases in organic enriched sediments, readers are referred to other studies (e.g. Banta et al. 1999; Heilskov et al. 2006).
Materials and methods
Sediment handling and experimental setup
Two types of sediment were collected in July 2007 at two sites located on the Western part of the island of Fyn, Denmark (Fig. 1). Medium sand (medium grain size = 491 μm) with a low content of iron and organic matter was collected at Svenstrup Strand (Fe-poor), and medium sand (medium grain size = 221 μm) relatively rich in iron and organic matter was collected at Odden (Fe-rich) close to the Sound of Fænø. The two sites are situated 5 km apart and share the same water body. Nine undisturbed sediment cores were collected at each site in shallow water (<1 m depth) with Plexiglas core liners (i.d. 8 cm) that were closed at both ends with rubber stoppers.

Location of Svenstrup Strand (Fe-poor) and Odden (Fe-rich)
Upon return to the laboratory, cores were placed in a freezer at −18°C for 3 days to kill macrofauna. The freezing process may also disturb the microbial community and influence chemical and physical profiles within the sediment, but this disturbance is considered short-term compared to the total 8 week duration of the experiment (Banta et al. 1999). After thawing cores were adjusted to a depth of ~20 cm, leaving a 10 cm water column, and split into two groups that were placed in separate tanks containing 70 L of 17‰ GF/F-filtered seawater from Svenstrup Strand. Cores were equipped with 4 cm long magnetic stirring bars located a few centimetres above the sediment surface, and were arranged around a central magnet rotating at 60 rpm. The water reservoir in each tank was mixed and aerated by air pumps. The cores were kept submerged in darkness at 15°C, and ~20% of the water in each tank was changed weekly throughout the experiment with 17‰ seawater to prevent accumulation or depletion of metabolites in the water column.
Cores were allowed to stabilize for 2 weeks, after which the measurements were initiated (t = 0). Six cores from each site were kept as controls (control), and three cores were subjected to weekly pulses of labile organic matter (enriched) in the form of commercially available fish food (Ecolife, Dansk Ørredfoder, 49.4% particulate organic C (POC) and 8.1% particulate organic N (PON)). The organic matter (0.29 g homogenized fish food) was thoroughly mixed into 10 g homogenized sediment from the site, corresponding to a 1 mm thick layer of sediment. The enriched sediment pellet was applied directly to the sediment surface with a spatula after removal of the water phase. Cores were refilled with water, avoiding resuspension, and returned to the incubation tanks. Each enrichment corresponded to 9.1, 4.5 and 0.73 g DW m−2 day−1 of OM (organic matter), organic C (OC) and organic N (ON), respectively or 375 and 52 mmol m−2 day−1 of C and N, respectively.
Flux measurements
Efflux of TCO2, sediment O2 uptake (SOU), and exchange of dissolved organic carbon (DOC), NH4 +, Fe2+ and TH2S were determined, in triplicates, on enriched cores twice a week throughout the experiment (day 2 and 6 after the enrichment) and once a week on control cores. Initial water samples were taken and cores were closed with rubber stoppers. Incubations were terminated after 2 h (enriched cores) and 5 h (control cores) and final water samples were taken. Samples for dissolved TCO2 were preserved with saturated HgCl2 (volume ratio of 9:1) in gas tight glass exetainers closed with Teflon coated screw caps and stored for no more than 14 days at 5°C before analysis by flow injection analysis (Hall and Aller 1992). Samples for NH4 + and DOC were stored frozen (−20°C) in precombusted (5 h, 520°C) glass exetainers until analysis. NH4 + was analysed by the salicylate-hypochlorite method described by Bower and Holm-Hansen (1980) and DOC was analysed on acidified samples on a Shimadzu TOC-5000 Analyzer. TH2S samples were preserved with 1 M zinc acetate (ZnAc ~ volume ratio of 9:1) and analyzed by the method of Cline (1969). Samples for dissolved Fe2+ were acidified with 0.5 M HCl (volume ratio of 1:1) and analyzed by the Ferrozine method (Stookey 1970). Changes in O2-concentration during incubations were detected by an O×100 mini electrode (Unisense).
Test for diffusive loss of DOC and DON
Since OM was applied directly at the sediment surface, a fraction of added C and N may have escaped to the water column as DOC and DON by leaching and diffusion without being mineralized. At one occasion the enrichment procedure was tested for initial leaching of DOC and DON. Three cores from each site were enriched as described above and gently refilled with a known volume of water. Samples for DOC and total N (TN) were transferred to precombusted exetainers at 1, 2, 4 and 24 h after the enrichment procedure and stored frozen until analysis. TN was determined as NO3 − by flow injection analysis on a Lachat Quickchem 8500 following oxidation of organic N with persulphate according to Delia et al. (1977), and DON was estimated as the difference between TN after persulfate oxidation and DIN (NO3 −, NH4 +) before persulfate digestion. DOC was analyzed as described above.
Core sectioning
Three control cores from each site were sectioned at time zero (t = 0) to establish the initial conditions within the sediment (initial). At the end of the experiment, three control cores (end, control) and three enriched cores (end, enriched) from each site were sectioned. Cores were sectioned into 0.5 cm intervals to 2 cm depth and into 1 cm intervals to 4 cm depth. The sediment from each interval was homogenized and density was obtained by the weight of a known volume of wet sediment and water content as the weight loss of wet sediment after drying (12 h, 105°C). TC and TN content were determined on dried sediment subsamples by elemental analysis on a Carlo Erba CHN EA1108 elemental analyzer. Accumulation of OC and ON was estimated as the difference between final and initial samples. A sediment subsample from each depth interval was preserved in 10 mL of 0.5 M ZnAc for later determination of acid volatile sulfides (AVS) and chromium reducible sulfur (CRS). Another subsample of sediment (~0.2 g) was transferred to preweighed centrifuge tubes containing 5 mL of 0.5 M HCl for later determination of total reactive Fe (RFe). The remaining sediment was transferred to double centrifuge tubes mounted with GF/F filters and centrifuged for 10 min at 1,500 rpm. The extracted porewater was sampled for analyses of dissolved metabolites: TCO2, NH4 +, DOC, Fe2+, TH2S and SO4 2−. Samples for SO4 2− were stored frozen and analyzed by liquid ion chromatography on a Dionex ICS-2000 system. The other solutes were stored and analyzed as described above. The slicing/centrifugation-procedure took on average 30 min during which sediment and porewater was exposed to the atmosphere and we recognize that some TH2S or Fe2+ was oxidized and TH2S lost due to evaporation. Subsequent tests showed that evaporation and oxidation corresponded to 40–50 μM h−1 TH2S loss.
Sulfate reduction essay
During the final sectioning, sulfate reduction rates (SRR) were measured down to 4 cm depth in subcores. After decanting the overlying water and before sectioning was initiated, a 60 mL cut-off syringe (2.6 cm i.d.) was gently pushed down the sediment to obtain 6 cm deep subcores. 35S-SO4 2−-tracer (~35 kBq pr. mL sediment) was injected vertically into the subcores at 0.25, 0.75, 1.25, 1.75, 2.5 and 3.5 cm depth, thus covering the same intervals as the bulk core sectioning. Subsequently, the syringes were closed with rubber stoppers and incubated for 1–3 h at 15°C in darkness. The incubation was terminated by slicing subcores into 0.5 cm intervals to 2 cm depth and into 1 cm intervals to 4 cm depth. Sediment was transferred to preweighed centrifuge tubes containing 10 mL of 0.5 M ZnAc and subsequently frozen until later analysis.
SR and sediment pools of AVS and CRS were determined in triplicates using the two-step distillation technique of Fossing and Jorgensen (1989). In brief, sediment subsamples were distilled for 30 min under anoxic conditions after adding 8 mL of 6 M HCl to release AVS, which is a heterogeneous mixture of reduced S-compounds consisting mainly of TH2S, mackinawite (FeS), greigite (Fe3S4) and some pyrite (FeS2) (Rickard and Morse 2005). Finally 16 mL of reduced chromium solution was added and distillation was continued for 30 min while boiling the slurry to release CRS, which consists mainly of pyrite and S0 (Rickard and Morse 2005). The AVS and CRS released during distillations were trapped in two separate traps containing 10 mL of 250 mM ZnAc. For the determination of SRR the 35S activity was determined in both supernatant, AVS-, and CRS-distillate on a Tri-Carb 2200 CA Liquid Scintillation Analyzer. The total pools of AVS and CRS were estimated after analysis of TH2S in the distillate by the method of Cline (1969).
Reactive Fe extraction
Reactive Fe was extracted on 0.2 g sediment subsamples by 5 mL of 0.5 M HCl for 30 min on a shaking table. After centrifugation (3,000 rpm, 10 min) and GF/F-filtration the supernatant was stored at room temperature until analysis. Reactive FeII (RFeII) was determined on the supernatant by the Ferrozine method (Stookey 1970) before (RFeII) and after (RFe) reduction with hydroxylamine (Lovley and Phillips 1987). Reactive FeIII (RFeIII) was estimated as the difference between RFe and RFeII. Furthermore, Fe bound in pyrite (pyriteFe) was estimated from CRS-pools, assuming that the majority of CRS is pyrite. Total reactive Fe (TRFe) was estimated as RFeIII + RFeII + pyriteFe (Raiswell and Canfield 1998).
Results
Sediment description
The initial freezing of sediment cores successfully killed all macrofauna, and no animals were observed in cores at any time. The two sediment types had similar density and porosity, but differed with respect to organic content, grain size and Fe-content (Table 1). Initially, the Fe-poor sediment was characterized by a 0.2 cm thick pale brown oxidized layer covering a 1 cm thick dark grey zone. Beneath 1.2 cm depth, the sediment was light gray with intermingled dark gray patches. The Fe-rich cores showed a distinct 0.3 cm thick rusty red band covering a 2–3 cm thick blackish gray zone. From 3.5 cm depth, the sediment gradually changed from dark gray to gray. Organic enrichments rapidly changed the appearance of the upper sediment. The oxidized layer quickly dwindled to a <1 mm thick oxidized film, and the upper 1 cm turned dark grey/black in Fe-poor cores and pitch black in Fe-rich cores. The darkened zone continued to grow downwards as the experiment progressed and reached 2–3 cm depth at the end. Furthermore, white patches of Beggiatoa-like bacteria (henceforth referred to as Beggiatoa) developed on the surface of enriched cores; most pronounced in Fe-poor cores, where 20–30% of the surface was covered within 1 week of the first enrichment. The cover increased to 60–80% within 2 weeks and 80–100% within 3 weeks. The same trend was observed in Fe-rich cores, but the development was much slower and total coverage never exceeded 50%.
In enriched cores C-content at the end was significantly (t-test) increased to 0.6% in the upper 1 cm of Fe-poor cores and to 0.75% in the upper 0.5 cm of Fe-rich cores when compared to initial cores (data not shown). N-content was only significantly increased in the upper 0.5 cm, to 0.09% in Fe-poor cores and to 0.13% in Fe-rich cores. There was no difference between enriched cores and control cores below 0.5 or 1 cm depth.
Effect of organic enrichment on solute exchange
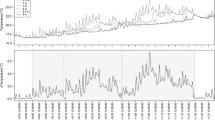
Sediment metabolism increased rapidly in response to organic matter additions (Fig. 2). TCO2 release and SOU in enriched Fe-poor sediment were of similar magnitude and ranged from 80 to 330 mmol m−2 day−1 compared to values of 22–46 mmol m−2 day−1 in control cores. Generally, there was a tight coupling between fluxes and organic matter additions, and the highest fluxes were measured right after enrichments (Table 2). In enriched Fe-rich cores TCO2 effluxes were always higher than SOU and showed a strong response to organic matter additions. NH4 + release was also stimulated by organic matter additions in both sediment types reaching up to 40 mmol m−2 day−1 in enriched cores compared to a maximum of 6 mmol m−2 day−1 in controls for both Fe-poor and Fe-rich sediment. No dissolved Fe2+ or TH2S escaped the sediments during the experiment for both sediment types. No relevant DOC-release was measured in any of the treatments. However, the test for initial leaching showed that a large burst of DOC and DON escaped the sediments in the first hour following the enrichment procedure with release rates of up to 2,000 and 350 mmol m−2 day−1 for DOC and DON, respectively. Release ceased rapidly to no detectable flux within 4 hours of the enrichment procedure, and only 3.5–5.6% and 2.1–2.4% of added C and N were released from the sediment through initial leaching (Table 3).

Fluxes of total CO2 (TCO2, ●), NH4
+ (Δ) and sediment oxygen uptake (SOU,  ) in enriched- and control cores (upper and lower panels, respectively). Note the different axes for TCO2-fluxes and SOU (left
y-axis) and NH4
+-fluxes (right
y-axis). Dotted vertical lines in upper panels indicate times of enrichment. Error bars indicate standard error (n = 3)
) in enriched- and control cores (upper and lower panels, respectively). Note the different axes for TCO2-fluxes and SOU (left
y-axis) and NH4
+-fluxes (right
y-axis). Dotted vertical lines in upper panels indicate times of enrichment. Error bars indicate standard error (n = 3)
There was a tight coupling between TCO2 fluxes and SOU in enriched cores resulting in significant linear regressions of TCO2 release vs. SOU in both Fe-poor (y = 0.92x + 50, R 2 = 0.67) and Fe-rich sediment (y = 1.87 − 40, R 2 = 0.72). The regressions were significantly different (t-test, p < 10−4), indicating that the ratio of SOU to TCO2-release was specific for each sediment type. Thus, average values of TCO2 release were higher in Fe-rich sediment, while no difference was apparent between sediments for SOU (Table 2). Average NH4 + release was always highest in Fe-poor sediment.
C- and N-budgets
Time-integrated excess mineralization in enriched cores was calculated as the flux enhancement of TCO2 and NH4 + in enriched cores. Whereas C-mineralization was highest in Fe-rich sediment N-mineralization was highest in Fe-poor sediment (Table 3). These tendencies were not supported by accumulation of OC and ON that, with a high intercore variability, suggested a similar degree of burial in the two types of sediment. Total time-integrated excess budgets based on fluxes, burial and accumulation accounted for most of the added C (74–93%) but less than half of the added N (36–37%) (Table 3), whereas effluxes suggested that only about half (41–56%) of added C was released as TCO2 and 22–29% of added N was released as NH4 + during the experiment. Thus the budget for N was not closed, indicating that N was not lost primarily through NH4 + release or that the frequency of flux-measurements was out of phase with NH4 +-release. The difference between added, lost and accumulated OC and ON suggested average mineralization rates of 253 and 236 mmol C m−2 day−1 C and 60 and 55 mmol N m−2 day−1 in Fe-poor and Fe-rich sediment, respectively.
Effect of organic enrichment on sulfate reduction rates
SRR in control cores were elevated in the uppermost 0.5 cm at 495 nmol cm−3 day−1 in both types of sediment (Fig. 3). Below this depth, SR was constant and low at an average rate of 56 and 74 nmol cm−3 day−1 in Fe-poor and Fe-rich sediments, respectively. SRR were stimulated in enriched sediments reaching up to 1,500 nmol cm−3 day−1 in enriched Fe-poor cores and up to 2,400 nmol cm−3 day−1 in enriched Fe-rich cores. The stimulation was, however, restricted to the uppermost part of the sediment horizon (1 and 1.5 cm in Fe-poor and Fe-rich cores) and resembled control values beneath this depth. Depth integrated SRR ± SE to 4 cm depth in control and enriched cores were 2.7 ± 0.5 and 14.2 ± 0.5 mmol m−2 day−1 in Fe-poor sediment and 2.3 ± 1.4 and 28.3 ± 9 mmol m−2 day−1 in Fe-rich sediment.

Sulfate reduction rates (SRR) in final control cores (○) and enriched cores (●). Error bars indicate standard error (n = 3)
Effect of organic enrichment on sediment chemistry
Porewater TCO2 was significantly increased in the upper 2 cm of enriched Fe-poor cores, where concentrations reached 8.5 mM compared to 2 mM in controls (Fig. 4). Below 2 cm depth there was no significant effect of OM enrichment on TCO2. The same trend was observed in Fe-rich cores where TCO2 differed in the upper 1 cm with 10 mM in enriched and 3–4 mM in control cores. Below 1 cm enriched cores showed no significant effect of OM enrichment on porewater TCO2 when compared to initial controls, but were significantly higher than final controls. DOC was elevated up to 25 and 9 mM in the upper 0.5 cm of enriched Fe-poor and Fe-rich cores, respectively, but was depleted to <0.9 mM below (Fig. 4). Porewater DOC, which in initial control cores was on average 4.1 and 5 mM in Fe-poor and Fe-rich sediment, was depleted to <2.5 mM in final control cores. Trends in porewater NH4 + were similar to TCO2, except for an excessive concentration of 4 mM in the top 1 cm of enriched Fe-poor cores compared to 0.280 mM in control cores. In enriched Fe-rich sediment NH4 +-concentrations were significantly increased in the upper 0.5 cm and reached 1.5 mM compared to 0.37 and 0.06 mM in initial and final controls. Below 0.5 cm enriched cores and final controls did not differ significantly and was ~1.4 mM in both sediment types, which was slightly lower than initial controls.

Porewater profiles of total CO2 (TCO2), dissolved organic C (DOC) and NH4
+ in the upper 4 cm of initial (○) and final ( ) control cores and enriched cores (●). Profiles from Fe-poor and Fe-rich sediments are shown in upper and lower
panels, respectively. Horizontal dashed line indicate the sediment-water interface, and + indicate the concentration of solute in the overlying water (2.6, 0.45 and 4.6*10−2 mM for TCO2, DOC and NH4
+, respectively). Error bars indicate standard error (n = 3)
) control cores and enriched cores (●). Profiles from Fe-poor and Fe-rich sediments are shown in upper and lower
panels, respectively. Horizontal dashed line indicate the sediment-water interface, and + indicate the concentration of solute in the overlying water (2.6, 0.45 and 4.6*10−2 mM for TCO2, DOC and NH4
+, respectively). Error bars indicate standard error (n = 3)
Porewater sulfate concentrations were initially 12–14 mM near the surface and decreased gradually to 8–9 mM at 3.5 cm depth. Sulfate was not depleted in enriched cores despite high mineralization rates (Fig. 5), but was reduced by up to 7 mM in the upper 1.5 cm of enriched cores compared to control cores. The two types of sediment behaved differently with respect to dissolved TH2S and Fe2+ in porewater (Fig. 5). TH2S in Fe-poor sediments was only detectable in the upper 1 cm in enriched cores at the end and reached 0.9 mM in the upper 0.5 cm. Porewater Fe2+ showed the same pattern in the final enriched cores with concentrations of 70 μM in the upper 0.5 cm. In Fe-poor controls 37 μM Fe2+ peak was evident at 0.75 cm depth but above and below Fe2+ did not exceed 18 μM. A different trend was observed in the final Fe-rich sediment where TH2S in enriched cores was barely detectable, except for around 10 μM in the upper 0.5 cm. Fe2+, on the other hand, was characterized by a distinct subsurface peak of 0.5 mM at 1–3 cm depth with low concentrations above and below. The controls initially showed a Fe2+ peak of 0.4 mM at 0.25 cm depth, but in final controls the Fe2+ peak had shifted downwards to 1.25 cm depth and was 0.9 mM.

Porewater profiles of SO4
2−, total dissolved sulfide (TH2S) and Fe2+ in initial (○) and final (
) control cores and enriched cores (●). Profiles from Fe-poor and Fe-rich sediments are shown in upper and lower panels, respectively. Dashed horizontal line indicate the sediment-water interface and + indicate the concentration of SO4
2− (13.5 mM) in the overlying water. No TH2S and Fe2+ were measured in the overlying water. Error bars indicate standard error (n = 3). Note the different scaling in individual graphs
AVS constituted a minor fraction (maximum 10.9%) of reduced S (Fig. 6) in Fe-poor sediment, but was increased by >58% in both final enriched and control cores (Table 4). CRS in enriched Fe-poor cores was not different from control cores (Table 4), but in the 0–0.5 cm depth interval there were trends towards higher CRS in enriched cores and lower CRS in final control cores (Fig. 6). AVS accounted initially for a larger fraction (up to 36%) of total reduced sulfur in Fe-rich sediment. AVS was not significantly different in final enriched cores (Table 4), but were significantly lowered in final control cores (average 6.5 μmol cm−3) when compared to enriched cores (average 15 μmol cm−3) and initial controls (average 9.8 μmol cm−3). CRS was more variable in Fe-rich sediment, and was significantly increased in the upper 0.5 cm of enriched cores (95 μmol cm−3) when compared to initial (24 μmol cm−3) and final (15 μmol cm−3) controls (Fig. 6). Depth integrated pools suggested significant CRS accumulation in both final controls and enriched cores (Table 4).

Cumulated content of acid volatile sulfides (AVS) and chromium reducible sulfides (CRS) to 4 cm depth in initial (a) and final (c) control cores (upper and lower panels, respectively) and enriched (b) cores (middle panels). Left panels represent Fe-poor sediment and right panels represent Fe-rich sediment. Dashed line indicate the ratio of reduced S bound in AVS (AVS/(AVS + CRS)). Error bars indicate standard error (n = 3)
RFeIII was initially 4 and 11 μmol cm−3 in the upper 0.5 cm of Fe-poor and Fe-rich sediment, respectively, but was depleted to <1 μmol cm−3 in enriched cores and increased (up to 41 μmol cm−3) in final control cores (Fig. 7). Depth integrated pools of TRFe and RFeII did not change during the experiment (Table 4), although there was a tendency to changes in the vertical distribution in Fe-rich sediment (Fig. 7) towards lower RFeII in enriched cores and higher ReFeII below 2 cm depth in controls. PyriteFe was significantly increased in enriched Fe-rich cores, with almost a doubling during the experiment (Fig. 7), particularly in the upper 1 cm.

Cumulated content of FeIII, FeII and Fe bound in pyrite (pyriteFe) to 4 cm depth in initial (a), final (c) control cores (upper and lower panels, respectively) and enriched (b) cores (middle panels). Left panels represent Fe-poor sediment and right panels represent Fe-rich sediment. Error bars indicate standard error (n = 3)
Discussion
S-cycling and TH2S-buffering capacity
The absence of oxidized sediment and the presence of sulfide oxidizing bacteria at the sediment surface in enriched cores indicate an anoxic sediment-water interface, as Beggiatoa position themselves where gradients of H2S and O2 meet (Preisler et al. 2007). Since ReFeIII was depleted in enriched cores, fermentation and sulfate reduction are assumed to be the dominant heterotrophic processes, as in other anoxic sediments without sulfate depletion (Canfield et al. 2005). However, SRR measured 5 days after final enrichments did not adequately account for the TCO2 released by the sediment. The discrepancy is probably due to a strong dependence and rapid response of SR to OM-input causing SR and TCO2 release to be high right after enrichments and then rapidly decrease as labile substrates were depleted, as indicated by the decreasing trends in both TCO2 and NH4 + release in response to OM enrichments (Table 2).
SRR in the upper enriched layer were similar to those measured in highly enriched fish farm sediments (Holmer and Kristensen 1992; Holmer et al. 2003; Holmer and Frederiksen 2007), and TH2S was therefore expected to accumulate to high concentrations as observed in these studies. However, the close contact between the enriched sediment and overlying water facilitated substantial TH2S reoxidation with O2 from the overlying water. Based on the rates of C-mineralization estimated from TCO2 production (Table 2) and the 1:2 stoichiometry between SO4 2− used and OC oxidized, TH2S-production was estimated to 105 and 131 mmol m−2 day−1 in Fe-poor and Fe-rich sediment, respectively. The close and linear relationship between TCO2 efflux and SOU in both sediments indicates that TH2S was reoxidized rapidly at the sediment-water interface (Preisler et al. 2007). However, the degree of TH2S oxidation appears different in the two sediment types. The close to unity relationship between TCO2-efflux and SOU in Fe-poor sediment suggests a complete oxidation of TH2S, which was also evident from the negligible TH2S accumulation. The twice as steep relationship between TCO2 efflux and SOU in Fe-rich sediment indicates TH2S storage by FexSy-precipitation since no associated build up of TH2S occurred. This is consistent with the almost doubling of the CRS-pool; corresponding to precipitation of 46% of produced TH2S.
Despite precipitation of FexSy in enriched Fe-rich cores much TH2S was still oxidized at the sediment-water interface. Pore water profiles suggest that Fe2+ was generated within the upper 2 cm (Fig. 5), but the upwards flux as predicted by molecular diffusion only correspond 0.9 mmol m−2 day−1. This suggests that Fe2+ produced locally in the enriched surface layer (0–1 cm depth) was responsible for most of the FexSy-precipitation. Thus if 50% of RFeII extracted by HCl is available for FexSy-precipitation (ratio similar to Heijs et al. 1999), then the potential for hampering TH2S build-up could be sustained for 120 days within the 0–4 cm interval, but only for 30 days if the 0–1 cm interval is considered. Similarly it has been estimated that precipitation could prevent TH2S build up for up to 160 days in a eutrophic coastal lagoon with high SR (Heijs et al. 1999, 2000), and prevent TH2S-release from sandy sediments for up to 14 days following an oxic to anoxic shift in the water column (Kristiansen et al. 2002). However, estimates of buffering capacity are in many cases derived from anoxic sediment slurries (e.g. Heijs et al. 2000; Giordani et al. 2008), which may overestimate the importance of FexSy precipitation compared with diffusion controlled sediments, where precipitation is restricted to a narrow surface and may be limited locally by availability of Fe2+. Furthermore, as FexSy-precipitation occurs down to 1.5 cm depth, downward transport of dissolved TH2S by diffusion must occur in Fe-rich sediment. This can only happen where SRR exceed Fe2+ availability, and is consistent with accumulation of TH2S near the surface of the same sediments in a parallel experiment (Valdemarsen et al., unpublished results). Similarly, Giordani et al. (2008) found that TH2S is present for several hours after addition to sediment slurries containing RFeII, which indicates that FexSy-precipitation is limited by Fe2+ generation. This observation should be considered for the concept of TH2S-buffering in Fe-containing reactive sediments, since TH2S accumulation may occur locally at high SRRs, even though pools of RFeII are not depleted.
Impact of enrichment on C- and N-mineralization
Fluxes of metabolites (i.e. CO2 and NH4 +) were stimulated within 2 days of the first enrichment, which clearly demonstrate the labile nature of added fish feed and fast response time of both fermentative and respiratory microbial communities. Furthermore the synchronous oscillatory pattern of fluxes upon OM-loading (Fig. 2) supports an initial rapid decay of the most labile OM-fraction (Westrich and Berner 1984; Kristensen and Hansen 1995; Kristensen and Holmer 2001). POC- and PON-data suggested a high degree of OM-mineralization as only 27–33% of added C and 4–13% of added N was buried in enriched sediment (Table 3). The fluxes of TCO2- and NH4 + did, however, not fully capture all the lost C and particularly N, which indicate that flux measurements were out of phase with the peak of metabolite release or that other processes were important for C and N removal. However, incorporation of C and N into bacterial biomass as well as coupled nitrification–denitrification were probably minor sinks compared to total added C and N, in accordance with the frequently observed limited potential for increased bacterial biomass and suppressed coupled nitrification–denitrification in organic enriched sediments (Sloth et al. 1995; Christensen et al. 2000; Kunihiro et al. 2008). Thus degradation of the most labile OC and ON probably resulted in much higher metabolite release than measured 2 days after enrichments resulting in substantial budget deficits. The much higher deficit for N than for C could be caused by a preferential degradation of ON (Kristensen and Hansen 1995) resulting in a different release pattern for TCO2 and NH4 +.
The shape of porewater profiles suggest that stimulation of mineralization processes was largely restricted to the upper 1 cm. From release of TCO2 and NH4 + over the duration of the experiment (Table 3) volume specific reaction rates must have been 21–26 and 1.5–2.2 μmol cm−3 day−1 for C and N, respectively, and have probably been much higher due budget deficits. These rates are extreme compared to some of the highest measured (0.2–5 (C) and 0.2–1 (N) μmol cm−3 day−1) in closed sediment incubations enriched with fish-feed (Holmer and Kristensen 1994a, b) or other types of labile organic matter (Kristensen and Hansen 1995). However, in these batch incubations the initial hydrolysis and fermentation was probably inhibited by accumulation of fermentation products (e.g. short chain fatty acids and H2) (Hoehler et al. 1998; Canfield et al. 2005). Our rates were also 2–5 times higher than initial rapid decay rates found in open thin-layer sediment plugs enriched with diatom or hay detritus (Kristensen and Holmer 2001), where rates actually may have been overestimated due to wall effects (Valdemarsen and Kristensen 2005). It is possible that the open nature, and thus close contact between stimulated sediment and overlying water in our experiment facilitates mineralization at maximum rates as suggested by Aller and Aller (1998). Accumulation of inhibiting substances and depletion of electron acceptors is counteracted by rapid diffusive transport due to the limited spatial scale. Accordingly, sulfate was never limiting despite the high SRR.
The OM that accumulated in our experiment may represent a refractory residue, which is only slowly degraded by hydrolyzing and fermenting enzymes. The fish feed used are composed of 47% proteins and 26% lipids by mass, which together constitute >98% of organic components. While proteins and amino acids are considered labile towards anaerobic degradation over days to weeks (Nunn et al. 2003), lipids are less reactive and degrade over periods of weeks to months (Canuel and Martens 1996; Ding and Sun 2005). However, OM-accumulation can also occur when the diagenetic capacity of the fermenting community has been exceeded. Since every sediment type has an upper limit for bacterial abundance (Deflaun and Mayer 1983; Schmidt et al. 1998), an upper limit for exoenzyme activity and substrate uptake may also exist, as indicated by the reported maximum potential for hydrolysis of proteins and carbohydrates in organic enriched sediments (Arnosti and Holmer 1999; Patel et al. 2001; Sakami et al. 2005). This may in turn lead to an upper limit for terminal mineralization, as indicated by previous studies of organically loaded fish-farm sediments where labile OM is supplied in excess and mineralization is not limited by metabolite build up or SO4 2−depletion. Here maximum SRRs fall within a narrow range equivalent to 6–34 μmol C cm−3 day−1 (Holmer and Kristensen 1996; Holmer et al. 2003; Holmer and Frederiksen 2007), which is similar to the diagenetic capacity of ~25 μmol cm−3 day−1 estimated in this study.
Consequences of a maximum diagenetic capacity
Decay of OC through time in sediments is usually modelled as first-order decay according to (Berner 1980; Westrich and Berner 1984):
where the inventory of organic matter is G (e.g. mol cm−3) and k is the decay constant (day−1). However, when high concentrations of labile OC are present, initial decay rates (e.g. mol cm−3 day−1) predicted by (1) become unrealistic. For example, in our enrichment experiment OC-concentration was ~2,400 μmol cm−3 in the enriched surface layer after enrichments and initial decay should be ~240 μmol cm−3 day−1, and thus one order of magnitude higher than measured rates when using a conservative estimate for k of 0.1 day−1 (Kristensen and Holmer 2001). To avoid such unrealistic high initial decay, it must be assumed that there exists a maximum diagenetic capacity R max (e.g. mol cm−3 day−1), when G is larger than a critical threshold G′, where decay is zero-order according to:
The decay then becomes first-order according to (1) only when G < G′. By integrating (1) and (2) we obtain:
where G 0 is initial pool of organic matter and G N is a residual non- or slow-degradable fraction of OC. By applying an upper diagenetic capacity, the decrease in labile OC-concentration above G′ will be slower, but the period where diagenesis occurs at high rates will be prolonged (Fig. 8). G′ can be deduced from the intercept of (3) and (4) when R max and k are known:
The critical threshold G′ is constant for any sediment type, but may vary among different sediments and depends on the sediment specific R max and OM-lability specified by k. The k-value of 0.1 day−1 used above for modeling purpose is in the low range of reported values from fresh or partly degraded phytoplankton debris (0.008–1 day−1) (Westrich and Berner 1984; Andersen 1996; Kristensen and Holmer 2001). The present experiment suggest that a more realistic k for fish feed is 0.15–0.2 d−1 since 65–75% of added fish feed is degraded between enrichments (calculated from (3)). By assuming a R max of 25 μmol cm−3 day−1, G′ in the present experiment is 125–167 μmol cm−3 according to (5), which is only 5–7% of G 0.

Exponential decay with R max. a. decay of an arbitrary organic matter pool, G 0, follows zero order kinetics until G′ is reached at time t′. Further decreases in the organic matter pool, G, when G < G′ follows first order kinetics and G will gradually approach G N as t approaches ∞. b. dG/dt is constant and equal to R max when G′ < G < G 0, and decreases linearly when G < G′
When deposition of OC is continuous and sediments are modeled as vertically accreting bodies, (3) and (4) are still valid, but t is substituted by \( {\frac{y}{\omega }} \), where ω is deposition rate (cm day−1) and y is depth (cm). When previously unexposed sediments are subjected to increased labile OC-loading, the exponential decay approach (Eq. 1) predicts unrealistically high near-surface rates followed by rapid cessation with depth and limited OC burial (Fig. 9). However, such prediction is inconsistent with the observations from our enrichment experiment. The exponential approach is too favorable towards near-surface processes, and molecular diffusion across the sediment-water interface will effectively counteract any SO4 2− depletion and metabolite build up over such short distances. The exponential approach does therefore not explain the commonly observed shift towards sulfate depletion, methanogenesis and OC-burial in rapidly accumulating sediments. If, on the other hand, the diagenetic capacity approach derived from (3) and (4) is considered, both mineralization processes and accumulation of degradable OC will occur at much greater depth (Fig. 9). Thus the diagenetic capacity approach facilitates depletion of e−-acceptors in the deeper sediment and explains the shift towards methanogenesis and increased OC-burial observed in sediments subjected to high OC-loading (e.g. Hall et al. 1990; Morrisey et al. 2000). However, the difference between the exponential and the diagenetic capacity approach diminishes when R max is very high or deposition rate is very low. The relatively narrow range of reported maximum SRRs and the up to 0.3 cm day−1 deposition rates reported from eutrophic coastal and fish farm sediments (Hall et al. 1990; Haddad et al. 1992) do, however, support the use of the diagenetic capacity approach. It is therefore important to determine if the maximum metabolic capacity is reached in sediments receiving high amounts of labile OM, as it would greatly alter the mechanism controlling e.g. OM-burial compared to sites with lower deposition rates.

The organic matter pool G (a) and dG/dt (b) modelled by first order kinetics (solid line) or combined first and zero order kinetics (dashed line). G and dG/dt was modelled for a loading rate of 0.4 mol m−2 day−1 assuming that 90% of added OC was labile and degradable with k = 0.1 day−1. Sediment accumulation rate = 0.04 cm day−1 and R max = 25 μmol cm−3 day−1
Environmental implications
In organic enriched sediments controlled by molecular diffusion where SR is the dominating heterotrophic pathway, the OM sedimentation threshold (TOM) can be defined as the situation where the O2 required for TH2S reoxidation is balanced by O2 diffusion from the overlying water. In this case TOM is equivalent to the upper limit for diffusive O2 uptake. If TOM is surpassed, the area-specific SR will increase above the reoxidation capacity, resulting in net TH2S accumulation and eventually TH2S release to the overlying water column. Thus TOM has the potential to serve as an operationally defined threshold for diffusion controlled sediments where SOU is confined by O2 availability and physical properties that control diffusive boundary layer thickness, e.g. water flow velocity may account for a ~5 times increase in diffusive SOU (from 400 to 1,400 mmol O2 m−2 day−1) (Jorgensen and Marais 1990; Findlay and Watling 1997). Furthermore, TOM could serve as a “safe” threshold for faunated sediments where O2-supply typically is stimulated by bioirrigation (Kristensen 2000). The recommendation that sediment metabolism should not exceed maximum SOU (Findlay and Watling 1997) implies that sedimentation of degradable organic matter can be increased to a certain threshold, above which sediment conditions deteriorate. Thus the extent by which deposited OM is degraded is a key parameter to understand deterioration of ecological quality and functioning in sediments enriched with OM. Our experiment showed that OM did accumulate at a loading rate of 375 mmol C m−2 day−1, and we propose two possible consequences: (1) the accumulated OM is not degradable and the simulated loading rate is sustainable in the sense that no TH2S will escape to the overlying water column (OM loading < TOM) or (2) the accumulated OM is degradable and continuous loading will through time lead to an increase in the volume of reactive sediment, and area specific SR will eventually reach higher levels. The latter case can only be sustainable without TH2S release if maximum diffusive O2-uptake is higher than the demand. Thus since water movement alone may account for a five-fold difference in maximum diffusive O2-uptake (Jorgensen and Marais 1990), similar loading rates may have widely different consequences in otherwise similar environments (Hargrave et al. 2008). Careful consideration is therefore needed when controlled discharges of OM is decided (e.g. location of fish farms and other point sources).
Our results show that Fe-driven TH2S-buffering is indeed an important process in OM-loaded sediments, which can decrease SOU by almost half when passive diffusion is the only transport mechanism. It therefore seems reasonable also to consider sediment Fe-content when planning the location of aquaculture facilities. However, it is important to emphasize that the TH2S-buffering capacity is limited by availability of Fe2+, which needs periodic regeneration (e.g. seasonal fallowing or resuspension events). Continuous OM enrichment will eventually lead to RFeII depletion and the TH2S-buffering capacity may never fully recover due to lack of transport processes other than diffusion. The importance of TH2S-buffering is greatly increased in bioturbated sediments where reduced FexSy-compounds are regularly exposed to oxidized conditions by sediment reworking and irrigation (Aller et al. 2001).
Whereas SR dominates at a wide range of deposition rates, there is an upper threshold for OM loading where SO4 2− availability diminishes and methanogenesis becomes the dominating heterotrophic pathway. This shift is unfortunate since it promotes increased burial of degradable OM, and a change back to SR dominance is somewhat irreversible. Even if OM loading is dramatically reduced, buried OM may sustain the methanogenesis-dominated situation as SR only occurs near the sediment surface where SO4 2− is high and upwards diffusing inhibiting metabolites (e.g. LM-DOC and H2) are low. First order kinetic modelling predicts a rapid decay of depositing OM and limited OM burial, and extreme OM-loading rates are needed to explain the shift towards methanogenesis. However, our results suggest that a maximum diagenetic capacity for degradation of surface deposited OM exists, which would greatly alter the prediction of OM-burial at high loading rates. OM-degradation in the enriched surface layer changes to zero order kinetics and both OM-burial and -mineralization with depth is greatly stimulated. Thus, by applying a maximum diagenetic capacity a more realistic prediction can be provided of the commonly observed rapid shifts towards methanogenesis and increased OM-burial in diffusion controlled sediments.
Conclusions
In this study we have demonstrated the initial effects of continuous OM enrichment at the sediment surface in diffusion controlled sediment cores. Our results showed that labile OM mineralization at the sediment-water interface was efficient, resulting in up to 26 and 2.2 μmol cm−3 day−1 C and N mineralization, respectively. OM additions stimulated microbial SR in enriched surface sediment, but TH2S only accumulated to low levels due to sulfide mitigating processes. In Fe-poor sediment spontaneous or microbial sulfide reoxidation prevented TH2S accumulation, whereas sulfide reoxidation and Fe-sulfide precipitation prevented TH2S accumulation in Fe-rich sediment. OM accumulated despite efficient mineralization at the sediment-water interface, and we speculate that the extent of OM burial is determining the environmental impact of OM enrichment. Thus the excessive TH2S accumulation frequently observed in organically enriched sediments is probably the result of prolonged periods of high OM deposition above the sustainable threshold. Hence TH2S accumulation may be markedly stimulated if microbial mineralization is governed by a maximum volume specific capacity for OM mineralization.
References
Aller RC, Aller JY (1998) The effect of biogenic irrigation intensity and solute exchange on diagenetic reaction rates in marine sediments. J Mar Res 56:905–936
Aller RC, Aller JY, Kemp PF (2001) Effects of particle and solute transport on rates and extent of remineralization in bioturbated sediments. In: Aller JY, Woodin SA, Aller RC (eds) Organism–sediment interactions. University of South Carolina Press, Columbia
Andersen FO (1996) Fate of organic carbon added as diatom cells to oxic and anoxic marine sediment microcosms. Mar Ecol Prog Ser 134(1–3):225–233
Arnosti C, Holmer M (1999) Carbohydrate dynamics and contributions to the carbon budget of an organic-rich coastal sediment. Geochim Cosmochim Acta 63(3–4):393–403
Banta GT, Holmer M, Jensen MH, Kristensen E (1999) Effects of two polychaete worms. Nereis diversicolor and Arenicola marina, on aerobic and anaerobic decomposition in a sandy marine sediment. Aquat Microb Ecol 19:189–204
Berner RA (1980) Early diagenesis—a theoretical approach. Princeton University Press, New Jersey
Bower CE, Holm-Hansen T (1980) A salicylate-hypochlorite method for determining ammonia in seawater. Can J Fish Aquat Sci 37:794–798
Callier MD, Weise AM, McKindsey CW, Desrosiers G (2006) Sedimentation rates in a suspended mussel farm (Great-Entry Lagoon, Canada): biodeposit production and dispersion. Mar Ecol Prog Ser 322:129–141
Canfield DE (1994) Factors influencing organic matter preservation in marine sediments. Chem Geol 114:315–329
Canfield DE, Jørgensen BB, Fossing H, Glud R, Gundersen J, Ramsing NB, Thamdrup B, Hansen JW, Nielsen LP, Hall POJ (1993) Pathways of organic carbon oxidation in three continental margin sediments. Mar Geol 113:27–40
Canfield DE, Thamdrup B, Kristensen E (2005) Aquatic geomicrobiology. Elsevier, San Diego
Canuel EA, Martens CS (1996) Reactivity of recently deposited organic matter: degradation of lipid compounds near the sediment-water interface. Geochim Cosmochim Acta 60:1793–1806
Christensen PB, Rysgaard S, Sloth NP, Dalsgaard T, Schwærter S (2000) Sediment mineralization, nutrient fluxes, denitrification and dissimilatory nitrate reduction to ammonium in an estuarine fjord with sea cage trout farms. Aquat Microb Ecol 21(1):73–84
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Deflaun MF, Mayer LM (1983) Relationships between bacteria and grain surfaces in intertidal sediments. Limnol Oceanogr 28:873–881
Delia CF, Steudler PA, Corwin N (1977) Determination of total nitrogen in aqueous samples using persulfate digestion. Limnol Oceanogr 22:760–764
Ding HB, Sun MY (2005) Biochemical degradation of algal fatty acids in oxic and anoxic sediment-seawater interface systems: effects of structural association and relative roles of aerobic and anaerobic bacteria. Mar Chem 93(1):1–19
Findlay RH, Watling L (1997) Prediction of benthic impact for salmon net-pens based on the balance of benthic oxygen supply and demand. Mar Ecol Prog Ser 155:147–157
Fossing H, Jorgensen BB (1989) Measurement of bacterial sulfate reduction in sediments—evaluation of a single-step chromium reduction method. Biogeochemistry 8:205–222
Fossing HV, Gallardo VA, Jorgensen BB, Huttel M, Nielsen LP, Schulz H, Canfield DE, Forster S, Glud RN, Gundersen JK, Kuver J, Ramsing NB, Teske A, Thamdrup B, Ulloa O (1995) Concentration and transport of nitrate by the mat-forming sulfur bacterium thioploca. Nature 374:713–715
Giles H, Pilditch CA, Bell DG (2006) Sedimentation from mussel (Perna canaliculus) culture in the Firth of Thames, New Zealand: impacts on sediment oxygen and nutrient fluxes. Aquaculture 261(1):125–140
Giordani G, Azzoni R, Viaroli P (2008) A rapid assessment of the sedimentary buffering capacity towards free sulphides. Hydrobiologia 611:55–66
Haddad RI, Martens CS, Farrington JW (1992) Quantifying early diagenesis of fatty-acids in a rapidly accumulating coastal marine sediment. Org Geochem 19(1–3):205–216
Hall POJ, Aller RC (1992) Rapid, small-volume, flow injection analysis for ∑CO2 and NH4 + in marine and freshwaters. Limnol Oceanogr 37:1113–1119
Hall POJ, Anderson LG, Holby O, Kollberg S, Samuelson M (1990) Chemical fluxes and mass balances in a marine fish farm. I. Carbon. Mar Ecol Prog Ser 61:61–73
Hargrave BT, Holmer M, Newcombe CP (2008) Towards a classification of organic enrichment in marine sediments based on biogeochemical indicators. Mar Pollut Bull 56(5):810–824
Heijs SK, van Gemerden H (2000) Microbiological and environmental variables involved in the sulfide buffering capacity along a eutrophication gradient in a coastal lagoon (Bassin d’Arcachon, France). Hydrobiologia 437(1–3):121–131
Heijs SK, Jonkers HM, van Gemerden H, Schaub BEM, Stal LJ (1999) The buffering capacity towards free sulphide in sediments of a coastal lagoon (Bassin d’Arcachon, France)—the relative importance of chemical and biological processes. Estuar Coast Shelf Sci 49(1):21–35
Heijs SK, Azzoni R, Giordani G, Jonkers HM, Nizzoli D, Viaroli P, van Gemerden H (2000) Sulfide-induced release of phosphate from sediments of coastal lagoons and the possible relation to the disappearance of Ruppia sp. Aquat Microb Sci 23:85–95
Heilskov AC, Alperin M, Holmer M (2006) Benthic fauna bio-irrigation effects on nutrient regeneration in fish farm sediments. J Exp Mar Biol Ecol 339:204–225
Hoehler TM, Alperin MJ, Albert DB, Martens CS (1998) Thermodynamic control on hydrogen concentrations in anoxic sediments. Geochim Cosmochim Acta 62(10):1745–1756
Holmer M, Frederiksen MS (2007) Stimulation of sulfate reduction rates in Mediterranean fish farm sediments inhabited by the seagrass Posidonia oceanica. Biogeochemistry 85(2):169–184
Holmer M, Kristensen E (1992) Impact of marine fish cage farming on metabolism and sulfate reduction of underlying sediments. Mar Ecol Prog Ser 80:191–201
Holmer M, Kristensen E (1994a) Organic-matter mineralization in an organic-rich sediment—experimental stimulation of sulfate reduction by fish food pellets. FEMS Microbiol Ecol 14(1):33–44
Holmer M, Kristensen E (1994b) Coexistence of sulfate reduction and methane production in an organic-rich sediment. Mar Ecol Prog Ser 107:177–184
Holmer M, Kristensen E (1996) Seasonality of sulfate reduction and pore water solutes in a marine fish farm sediment: the importance of temperature and sedimentary organic matter. Biogeochemistry 32(1):15–39
Holmer M, Duarte CM, Heilskov A, Olesen B, Terrados J (2003) Biogeochemical conditions in sediments enriched by organic matter from net-pen fish farms in the Balinao area, Phillipines. Mar Pollut Bull 46:1470–1479
Holmer M, Marba N, Diaz-Almela E, Duarte CM, Tsapakis M, Danovaro R (2007) Sedimentation of organic matter from fish farms in oligotrophic Mediterranean assessed through bulk and stable isotope (δ13C and δ15N) analyses. Aquaculture 262(2–4):268–280
Jorgensen BB, Marais DJD (1990) The diffusive boundary-layer of sediments—oxygen microgradients over a microbial mat. Limnol Oceanogr 35:1343–1355
Karle IM, Hall POJ, Dahllof I (2007) Biogeochemical response of an intact coastal sediment to organic matter input: a multivariate approach. Mar Ecol Prog Ser 342:15–25
Kristensen E (2000) Organic matter diagenesis at the oxic/anoxic interface in coastal marine sediments, with emphasis on the role of burrowing animals. Hydrobiologia 426:1–24
Kristensen E, Hansen K (1995) Decay of plant detritus in organic-poor marine sediment: production rates and stoichiometry of dissolved C and N compounds. J Mar Res 53:675–702
Kristensen E, Holmer M (2001) Decomposition of plant materials in marine sediment exposed to different electron acceptors (O2, NO3 −, and SO4 2−), with emphasis on substrate origin, degradation kinetics, and the role of sulfate reduction. Geochim Cosmochim Acta 65:419–433
Kristensen E, Ahmed SI, Devol AH (1995) Aerobic and anaerobic decomposition of organic matter in marine sediment: which is fastest? Limnol Oceanogr 40:1430–1437
Kristiansen KD, Kristensen E, Jensen MH (2002) The influence of water column hypoxia on the behaviour of manganese and iron in sandy coastal marine sediment. Estuar Coast Shelf Sci 55:645–654
Kunihiro T, Miyazaki T, Uramoto Y, Kinoshita K, Inoue A, Tamaki S, Hama D, Tsutsumi H, Ohwada K (2008) The succession of microbial community in the organic rich fish-farm sediment during bioremediation by introducing artificially mass-cultured colonies of a small polychaete, Capitella sp I. Mar Pollut Bull 57:68–77
Kutti T, Ervik A, Høisæter T (2008) Effects of organic effluents form a salmon farm on a fjord system. III. Linking deposition rates of organic matter and benthic productivity. Aquaculture 282:47–53
Lovley DR, Phillips EJP (1987) Rapid assay for microbially reducible ferric iron in aquatic sediments. Appl Environ Microbiol 53:1536–1540
Macleod CK, Crawford CM, Moltschaniwskyj NA (2004) Assesment of long term change in sediment condition after organic enrichment: defining recovery. Mar Pollut Bull 49:79–88
Mascaro O, Valdemarsen T, Holmer M, Perez M, Romero J (2009) Experimental manipulation of sediment organic content and water column aeration reduces Zostera marina (eelgrass) growth and survival. O. Mascaró. J Exp Mar Biol Ecol 373:26–34
Morrisey DJ, Gibbs MM, Pickmere SE, Cole RG (2000) Predicting impacts and recovery of marine-farm sites in Stewart Island, New Zealand, from the Findlay-Watling model. Aquaculture 185:257–271
Nickell LA, Black KD, Hughes DJ, Overnell J, Brand T, Nickell TD, Breuer E, Harvey SM (2003) Bioturbation, sediment fluxes and benthic community structure around a salmon cage farm in Loch Ceran, Scotland. J Exp Mar Ecol Biol 285–286:221–233
Nunn BL, Norbeck A, Keil RG (2003) Hydrolysis patterns and the production of peptide intermediates during protein degradation in marine systems. Mar Chem 83(1–2):59–73
Papaspyrou S, Thessalou-Legaki M, Kristensen E (2004) Impact of Pestarella tyrrhena on benthic metabolism in sediment microcosms enriched with seagrass and macroalgal detritus. Mar Ecol Prog Ser 281:165–179
Patel AB, Fukami K, Nishijima T (2001) Extracelluar proteolytic activity in the surface sediment of a eutrophic inlet. Microb Environ 16(1):25–35
Porrello S, Tomassetti P, Manzueto L, Fionia MG, Persia E, Mercatali I, Stipa P (2005) The influence of marine cages on the sediment chemistry in the Western Mediterranean Sea. Aquaculture 149:145–158
Preisler A, de Beer D, Lichtschlag A, Lavik G, Boetius A, Jorgensen BB (2007) Biological and chemical sulfide oxidation in a Beggiatoa inhabited marine sediment. ISME J 1(4):341–353
Raiswell R, Canfield DE (1998) Sources of iron for pyrite formation in marine sediments. Am J Sci 298(3):219–245
Rickard D, Morse JW (2005) Acid volatile sulfide (AVS). Mar Chem 97(3–4):141–197
Sakami T, Yokoyama H, Ishihi Y (2005) Microbial hydrolytic enzyme activity in the sediments of a net cage aquaculture area. Fish Sci 71(2):271–278
Schippers A, Jorgensen BB (2002) Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments. Geochim Cosmochim Acta 66(1):85–92
Schmidt JL, Deming JW, Jumars PA, Keil RG (1998) Constancy of bacterial abundance in surficial marine sediments. Limnol Oceanogr 43:976–982
Sloth NP, Blackburn H, Hansen LS, Risgaard-Petersen N, Lomstein BA (1995) Nitrogen cycling in sediments with different organic loading. Mar Ecol Prog Ser 116:163–170
Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42:779–781
Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments. In: Schink B (ed) Advances in Microbial Ecology, vol 16. Kluver, New York, pp 41–84
Vaalgamaa S (2004) The effect of urbanisation on Laajalahti Bay, Helsinki City, as reflected by sediment geochemistry. Mar Pollut Bull 48(7–8):650–662
Valdemarsen T, Kristensen E (2005) Diffusion scale dependent change in anaerobic carbon and nitrogen mineralization: true effect or experimental artifact? J Mar Res 63:645–669
Westrich JT, Berner RA (1984) The role of sedimentary organic matter in bacterial sulfate reduction: the G-model tested. Limnol Oceanogr 29:236–249
Acknowledgments
A special thanks to Birthe Christensen and Maria Jensen for assistance in the laboratory. This work was funded by EU-project Thresholds, contract no. 003933.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Valdemarsen, T., Kristensen, E. & Holmer, M. Metabolic threshold and sulfide-buffering in diffusion controlled marine sediments impacted by continuous organic enrichment. Biogeochemistry 95, 335–353 (2009). https://doi.org/10.1007/s10533-009-9340-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-009-9340-x