
Abstract
The muddy sediments of the central South Yellow Sea (SYS) are not only an important depocenter, but also an important area for early diagenesis. A systematic study of the interlinked sulfur (S), iron (Fe), and phosphorus (P) in sediments of the sea could provide important information on the diagenetic cycle of these elements; however, their interactions have not been well documented. In this study, an in situ, high-resolution diffusive gradient in thin films (DGT) technique, together with solid-phase speciation was used to reveal the diagenesis of S, Fe, and P in muddy sediments, and to quantify the benthic flux of dissolved inorganic phosphate (DIP). Solid-phase and porewater chemistry indicate that ferruginous conditions prevail in the sediments, dissimilatory iron reduction (DIR) dominated Fe reduction, and the sulfate reduction rate is generally low due to the low lability of sedimentary organic carbon. Two-dimensional porewater distributions provide direct evidence of high spatial heterogeneity of sulfide and DIP on a microscale, and spatial coexistence of DIR and sulfate reduction. A positive linear correlation (R2 > 0.75) between porewater Fe and DIP measured by the DGT over the depth interval of rapid increase in DGT-labile Fe suggests that DIP release is primarily driven by DIR. Overall, the muddy sediments are a weak source of DIP, with an average benthic flux of 0.422 ± 1.004 × 10−3 mmol/m2/day, which accounts for only 0.02−0.04% of DIP required for the primary production of the SYS, and thus has only minor impact on P budget in the sea.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In marine sediments, microbially mediated mineralization of organic carbon (OC) is the driving force of early diagenesis (Berner 1980), which governs the cycling of almost all redox-sensitive elements. Thermodynamically, microbially mediated OC mineralization preferentially utilizes the electron acceptor with which the reaction yields the largest Gibbs free energy change (ΔrG). When the electron acceptor becomes depleted, the electron acceptors with the next highest ΔrG are utilized sequentially (Burdige 2006; Froelich et al. 1979). Typically, oxygen is firstly used for OC mineralization, followed by nitrate, manganese oxides, iron (Fe) oxides, and sulfate, with increasing depth of the sediments. The rates and relative contributions of the individual mineralization pathways vary widely, depending on specific depositional environments. Due to its high availability, sulfate is usually the dominant electron acceptor for OC mineralization in OC-rich sediments of marginal seas (Jørgensen 1982), despite the low ΔrG of this pathway. Dissolved sulfide produced by sulfate reduction readily reacts with reactive iron oxides, forming various Fe sulfides, among which pyrite (FeS2) is the most dominant form of reduced Fe and sulfur (S) permanently buried (Berner 1985). In such sulfidic sediment conditions, dissolved inorganic phosphorus (DIP) released from the regeneration of organic P, as well as mobilization of Fe oxide-bound P due to reductive dissolution of Fe oxides, can accumulate in porewater to high concentrations, to diffuse upward to the overlying water (Lehtoranta et al. 2009) due to low sorption capacity of Fe sulfides for DIP (Bebie et al. 1998).
In sediments rich in reactive Fe oxides but poor in labile OC, Fe oxides also serve as an important electron acceptor coupled to OC mineralization (i.e., dissimilatory iron reduction, DIR) (Thamdrup 2000). Under this circumstance, sulfate reduction is largely inhibited, the majority of Fe2+ generated by the DIR is not precipitated as Fe sulfides, but diffuses upward and then reoxidized to Fe (hydr)oxides near the sediment surface. The accumulation of the newly formed Fe (hydr)oxides can serve as a “geochemical barrier” for the upward diffusing DIP, and also as an important sink for DIP in the overlying water (Lehtoranta et al. 2009; Rozan et al. 2002). Thus, the diagenetic cycle and fates of C–S–Fe–P in marine sediments are intricately interwoven and are regulated by different pathways of OC mineralization. Therefore, simultaneous characterization of porewater sulfide, Fe, and DIP, together with detailed speciation of solid-phase S, Fe, and P can provide insightful information on diagenetic interactions of C–S–Fe–P and the factors governing the benthic fluxes of DIP.
Since sulfide, Fe, and DIP concentrations in porewater of marine sediments can display steep gradients, on centimeter or even millimeter scales, the low resolution (larger than 1−2 cm) of conventional porewater extraction techniques is not usually able to capture small-scale signals. More importantly, conventional sampling suffers from potential oxidation of highly redox-sensitive components such as sulfide, Fe2+, and Mn2+, which is a big challenge for reliable measurements of their concentrations. In addition, without in situ preconcentration, the conventional sampling techniques do not allow the detection of some redox-sensitive components of low concentrations. The application of in situ, high-resolution diffusion gradients in thin films (DGT) technique can effectively overcome these shortcomings and allows more detailed depiction of porewater profiles on a millimeter scale (Davison and Zhang 1994; Ding et al. 2016; Ma et al. 2019; Meng et al. 2019).
The semi-enclosed South Yellow Sea (SYS) is an important channel for the transport of land-derived materials from East Asia to the western Pacific Ocean. Sediments in this sea are important locations for OC deposition and mineralization, and thus play a key role in the budget of carbon and nutrients of the water (Qi et al. 2021; Tao et al. 2016). In addition, the complex hydrodynamic conditions of the SYS, particularly the cold water mass, exert an important influence on the depositional and diagenetic regimes of the sea (Ma et al. 2018; Ren et al. 2022; Yang et al. 2003; Zhou et al. 2014), and thus on the cycle and fates of biogeochemically important elements in the sediments. Porewater chemistry of nutrients (N, P, Si) and solid-phase chemistry of S, Fe, and P in the SYS sediments have been extensively studied (Liu et al. 2003; Wu et al. 2019; Zhou et al. 2022). However, the diagenetic interactions of S–Fe–P in the sediments have not been well documented, and a mechanistic understanding of P mobilization is still lacking. Furthermore, in all previous studies in the SYS, only low-resolution sampling was used to characterize porewater profiles of the sediments, which may miss some millimeter-scale details indicating the interactions of S–Fe–P (Zhou et al. 2022). In this study, we combined the in situ DGT technique with solid-phase chemistry to reveal S–Fe–P diagenesis, the mechanism of P mobilization, and benthic fluxes of P in the muddy area of the central SYS.

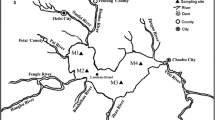
Sampling sites and circulation currents in the South Yellow Sea. KC Kuroshio Current, YSWC Yellow Sea warm current, LCC Liaodong coastal current, BCC Bohai coastal current, JCC Jiangsu coastal current, YSCC Yellow Sea coastal current. Oblique line area indicates muddy sediment in the central South Yellow Sea
Sampling and analytical methods
Sediment sampling, porewater collection, and total organic carbon analysis
Sediments were collected at five sites (A3, A4, B3, B4, and C4) of the muddy area of the central SYS from 18 April to 5 May 2021 (Fig. 1). Upon retrieval by a box corer, sediments were subcored by two polyvinyl chloride (PVC) tubes by vertical insertion into the box corer. The subcores were immediately sealed, leaving no headspace to avoid disturbance of the surface layer, and then stored at 4 °C. After being transported to the laboratory, one core from each site was sliced at 2 cm intervals in an N2 atmosphere. After sectioning, all subsamples were sealed in zip-lock plastic bags and frozen at −20 °C for later analyses of solid-phase chemistry. The other core was allowed to acclimatize at ambient temperature for 5 d after the addition of 500 mL of bottom water collected at the sampling site, then the DGT devices were inserted into the core to collect porewater sulfide, Fe, and DIP.
For analysis of total organic carbon (TOC), preweighed subsamples (about 0.5 g) were leached with 1 M HCl for 24 h to remove carbonates. After washing three times with deionized water, the residues were freeze dried and ground again for TOC analysis by a Vario EA1112 Cube elemental analyzer, with relative deviations between 1% and 5%.
Extraction of solid-phase Fe
After freeze drying and grinding, sediment samples were treated with 0.5 M HCl for 1 h to extract labile Fe, which includes amorphous and poor crystalline Fe oxides (such as ferrihydrite and lepidocrocite), acid soluble iron sulfides (such as FeS, Fe3S4), and unsulfidized Fe2+ initially adsorbed on sediment surfaces (Cornell and Giovanoli 1988; Burdige and Christensen 2022). After centrifugation (4800 rpm, 15 min) and filtration (0.22 µm), total Fe and Fe2+ were immediately determined by ferrozine colorimetry (Stookey 1970). For the determination of total Fe, hydroxylamine was used to reduce Fe3+ to Fe2+, while hydroxylamine was omitted in the determination of Fe2+. Labile Fe(III) was estimated by the difference between the total Fe and Fe2+. The extracted total Fe, Fe3+, and Fe2+ were termed LFe, LFe(III), and LFe(II), respectively. Contents of LFe(III) and LFe(II) are expressed in µmol per gram dry sediment weight (µmol/g). Contents of solid-phase sulfide and phosphorus in the following subsections are also expressed in µmol/g.
Analyses of solid-phase sulfides
Three solid-phase sulfur forms were qualified by sequential extraction: acid volatile sulfide (AVS), elemental sulfur (S0), and pyrite-S (Spy). Frozen sediments were directly used for extraction of AVS with a mixture of 6 M HCl and 1 mL ascorbic acid (0.1 M) in a sealed reactor under an N2 atmosphere (Burton et al. 2008). Ascorbic acid was used to inhibit the oxidation of extracted sulfide by concomitantly extracted Fe3+ (Burton et al. 2008). Evolved H2S gas was precipitated as ZnS by alkaline ZnAc solution in a separate vial mounted in the reactor. After 24 h, trapped sulfide content was immediately determined by the methylene blue method (Cline 1969).
Residual sediments after AVS extraction were washed twice with deoxygenated deionized water and then used to extract S0 with acetone under an N2 atmosphere (Kallmeyer 2004). After centrifugation and filtration, separated acetone extracts were left in the fume hood for evaporation of acetone to near dryness. Extracted S0 was reduced to H2S by an acid chromium(II) reductant and then precipitated as ZnS following the procedure of AVS extraction. Residual sediments after S0 extraction were washed twice with deoxygenated deionized water, and then extracted with acid chromium(II) reductant to reduce Spy to H2S and precipitated as ZnS following the procedure of AVS extraction (Burton et al. 2008; Zhu et al. 2013). The method for measurements of extracted S0 and Spy was the same as that for the AVS determination. Total reduced inorganic sulfide (TRIS) is defined as the sum of AVS, S0, and Spy (i.e., TRIS = AVS + S0 + Spy).
Analyses of solid-phase P
A three-step sequential extraction was used for solid-phase P speciation (Zhang et al. 2010): (1) exchange and loosely adsorbed P (Ex–P) was extracted with 1 M MgCl2 (pH 8); (2) Fe bound P (Fe–P) was extracted with a mixture of 0.11 M NaHCO3 and 0.11 M Na2S2O4 (pH 7); (3) authigenic P (Au–P) was extracted with 1 M NaAc–HAc mixture (pH 4). Note that sediment residues after the previous extraction step were rinsed with 1 M MgCl2 solution to purge remaining dissolved P before we moved on to the next extraction step. Extracted P was determined by the molybdenum blue method. Also note that prior to the determination of Fe–P, the extracts were exposed to air for more than 20 h to ensure complete oxidization of residual Na2S2O4. The sum of Ex–P, Fe–P, and Au–P is defined as total reactive inorganic P (i.e., TRIP = Ex−P + Fe−P + Au−P).
Application of the DGT technique
The DGT probe is a device for in situ, high-resolution passive porewater sampling based on the Fick’s law, which consists of an inert diffusive layer and a binding gel for fixation of given porewater components (Davison and Zhang 1994). The DGT devices used in this study were purchased from Nanjing Zhigan Environmental Technology Co. Ltd. When the DGT device is inserted into sediment, target components are fixed on the binding gel after diffusion through the diffusive layer of a certain thickness. The accumulation on the binding gel represents the time-averaged contents during the sampling period. In this study, a zirconia (ZrO) probe was used as the binding gel for DIP collection, and a composite ZrO-Chelex-100-AgI (ZrO−CA) probe was used as the binding gel for sulfide and Fe collection (Wang et al. 2019). Prior to employment, the DGT devices were immersed in 0.03 M NaNO3 solution and purged with N2 for 16 h for deoxygenation. Upon retrieval from sediment, the DGT devices were immediately disassembled in the dark for analyses of accumulated sulfide, Fe, and DIP.
High-resolution two-dimensional distributions of sulfide and DIP accumulated on the binding gel were determined. For sulfide, Ag2S accumulated on the binding gel was directly scanned using a flat-bed scanner (dpi 600), and resultant images were converted to grayscale density using a computer-imaging densitometry technique. Sulfide content (x) per unit area (µg/cm2) was calculated according to the corrected grayscale standard curve (Eq. 1). DIP accumulated on the ZrO binding gel was colored by the molybdenum blue method, and the colors were converted to grayscale for the quantification of DIP content (µg/cm2) with the grayscale standard curve (Eq. 2).
where x is the accumulated amount per unit area on the binding gel (µg/cm2) and y is the grayscale. The total accumulation of sulfide and DIP on the binding gel M (µg) is the product of x and area A (cm2) of the binding gel (M = xA).
After scanning, the composite ZrO–CA binding gels were cut at 3 mm intervals along the depth. The cut pieces were placed in centrifuge tubes containing 1.0 M HNO3 for Fe extraction (24 h). Extracted Fe was determined with an Epoch microporous plate spectrophotometer. Total Fe accumulated on the binding gel was calculated with Eq. (3).
where Ce is the Fe concentration in the eluates, Ve and Vg are the volume of eluates and gels, respectively, and fe is the elution efficiency of Fe (0.88). The time-averaged concentrations of P (CDGT–P) and Fe (CDGT–Fe) are calculated by Eq. (4).
where M (µg) is the total cumulative amount of DIP and Fe in the binding gels, Δg (cm) is the thickness of the diffusion layer, D (cm2/s) is the diffusion coefficient of Fe in the DGT diffusion layer, A (cm2) is the area of the binding gel, and t (s) is sampling time. Note that DGT measures only DGT-labile species and will exclude kinetically inert organic species and large colloids (Davison and Zhang 1994). Therefore, it is expected that the measured concentrations of sulfide, Fe, and DIP are somewhat lower than the concentrations of operationally defined dissolved components in the porewater. Because of the difference, we denoted hereafter the DGT-labile concentration as CDGT (CDGT–S, CDGT–Fe, CDGT–P). In addition, we assumed that all DGT-labile Fe was Fe2+, considering that Fe(III) hydroxides are almost insoluble in the pH condition of seawater.
Calculation of apparent benthic fluxes of DIP
The CDGT–P gradient can be used to estimate apparent benthic flux of DIP according to Fick’s first law (Eq. 5) (Ma et al. 2019),
where J is apparent benthic flux, (dCDGT/dz)0 is solute gradient from the sediment–water interface to depth z (cm), φ is the average porosity over the depth interval. Ds is H2PO4‒ diffusion coefficient in sediments, estimated from Ds = Dw/(1 ‒ lnφ2) (Boudreau 1997), where DW is H2PO4− diffusion coefficient in seawater (8.46 × 10−6 cm2/s) (Li and Gregory 1974). A positive value of J indicates upward diffusion from the sediment–water interface, and a negative value downward diffusion. The high-resolution CDGT‒P profiles allow more accurate determination of the gradient and flux than the profiles from the conventional porewater sampling. Due to its low resolution, the conventional porewater sampling could only roughly depict the porewater profiles, which may give rise to a large uncertainty in determination of DIP gradients and flux.
Results
TOC contents and speciation of S and reactive Fe
Contents of TOC (wt%) at A3, A4, B3, B4, and C4 were in the ranges of 1.03–1.47%, 1.04–1.32%, 0.75–0.89%, 0.85–1.16%, and 0.52–0.77%, respectively (Fig. 2). The contents displayed either a slight decrease or no decrease with depth, except a rapid decrease at B4 over the surficial 3 cm.

Vertical profiles of TOC contents
In general, LFe(II) contents increased rapidly with increasing depth over the upper 2−4 cm of the cores (except B4) (Fig. 3), whereas LFe(III) contents decreased with depth, indicating its reductive consumption and corresponding accumulation of LFe(II). LFe(III) contents at C4 were significantly higher than those at the other sites, which may suggest that sediments at this site have been superimposed by reactive iron-rich sediments delivered from the Yangtze River (Zhu et al. 2012). Below the depth of 8−10 cm, both LFe(III) and LFe(II) contents had no significant down-core changes, which indicates no further reduction of LFe(III).

Vertical profiles of labile Fe(III) (LFe(III) and Fe(II) (LFe(II))
AVS and S0 contents at five sites were extremely low, with the sum of the two in the range of 0−1.2 µmol/g (Fig. 4). Spy contents at each site displayed a down-core increase, but varied in a wide range among the five sites, with the maximum value only 5.41 µmol/g at B4 and 21.9−24.8 µmol/g at A3 and B3, but up to 117 µmol/g at A4. The maximum TRIS contents (118 µmol/g) at A4 was much higher than those at A3, B3, B4 and C4 (5.5−25 µmol/g), which are at the lower end for sediments of global continental margins (Goldhaber 2004; Rickard and Morse 2005). Regardless of its contents, TRIS was always dominated by Spy (85.4−99.7%).

Vertical profiles of acid volatile sulfide (AVS), elemental sulfur (S0), and pyrite–S (Spy)
Speciation of inorganic phosphorus
Ex–P contents at five sites were in a narrow range of 0.47−1.25 µmol/g (Fig. 5), and displayed no clear depth trend. This P pool accounts for 7.4−21.7% (14.6 ± 3.6%) of the TRIP. Au−P contents at the sites were 2.10−6.56 µmol/g, with the contents at C4 (3.40−6.56 µmol/g) much higher than at the other sites, and this pool accounted for 32.5−82.9% (57.9 ± 11.4%) of the TRIP and thus was the major P pool. Fe–P contents were in the range of 0.65−3.82 µmol/g, accounting for 9.7−53.3% (27.8 ± 10.3%) of the TRIP. In general, Fe−P contents displayed a significant down-core decrease over the upper 8 cm depth, corresponding to the down-core decrease in LFe(III) (Fig. 3), which implies that Fe−P release might be largely from reductive dissolution of Fe oxides.

Vertical profiles of adsorbed inorganic and exchangeable organic P (Ex−P), Fe-bound inorganic P (Fe−P), and authigenic phosphate (Au−P)
C DGT–S, C DGT–Fe, and C DGT–P concentrations and apparent benthic fluxes of DIP
As shown in Fig. 6, the 2-D distributions of CDGT–S and CDGT–P in porewater at the five sites were obviously spatially heterogeneous. CDGT–S concentrations at the five sites were constantly very low. For example, CDGT–S at B3 was lower than the detection limit of the DGT device, and was only sporadically detectable at A4 and B4, with CDGT–S concentrations less than 0.27 µmol/L. CDGT–S at A3 and C4 occurred mainly below 4−6 cm depth, at a concentration of less than 2.11 µmol/L. There were obvious differences in concentrations and spatial distributions of CDGT–P among the five sites. At A4, CDGT–P concentration in bottom water immediately above the sediment was relatively high and uniform, while the bottom-water concentrations at the other sites were low.
The vertical profiles of CDGT–S and CDGT–P obtained from lateral averaging of their 2-D distributions can be used for comparison with the vertical profiles of CDGT–Fe (Fig. 7) to reveal the interplay of the three components. CDGT–Fe concentrations were close to 0 in the uppermost layer (2−3 cm) (except for a peak value at the sediment-water interface at A4), below which all the concentrations increased rapidly with varying gradients. For example, CDGT–Fe concentrations at A3, B4, and C4 reached their maxima within the 2 cm depth interval; at A4 and B3, however, the concentrations increased to their maxima within the 4 cm depth interval. Generally, there was a rapid increase in CDGT–P concentrations in tandem with the rapid increase in CDGT–Fe concentrations (Fig. 7). A rapid increase in CDGT–S generally started below the depths where the CDGT–Fe reached its maximum (except for B3), but the two components coexisted within a certain depth interval (Fig. 7).
Figure 8 shows the apparent benthic fluxes of DIP at five sites. The fluxes were positive at A3, B3, B4, and C4, but negative at A4. The average flux of the five sites was 0.422 ± 1.004 × 10−3 mmol/m2/day.

Two-dimensional distributions of porewater concentrations of CDGT–S and CDGT–P

Vertical profiles of porewater concentrations of CDGT–S, CDGT–P, and CDGT–Fe

Benthic fluxes of DIP across sediment-water interfaces
Discussion
Diagenetic geochemistry of S and Fe
The profiles of porewater chemistry in marine sediments are a combined result of multiple diagenetic processes and usually display high spatial heterogeneity (Berg et al. 2003). High-resolution CDGT–S and CDGT–Fe profiles can provide more detailed information on S and Fe diagenetic cycles than the porewater profiles from the conventional sampling, though the DGT-labile concentrations do not represent the total porewater concentrations (Ma et al. 2017, 2019). A rapid increase in CDGT–Fe concentrations below 2−3 cm depth (Fig. 7) indicates a rapid reductive dissolution of Fe oxides, which is confirmed by a concomitant decrease in LFe(III) content and a corresponding increase in LFe(II) content over the same depth interval (Fig. 3). The reduced Fe occurs mainly as pyrite (FeS2) (Fig. 4) and acid-extractable solid-phase LFe(II) (Fig. 3), leaving only a minor fraction in the porewater. Very low AVS contents at all sites suggest that the contents of metastable Fe sulfides (FeS, Fe3S4) are negligible. Thus, we can assume that almost all the LFe(II) remains unsulfidized. In addition, Fepy (Fepy = 0.5 Spy) content accounts for only 14%, on average, of the total Fe(II) (i.e., Fe(II)T = LFe(II) + Fepy), which means that the majority of Fe(II)T occurs as HCl-extractable unsulfidized LFe(II).
In labile OC-rich sediments, microbially mediated oxidation of OC can result in a quick depletion of oxygen, nitrate, manganese oxides, and Fe oxides, such that sulfate is usually left as the solely important electron acceptor for OC oxidation. This usually results in highly sulfidic porewater conditions (Jørgensen 1977), in which Fe oxides are reduced dominantly by dissolved sulfide (i.e., abiotic reduction) and, as a result, Fe(II) occurs mainly as Fe(II) sulfides. This is because abiotic reduction is thermodynamically and kinetically more favored than DIR (Koretsky et al. 2003). Furthermore, biotoxicity of abundant dissolved sulfide to Fe-reducing bacteria also inhibits the prevalence of DIR (Canfield et al. 2005). However, the abundant presence of unsulfidized LFe(II) and overall low TRIS contents in the present study strongly suggest that DIR is prevailing in the sediments, and the sulfate reduction rate is generally low, such that the majority of dissolved sulfide is efficiently buffered by reaction with Fe oxides. Below the depth of its maximum, the CDGT–Fe concentrations were at least one order of magnitude higher than those of the CDGT–S (Fig. 7), which further supports the ferruginous porewater environment. This diagenetic regime suggests that OC in the sediments is overall low in lability, and thus its degradation could not maintain high rates of sulfate reduction, but favor DIR (Thamdrup 2000). This speculation is supported by only a slight down-core decrease in TOC contents at the five sites (Fig. 2) and also by our diagenetic modeling (Ren et al. 2022), which suggests that labile OC pool (with degradation rate constants of 0.6−1.65 year−1) in the muddy sediments of the central SYS is small (< 0.2 wt%), and quickly becomes depleted within the upmost 4 cm, while the remaining OC pool is barely degradable over the upper 40 cm depth interval.
At a certain depth (except at B3), a rapid increase in CDGT–S concentrations (Fig. 7) indicates a significant enhancement in sulfate reduction rate. These depths are greater than those of the rapid increase in CDGT–Fe concentrations, which is consistent with the thermodynamic prediction that sulfate reduction occurs following DIR (Burdige 2006). However, the coexistence of dissolved sulfide and Fe2+ over a wide depth interval suggests that DIR and sulfate reduction are not completely mutually exclusive, which implies that the classic geochemical zonation of early diagenesis is only an ideal paradigm, while the real situation is actually much more complicated. It is noted that the two-dimensional distributions of CDGT–S are highly spatially heterogeneous, probably reflecting millimeter-scale hotspots of sulfate reduction. Such spatial heterogeneities could only be revealed by in situ, high-resolution sampling such as DGT, but not easily captured by the conventional porewater sampling (Ding et al. 2016; Ma et al. 2017, 2019). In addition, low-concentration sulfide and its spatial heterogeneity are undetectable with the method of the conventional porewater sampling because of the high detection limit (1 µmol/L) of the traditional methylene blue method of dissolved sulfide determination (Cline 1969), but could be characterized by the DGT technique with in situ preconcentration.
In labile OC-rich sediments, rapid sulfate reduction, and thus the formation of highly sulfidic porewater conditions, usually result in rapid depletion of either sulfate or reactive Fe oxides within the upper sediment layer. By corollary, the rapid accumulation of Spy is restricted to the shallow sediment layer (Goldhaber 2003). AVS and S0 levels (Fig. 4) and CDGT–S concentrations (Fig. 7) at five sites are generally low, and CDGT–S concentrations at B3 are even undetectable with the DGT technique, while Spy content progressively increases with depth, especially in the deep layer, which strongly suggests that sulfide is rapidly consumed by reactive Fe and is efficiently converted to FeS2. This feature is just the reflection of the low content of labile OC, together with the slow but continuous sulfate reduction, which once again confirms the overall low lability of the sedimentary OC. It is worth pointing out that the maximum CDGT–S concentration at A4 is only slightly higher than that at B3 (Fig. 7), but the maximum Spy content at the former is 4.7−21.6 times those at the other sites (Fig. 4). In contrast, CDGT–S concentrations at C4 are relatively high and uniform at depth, while Spy contents are low (< 11.4 µmol/g), which suggests that porewater and solid-phase chemistry are not necessarily coupled. This is not unexpected considering that porewater chemistry is a combined result of ongoing diagenetic processes, while solid-phase chemistry is a time-integrated result of prolonged diagenesis. Therefore, porewater and solid-phase chemistry should be combined to well understand the intensity of prolonged diagenesis and the fate of involved elements.
Coupling of Fe and P diagenesis and benthic flux of DIP
The two-dimensional distributions of CDGT–P concentrations at five sites (Fig. 6) indicate high spatial heterogeneity of DGT-labile DIP in the porewater. Compared with the upper layer, the higher CDGT–P concentrations in the deep part of the sites (except A4) can be ascribed to be a result of a net accumulation of DIP. At A4, both bottom water and porewater close to the sediment–water interface (< 2 cm) have high CDGT–P concentrations (Fig. 6). In addition, CDGT–Fe peaks (Fig. 7) and high AVS and Spy contents over the thin layer of the site (Fig. 4) indicate the concurrence of active Fe reduction and sulfate reduction. As a result, effective mobilization of Fe–P together with regeneration of organophosphorus may have resulted in high CDGT–P concentrations at the site.
As shown in Fig. 9, CDGT–Fe concentrations at five sites are well linearly correlated with CDGT–P concentrations over the depth intervals of rapid down-core increase in CDGT–Fe, which strongly suggests that DIP release is mainly driven by DIR. This is because under ferruginous conditions maintained by prevailing DIR, reductively dissolved Fe2+ and concomitantly released DIP from Fe−P are accumulated mainly in the porewater, resulting in coupling of the two components (Lehtoranta et al. 2009). Under highly sulfidic conditions, however, the majority of Fe2+ produced mainly by reaction with abundant porewater sulfide is quickly precipitated as Fe sulfides, while released DIP remains in the porewater due to low sorption affinity of Fe sulfides for DIP (Bebie et al. 1998), rendering the two components spatially decoupled (Lehtoranta et al. 2009; Rozan et al. 2002). The decrease in Fe−P from the sediment surface to a depth of 6−8 cm (Fig. 5) further confirms the notion of DIR-driven mobilization of Fe−P. It should be pointed out that OC mineralization coupled to DIR also regenerates DIP. If the chemical compositions of the sedimentary organic matter are assumed to follow the Redfield ratio of C/N/P, the resulting DIP/Fe2+ ratio would be 1/424 (Eq. 6), which is much lower than the determined CDGT–P/CDGT–Fe ratios (0.02−2.94). Thus, we can hypothesize that the relative contribution of organic matter mineralization to DIP release is very limited.

Correlations between porewater CDGT–Fe and CDGT–P
As shown in Fig. 8, the positive benthic fluxes of DIP at A3, B3, B4, and C4 suggest that these sites are the source of DIP to the overlying water. The negative flux of DIP at A4 is largely due to the high DIP concentration in the bottom water (Fig. 6), the reason for which remains unclear, but may be ascribed to efficient regeneration of organophosphorus in the water column around the site. The average of the benthic fluxes at the five site (0.422 ± 1.004 × 10−3 mmol/m2/day) is at the lower end of DIP fluxes reported in different areas of China’s marginal seas (Fig. 8, Table S1). This indicates that the muddy area of the central SYS is only a weak source of DIP to the overlying water. The low DIP flux could be attributed to the following two reasons: (1) the DIR-prevailing condition favors upward diffusion of both Fe2+ and mobilized DIP; on the other hand, the subsequent reoxidation of Fe2+ close to the sediment surface can effectively sequestrate the upward diffusing DIP, resulting in the overall low DIP fluxes; (2) even though DIR is a prevailing pathway of Fe reduction, generally low DIR rates may have resulted in low release of Fe−P and thus low DIP flux. This speculation is inferred from the comparison with our previous study in Jiaozhou Bay (Ma et al. 2019). DIP release from Jiaozhou Bay sediments is also driven mainly by DIR, but CDGT–Fe concentrations and DIP fluxes in the bay sediments are much higher than those in the present study, which may suggest lower DIR rates in the muddy area of the central SYS.
Based on the area (3.09 × 105 km2) of the muddy sediments of the central SYS (Ren et al. 2022) and the average DIP flux in the area, the annual flux of DIP of the area is calculated to be 1.82 ± 3.92 × 1010 mol. The average primary production of the whole SYS (8.08 × 105 km2) is reported to be about 515−949 mg C/m2/day (Ning et al. 1995), which is equivalent to DIP demand of 0.40−0.75 mmol/m2/day, in terms of the Redfield ratio of planktonic C/N/P. It is estimated that DIP released from the muddy sediments of the central SYS accounts for only 0.02−0.04% of the total DIP required for the primary production of the whole SYS, and thus the DIP release plays only a minor role in P budget of the sea.
Conclusions
The two-dimensional distributions of CDGT–S concentrations reveal high spatial heterogeneity of dissolved sulfide in the sediments, suggesting highly varying sulfate reduction rates on millimeter scales, which usually could not be captured by the conventional low-resolution porewater sampling. CDGT–Fe concentrations are much higher than those of CDGT–S, and the majority of Fe(II) (86%) occurs as unsulfidized LFe(II), which strongly suggests that DIR is prevailing in ferruginous porewater conditions. Despite the prevalence of DIR, sulfate reduction can coexist with DIR over a wide depth interval.
The profiles of AVS, S0, and Spy suggest slow but progressive proceeding of sulfate reduction toward the depth of the sediments, which is a manifestation of low lability of the sedimentary OC. The spatial decoupling of CDGT–S and Spy profiles in the sediments highlights the importance of the combination of porewater and solid-phase chemistry to better elucidate the fate and intensity of the diagenetic processes.
A good positive linear correlation between CDGT–Fe and CDGT–P (R2 > 0.75) over the depth intervals of rapid down-core increase in CDGT–Fe strongly suggests that DIP release is mainly driven by DIR. Overall, the muddy sediments are a weak source of DIP to the water column, with an averaged benthic flux of 0.422 ± 1.004 × 10−3 mmol/m2/day. The flux accounts for only 0.02−0.04% of DIP required for the primary production in the whole SYS, which is a minor contributor to P budget of the sea.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. Much of the data is distilled within the manuscript or supplementary material.
References
Bebie J, Schoonen MA, Fuhrmann M, Strongin DR (1998) Surface charge development on transition metal sulfides: an electrokinetic study. Geochim Cosmochim Acta 62:633–642
Berg P, Rysgaard S, Thamdrup B (2003) Dynamic modeling of early diagenesis and nutrient cycling. A case study in an artic marine sediment. Am J Sci 303:905–955
Berner RA (1980) Early Diagenesis: A Theoretical Approach. Princeton University Press, Princeton
Berner RA (1985) Sulphate reduction, organic matter decomposition and pyrite formation. Philosophical Trans Royal Soc Lond Ser Math Phys Sci 315:25–38
Boudreau BP (1997) Diagenetic Models and Their Implementation. Springer, Berlin, Berlin
Burdige DJ (2006) Geochemistry of Marine Sediments. Princeton University Press, Princeton
Burdige DJ, Christensen JP (2022) Iron biogeochemistry in sediments on the western continental shelf of the Antarctic Peninsula. Geochim Cosmochim Acta 326:288–312
Burton ED, Sullivan LA, Bush RT, Johnston SG, Keene AF (2008) A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils. Appl Geochem 23:2759–2766
Canfield DE, Kristensen E, Thamdrup B (2005) Aquatic Geomicrobiology. Elsevier, Amsterdam
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Cornell RM, Giovanoli R (1988) Acid dissolution of akaganiéite and lepidocrocite: the effect on crystal morphology. Clays Clay Miner 36:385–390
Davison W, Zhang H (1994) In situ speciation measurements of trace components in natural water using thin-film gels. Nature 367:546–548
Ding S, Wang Y, Wang D, Li YY, Gong M, Zhang C (2016) In situ, high-resolution evidence for iron-coupled mobilization of phosphorus in sediments. Sci Rep 6:1–11
Froelich P, Klinkhammer GP, Bender ML, Luedtke NA, Heath GR, Cullen D, Dauphin P, Hammond D, Hartman B, Maynard V (1979) Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochim Cosmochim Acta 43:1075–1090
Goldhaber MB (2003) Sulfur-rich sediments. In: MacKenzie FT (ed) Treatise on Geochemistry. Asmterdam, Elsevier, pp 257–288
Jørgensen BB (1977) The sulfur cycle of a coastal marine sediment (Limfjorden, Denmark) 1. Limnol Oceanogr 22:814–832
Jørgensen BB (1982) Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature 296:643–645
Kallmeyer J, Ferdelman TG, Weber A, Fossing H, Jørgensen BB (2004) A cold chromium distillation procedure for radiolabeled sulfide applied to sulfate reduction measurements. Limnol Oceanography: Methods 2:171–180
Koretsky CM, Moore CM, Lowe KL, Meile C, DiChristina TJ, Van Cappellen P (2003) Seasonal oscillation of microbial iron and sulfate reduction in saltmarsh sediments (Sapelo Island, GA, USA). Biogeochemistry 64:179–203
Lehtoranta J, Ekholm P, Pitkänen H (2009) Coastal eutrophication thresholds: a matter of sediment microbial processes. AMBIO: A Journal of the Human Environment 38:303–308
Li YH, Gregory S (1974) Diffusion of ions in sea water and in deep-sea sediments. Geochim Cosmochim Acta 38:703–714
Liu SM, Li LW, Zhang Z (2011) Inventory of nutrients in the Bohai. Cont Shelf Res 31:1790–1797
Liu SM, Zhang J, Chen SZ, Chen HT, Hong GH, Wei H, Wu QM (2003) Inventory of nutrient compounds in the Yellow Sea. Cont Shelf Res 23:1161–1174
Ma WW, Zhu MX, Yang GP, Li T (2017) In situ, high-resolution DGT measurements of dissolved sulfide, iron and phosphorus in sediments of the East China Sea: insights into phosphorus mobilization and microbial iron reduction. Mar Pollut Bull 124:400–410
Ma WW, Zhu MX, Yang GP, Li T (2018) Iron geochemistry and organic carbon preservation by iron (oxyhydr) oxides in surface sediments of the East China Sea and the south Yellow Sea. J Mar Syst 178:62–74
Ma WW, Zhu MX, Yang GP, Li WJ, Meng T, Li T (2019) Diagenesis of sulfur, iron and phosphorus in sediments of an urban bay impacted by multiple anthropogenic perturbations. Mar Pollut Bull 146:366–376
Meng T, Zhu MX, Ma WW, Gan ZX (2019) Sulfur, iron, and phosphorus geochemistry in an intertidal mudflat impacted by shellfish aquaculture. Environ Sci Pollut Res 26:6460–6471
Ning XR, Liu ZL, Shi JX (1995) Assessment of primary productivity and potential fishery production in the Bohai Sea, Yellow Sea and East China Sea. Haiyang Xuebao 17:72–84 (In Chinese with English abstract)
Qi L, Wu Y, Chen S, Wang X (2021) Evaluation of abandoned Huanghe Delta as an important carbon source for the Chinese marginal seas in recent decades. J Geophys Res 126:e2020JC017125. https://doi.org/10.1029/2020JC017125
Ren JH, Zhu MX, Wang DY, Tan TT, Li T, Zhao M, Pan DW (2022) Organic carbon mineralization pathways in the muddy sediments of the South Yellow Sea: insights from steady-state modeling of porewater. Appl Geochem 138:105237
Rickard D, Morse JW (2005) Acid volatile sulfide (AVS). Mar Chem 97:141–197
Rozan TF, Taillefert M, Trouwborst RE, Glazer BT, Ma S, Herszage J, Valdes LM, Price KS, Luther GW III (2002) Iron-sulfur‐phosphorus cycling in the sediments of a shallow coastal bay: Implications for sediment nutrient release and benthic macroalgal blooms. Limnol Oceanogr 47:1346–1354
Stookey LL (1970) Ferrozine-a new spectrophotometric reagent for iron. Anal Chem 42:779–781
Tao S, Eglinton TI, Montluçon DB, McIntyre C, Zhao M (2016) Diverse origins and pre-depositional histories of organic matter in contemporary Chinese marginal sea sediments. Geochim Cosmochim Acta 191:70–88
Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments. Adv Microb Ecol 16:41–84
Wang Y, Ding S, Ren M, Li C, Xu S, Sun Q, Xu L (2019) Enhanced DGT capability for measurements of multiple types of analytes using synergistic effects among different binding agents. Sci Total Environ 657:446–456
Wu Z, Yu Z, Song X, Wang W, Zhou P, Cao X, Yuan Y (2019) Key nitrogen biogeochemical processes in the South Yellow Sea revealed by dual stable isotopes of nitrate. Estuar Coast Shelf Sci 225:106222. https://doi.org/10.1016/j.ecss.2019.05.004
Yang SY, Jung HS, Lim DI, Li CX (2003) A review on the provenance discrimination of sediments in the Yellow Sea. Earth Sci Rev 63:93–120
Zhang JZ, Guo L, Fischer CJ (2010) Abundance and chemical speciation of phosphorus in sediments of the Mackenzie River Delta, the Chukchi Sea and the Bering Sea: importance of detrital apatite. Aquat Geochem 16:353–371
Zhou L, Liu J, Saito Y, Zhang Z, Chu H, Hu G (2014) Coastal erosion as a major sediment supplier to continental shelves: example from the abandoned Old Huanghe (Yellow River) delta. Cont Shelf Res 82:43–59
Zhou N, Zhang GL, Liu SM (2022) Nutrient exchanges at the sediment-water interface and the responses to environmental changes in the Yellow Sea and East China Sea. Mar Pollut Bull 176:113420. https://doi.org/10.1016/j.marpolbul.2022.113420
Zhu MX, Shi XN, Yang GP, Hao XC (2013) Formation and burial of pyrite and organic sulfur in mud sediments of the East China Sea inner shelf: constraints from solid-phase sulfur speciation and stable sulfur isotope. Cont Shelf Res 54:24–36
Zhu MX, Hao XC, Shi XN, Yang GP, Li T (2012) Speciation and spatial distribution of solid-phase iron in surface sediments of the East China Sea continental shelf. Appl Geochem 27:892–905
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers: 42176041 and 41776085) and the National Key Research and Development Program of China (grant number 2016YFA0601301). Data and samples were collected onboard of R/V Dongfanghong 3. All crews of the cruise were thanked for their assistance in sampling.
Author information
Authors and Affiliations
Contributions
ZX and M-XZ wrote the main manuscript text and ZX prepared Figures 1−8. W-XS, ZX, Q-QL, and Y-JH undertook field investigation. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xu, Z., Sun, WX., Li, QQ. et al. Characterization of iron, sulfur, and phosphorus diagenesis in muddy sediments of the South Yellow Sea using the diffusive gradients in thin films (DGT) technique. Aquat Sci 85, 69 (2023). https://doi.org/10.1007/s00027-023-00967-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00027-023-00967-7