
Abstract
In the eutrophic coastal ocean, quick formation of iron (Fe) sulfide is environmentally important to effectively prevent accumulation of dissolved sulfide and its detrimental effects on the benthic ecosystem. In this study, 0.5 N HCl-extractable labile Fe (LFe), acid volatile sulfide, and pyrite in the East China Sea inner shelf sediments were examined to investigate the mechanisms of quick sequestration of dissolved sulfide and potential impacts of frequent algal blooms on the capacity of quick sulfide-buffering in eutrophic coastal areas of the large-sized continental shelf subject to massive terrestrial input. The results indicate that sulfate reduction has been competitively suppressed by dissimilatory Fe reduction due to limited availability of labile organic matter. Dissolved sulfide can be quickly buffered by reaction with LFe and, therefore, is difficult to accumulate to a high level. The quick sulfide-buffering capacity has not become exhausted partly because of the formation of un-sulfidized LFe(II) via dissimilatory reduction of less reactive Fe oxides. It is expected that dissolved sulfide will not pose detrimental effects on the benthic ecosystem in the near future if the current biogeochemical/ecological state remains.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In coastal marine sediments, anaerobic metabolism is generally the most important pathway for organic matter (OM) oxidation due to quick depletion of O2 and nitrate within several millimeters below the sediment–water interface. Among the anaerobic processes, sulfate reduction usually plays the predominant role, accounting for about 50 %, on average, of total OM mineralization (Jørgensen 1982). Free sulfide produced via sulfate reduction can be removed by three types of reactions (Azzoni et al. 2005; Sell and Morse 2006): (1) oxidation by O2 predominantly through microbial process, (2) abiotic oxidation by Fe oxides (Eq. 1), (3) precipitation as Fe sulfide minerals (Eq. 2), which can convert to thermodynamically more stable pyrite (FeS2) in the presence of elemental S0 (mainly occurring as polysulfide, S 2− n , in the presence of dissolved sulfide) in suboxic and anoxic conditions (Eqs. 3, 4) (e.g., Rickard 1975; Luther 1991).
A combination of Eqs. 1 and 2 yields Eq. 5:
The removal process expressed by Eq. 5 is termed chemical buffering of free sulfide (Heijs et al. 1999; Heijs and van Gemerden 2000), which is strongly dependent on the amount of Fe(III) oxides (Giordani et al. 2008; Valdemarsen et al. 2009, 2010; Zhu et al. 2012a). Iron oxides in marine sediments have wide spectrum of mineralogy, crystallinity, morphology, and chemical composition, and therefore display highly variable reactivity towards reaction with free sulfide (Canfield 1989). Amorphous and poorly crystalline Fe oxides, for example, ferrihydrite and lepidocrocite, react very quickly with dissolved sulfide (with half-lives from hours to days); well crystalline Fe oxides, for example, goethite and hematite, react slightly more slowly (with half-lives less than 31 days), poorly reactive sheet silicate Fe(III) reacts very slowly with dissolved sulfide over geological timescales (with half-lives from 102 to 105 a) (Canfield et al. 1992; Raiswell and Canfield 1998; Poulton et al. 2004). Thus, the availability of highly reactive Fe oxides plays a critical role in quick sulfide buffering.
Sediments enriched in OM in areas of highly eutrophic, mariculture, and hypoxic waters usually have much higher sulfate reduction rate in comparison with other coastal sediments (Cooper and Brush 1991; Otero et al. 2006). Enhanced sulfide production may result in accumulation of free sulfide in the porewater and overlying bottom water if it cannot be effectively buffered by sedimentary Fe oxides (Sorokin and Zakuskina 2012). For areas with pulsed inputs of labile OM associated with seasonal eutrophication and algal blooms, quick buffering of free sulfide by highly reactive Fe oxides is particularly important for the protection against accumulation of dissolved sulfide. Otherwise enhanced free sulfide will cause detrimental effects on the benthic ecosystem.
Chemical buffering capacity and its saturation degree are important parameters for assessing health status of benthic ecosystems and its resilience upon pulsed inputs of OM to sediments (Kanaya and Kikuchi 2004; Giordani et al. 2008; Valdemarsen et al. 2009). 0.5 N HCl extractable Fe has been used as an estimate of the Fe pool most readily to buffer dissolved sulfide over short-time scales (Azzoni et al. 2005; Giordani et al. 2008; Zhu et al. 2012a). The quick sulfide-buffering capacity, β L, has been defined by Eq. 6 (Azzoni et al. 2005).
where [LFe(II) − AVS] is buffering capacity of Fe(II) that has not been sulfidized, 1.5 LFe(III) is buffering capacity of reactive Fe(III) oxides according to the stoichiometry between S2− and FeS (3:2) in Eq. 5.
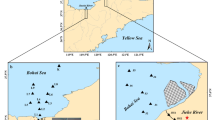
Many coastal areas of the East China Sea (ECS) have been influenced by eutrophication due to increasing mariculture and discharge of terrestrial nutrients from mainly the Yangtze River, the third largest river in the world. As a result, algal blooms have occurred, mainly around the Yangtze Estuary and Zhoushan Fishery (Fig. 1), since the 1980s with increasing frequency and intensity (Wang et al. 2004; Chai et al. 2006), and, as a result, the sea has become one of the largest low-oxygen waters in the world (Chen et al. 2007). The eutrophication and algal blooms may have enhanced OM burial and stimulated sulfate reduction at least locally and episodically. Numerous studies have been undertaken on burial and source of OM (Wang et al. 2008), diagenetic cycle of nutrients (Aller et al. 1985; Fang et al. 2007), and contents and distribution of contaminants in the sea (Lin et al. 2002a; Liu et al. 2011). Up to date, however, little attention has been paid on early diagenesis of S and Fe in the sediments (Huang and Lin 1995; Lin et al. 2000, 2002b; Zhu et al. 2012a). Yet not assessed are quick sulfide-buffering capacity of the sediment and potential impacts of eutrophication and algal blooms on the buffering capacity. To elucidate the above issues, we investigated the interactions between Fe and S and quantified quick sulfide-buffering capacity of the ECS inner shelf sediments.

Regional ocean circulation patterns and sampling stations (702, 802, and 803) in the East China Sea (ECS). JCC Jiangsu Coastal Current, KC Kuroshio Current, TWC Taiwan Warm Current, ZFCC Zhejiang-Fujian Coastal Current, YDW Yangtze Diluted Water. The location of Zhoushan Fishery is indicated by the dashed-line rectangle
Materials and methods
Background of study area
The ECS is one of the largest shelf seas in the world. It receives annually 5 × 108 t of terrestrial particulates (Liu et al. 2007; Wang et al. 2008) including 2–5 × 106 t of particulate OM (Wu et al. 2007). Driven by the Zhejiang-Fujian Coastal Current (ZFCC) (Fig. 1), southward dispersal of the Yangtze sediments occurs mainly along the inner shelf, developing an elongated mud wedge from the Yangtze mouth into the Taiwan Strait (for details, see Zhu et al. 2012b). Consistent with this dispersal system, sedimentation rates in the ECS decrease rapidly from the Yangtze Estuary southward along the inner shelf and eastward offshore (DeMaster et al. 1985; Huh and Su 1999). Sediment in the inner shelf sea is not only an important sink for terrestrial Fe and OM, but also an important site for OM mineralization. Sulfate reduction is believed to be an important anaerobic pathway for OM mineralization in the sediments (Aller et al. 1985; Huang and Lin 1995; Lin et al. 2000), while dissimilatory Fe reduction also plays a role, according to previous biogeochemical and microbiological studies (Bao 1989; Lü et al. 2011; Zhu et al. 2012b). Poorly reactive sheet silicate Fe pool is characteristically high in the surface sediments due probably to reverse weathering. Magnetite, albeit not one of the main Fe components, has much higher content than in many other marine sediments around the world, probably indicative of a product of dissimilatory Fe reduction (Zhu et al. 2012b). Spatial distributions of main solid Fe forms in the shelf sediments are strongly controlled by terrestrial inputs from the Yangtze River and the spatial distribution of clay fraction (see Zhu et al. 2012b, for details).
Sampling methods
Three sites, 702 (122.45°E, 29.21°N), 802 (121.44°E, 27.75°N), and 803 (121.65°E, 27.64°N), influenced by anthropogenic perturbations to variable extents were selected for this study. Water depths at 702, 802, and 803 are 40, 29, and 45 m, respectively, and sedimentation rates, estimated on basis of Huh and Su (1999), are 1.1, 0.65, and 0.5 cm/a, respectively. Site 702 is within direct impacts of frequent algal blooms around the Zhoushan Fishery (Fig. 1), and the impacts on the sediments are estimated to reach a depth of ~30 cm on basis of a steady-state sedimentation rate at this site, while both sites 802 and 803 are beyond the reach of the direct impacts.
Sediment cores were collected during the late spring cruise (late April to mid May) of 2009 using a box-corer. Immediately upon retrieval of the box-corer onboard, PVC tubes (inner diameter 8 cm) were vertically inserted for tube coring. The two ends of every core were covered and clasped with fitted tops leaving no headspace, and then the cores were frozen at −20 °C. All the cores were covered with ice to keep them cool during the transportation to our home laboratory (within 2 h) and then stored at −20 °C in the laboratory until further handling within 3 months.
After thawing in a N2 glove box at room temperature, the cores were extruded and sectioned in 2–3 cm interval and subsamples were immediately used for various analyses below.
Analytical methods
Wet sediment subsamples of known weight were dried at 105 °C until constant weight for determination of wet/dry weight ratios, which was used for calculation of Fe and acid volatile sulfide (AVS) contents per gram dry sediment. For organic carbon (OC) analysis, sediment subsamples of known weight (~1 g) were treated with 1 N HCl overnight and washed twice with deionized water to remove carbonates, then the samples were dried at ~60 °C for 12 h and ground to ~100 mesh for OC analysis using a PE2400II CN element analyzer, with variability between duplicates better than 8.0 %.
The most reactive amorphous and poorly crystalline Fe pool [referred to as labile Fe (LFe) hereafter] that quickly reacts with dissolved sulfide over a short term was extracted by 0.5 N HCl, following Azzoni et al. (2005) and Giordani et al. (2008). Wet sediment subsamples of known weight (~1 g) were loaded into centrifuge tubes containing 25 mL 0.5 N HCl for a 12-h extraction while shaking. After centrifuge (4,800 rpm), the resultant supernatants were filtrated (0.2 μm) for analysis of Fe. Extracted Fe was measured for ferrous Fe (Fe(II)) and ferric Fe (Fe(III)). Total LFe was measured by the ferrozine colorimetry (Hewlett-Packard 8453 UV–VIS spectrophotometer) with hydroxylamine reductant (Stookey 1970). LFe(II) was measured by the same method but without use of hydroxylamine. The content of LFe(III) was calculated by difference between LFe and LFe(II). Relative deviations between duplicates are <5.6 % for LFe and <5.7 % for LFe(II).
An attempt was made to determine porewater sulfide (by centrifuging in N2) in the subsurface (4–5 cm), mid (20 cm), and bottom (35–37 cm) layers of the three cores; however, sulfide concentrations were all undetectable (<1 μM). Thus, no further efforts were devoted to the determination in other layers. AVS in all the subsamples was released by 6 N HCl, evolving H2S gas was collected using the cool diffusion method (Hsieh and Shieh 1997; Burton et al. 2008). Twenty milliliters of HCl (final concentration ~4 N) together with 1 mL ascorbic acid (0.1 M) was loaded into Erlenmeyer reactors containing preweighed wet sediments (~5 g) and then immediately sealed. Ascorbic acid was used to inhibit oxidation of sulfide by concomitantly extracted Fe3+ (Burton et al. 2008). Evolving H2S gas was absorbed and trapped as ZnS precipitant in a separate vial containing 15 mL alkaline ZnAc solutions (20 % ZnAc + 2 M NaOH), which was attached to the inner wall of the reactor just above the surface of the extractant (Zhu et al. 2012a). After 24 h (stirring once a while), sulfide trapped was measured by the Cline method (Cline 1969) using a Hewlett-Packard 8453 UV–VIS spectrophotometer. The variability between duplicates in most cases is better than 8.8 %, but up to 18 % when AVS contents are extremely low in the upper 10 cm of C702 and C803.
Residual sediment pellets after the AVS extraction above were rinsed twice using deionized water and then immediately treated with cold acid Cr(II) solution to reduce pyrite-S to sulfide (48 h, stirring once a while) (Kallmeyer et al. 2004; Burton et al. 2008). Evolving H2S gas was collected and measured by the same methods for the AVS analysis above. The variability between duplicates in most cases is better than 7.6 %, but up to 17 % when pyrite contents are very low in the upper 10 cm of C702.
Results and discussion
Depth pattern of organic carbon
Organic carbon contents are 0.6–0.9 % (average 0.77 %) in C702, 0.6–0.7 % (average 0.67 %) in C802, and 0.67–0.86 % (average 0.77 %) in C803 (Fig. 2). Average OC contents for the three cores are slightly higher than the average for the inner shelf sediments (0.61 %), but significantly higher (t test, p < 0.01) than the average for the mid-outer shelf sediments (0.28 %) and for the entire ECS shelf sediments (0.34 %) (Kao et al. 2003). Higher OC content in the inner shelf sediments can be ascribed to high clay fraction of the sediments in comparison with the mid-outer shelf sediments (Lin et al. 2002a; Kao et al. 2003; Fang et al. 2009; Zhu et al. 2012b).

Depth profiles of organic carbon (OC) in cores C702, C802, and C803
Almost constant OC contents over the entire intervals of C802 and C803 indicate that OM mineralization has proceeded to a low extent due probably to refractory nature of the OM. High fraction of terrestrial OM in the inner shelf sediments (up to 54 %) estimated by OC isotope mass balance (Zhu et al. 2012b) may have imparted the bulk OM with low decomposability. Furthermore, the fluidized surface mud, an efficient incinerator of OM (Aller 1998, 2004), may have intensified the OM degradation, leaving eventually buried OM with low degradability. In comparison with C802 and C803, slightly higher OC contents in the upper 14 cm of C702 and a continuous decrease from this depth to the bottom may suggest somewhat higher reactivity of the OM. This difference is due probably to a direct impact of C702 by frequent algal blooms around the Zhoushan Fishery, while C802 and C803 are only marginally impacted by the events (Zhu et al. 2013). Note that, albeit the direct impacts of algal blooms on site C702, the small difference of OM among the three cores suggests that enhanced marine labile OM associated with pulsed input has been largely decomposed before burial. Quick decomposition in water column appears not to be a potential pathway due to shallow water depths (<50 m), instead, fluidized surface mud should have played an important role in the effective decomposition.
Vertical distributions of labile Fe
Figure 3 shows vertical distributions of LFe, LFe(II), and Fe(III) in the sediments. LFe contents in the three cores are at the range of 65–150 μmol/g (Fig. 3a). For C702 and C802, LFe contents increased significantly (t test, p < 0.05) with depth in the upper 20 cm, below which the LFe contents displayed only a small variability. For C803, LFe displayed only a small increase with depth mainly occurring between 5 and 10 cm.

Depth profiles of labile iron (LFe) (a), ferrous LFe (LFe(II)) (b), and ferric LFe (LFe(III) (c) in cores C702, C802, and C803
LFe(II) contents in the three cores are at the range of 31–131 μmol/g, accounting for 49 to nearly 100 % of the LFe (Fig. 3b). For C702 and C802, there is a significant downcore increase (t test, p < 0.05) in LFe(II) over the upper 20 cm. Below a depth of 25 cm, no obvious LFe(II) increase was seen. LFe(II) in C803 displayed a small downcore increase mainly occurring between 5 and 10 cm, similar to LFe in the core.
LFe(III) contents in C702 and C802 displayed a significant downcore decrease (t test, p < 0.05) to very low values at depth (Fig. 3c), which are coupled to the increase in LFe(II) at the corresponding depths. LFe(III) contents in C803 remain consistently low from the top layer to the bottom. Probably, a rapid decrease in LFe(III) was restricted to the uppermost millimeters, and therefore cannot be resolved by 2–3 cm sectioning of the core in the present study.
Vertical distributions of AVS and pyrite
Acid volatile sulfide in the three cores displays roughly a similar depth pattern (Fig. 4a). The contents maintained very low (~0.04–1 μmol/g) in the upper 5–8 cm, but increased obviously (t test, p < 0.05) from 8–10 to 25–30 cm, reaching their peak values (peak value 4.04 μmol/g for C702, 4.85 μmol/g for C802, and 4.89 μmol/g for C803). Below the depths of the peak values, AVS contents started to decrease.

Depth profiles of acid volatile sulfide (AVS) (a) and pyrite-S (b) in cores C702, C802, and C803
The three cores display different depth patterns of pyrite-S (Fig. 4b). C702 and C803 have similar pyrite-S contents at the corresponding depths, and their contents were much lower (t test, p < 0.01) than in C802 at a depth below 10 cm. Pyrite-S contents remained consistently low in the upper 18 cm of C702 and C803, but started to increase below the depth until a peak value of 17.9 μmol/g at 28 cm in C702, and 27.8 μmol/g at 27 cm in C803. Below the depths of their peak values, pyrite-S contents in the two cores started to decrease with depth. Pyrite-S contents in C802 increased from the uppermost layer to a depth of 20 cm, and then stayed nearly constant to the bottom of the core.
Iron sulfide formation and quick sulfide-buffering by labile Fe
Porewater sulfide was undetectable (<1 μM) in the subsurface, mid, and bottom layers of the three cores. It is reasonable to assume that free sulfide is very low or absent over the entire cores. Very low porewater sulfide concentration has also been reported in sediments off the Yangtze Estuary (Aller et al. 1985) and in the southern ESC (Huang and Lin 1995; Lin et al. 2000). Extremely low AVS contents in the uppermost zones of the three cores (Fig. 4a) can be ascribed to low sulfate reduction rate together with high sulfide oxidation at the redox interfaces because surface mud in the ECS inner shelf is under a very dynamic condition (McKee et al. 1983; DeMaster et al. 1985; Aller et al. 1985). Dynamic sediment resuspension can maintain redox potential relatively high in the topmost layer, which can substantially suppress sulfate reduction but encourage sulfide oxidation. High redox potential in surface layer of the ECS inner shelf sediments has been reported (Aller et al. 1985). Given the undetectable level of porewater sulfide in the three cores, AVS can be considered as mainly Fe sulfide minerals (pyrite excluded because of HCl nonextractability). Downcore increase in AVS below the upper zones (5–10 cm) points to the formation of Fe sulfides. Low porewater sulfide along with obvious accumulation of Fe sulfides below the subsurface suggests that free sulfide has been effectively buffered by reaction with Fe oxides. The quick sulfide-buffering has proceeded via un-sulfidized LFe pool, as demonstrated by previous studies (Azzoni et al. 2005; Giordani et al. 2008; Zhu et al. 2012a), and will be reconfirmed by this study.
The decrease in AVS from the depths of AVS peak values (29 cm for C702, 26 cm for C802, and 24 cm for C803) to the bottom of the cores is attributable to two reasons: (1) gradually decreasing activity of sulfate reducer and in turn low sulfide production due to limited availability of metabolizable OM at depth (Burdige 2006), (2) pyrite formation at the expense of AVS (Eqs. 3, 4). As a result, it is commonly observed that pyrite formation reaches its maximum near the zone where AVS peaks.
Downcore decrease in LFe(III) in the three cores (Fig. 3c) roughly corresponds to the increase in LFe(II), AVS, and pyrite-S, indicating reductive consumption of Fe oxides, formation of FeS, and its conversion to pyrite. However, AVS contents in the uppermost zone of the three cores remain very low in spite of adequate LFe(II) to precipitate free sulfide, suggesting that the availability of sulfide has been limiting FeS formation. Much higher LFe(II) than sulfide contents are due probably to the following reasons: (1) upward-diffusing sulfide is more readily oxidized than Fe(II) by O2 and other thermodynamically favorable electron acceptors such as nitrate and manganese oxides (Stumm and Morgan 1996), (2) besides formation via chemical reduction (i.e., Fe oxide reduction by sulfide), Fe(II) can also be formed via dissimilatory Fe reduction in suboxic environments, which can competitively suppress sulfate reduction and in turn sulfide formation (Thamdrup 2000; Canfield et al. 2005). Low to intermediate availability of labile OM and high availability of reactive Fe oxides are generally required to ensure the prevalence of dissimilatory Fe reduction in marine sediments (Jensen et al. 2003; Nickel et al. 2008). The two conditions are reasonably secured in the ECS inner shelf sediments due to massive input of terrestrial OM as well as reactive Fe from the Yangtze River and then dispersion along the inner shelf (Lin et al. 2000; Kao et al. 2003; Zhu et al. 2012b). Enhanced flux of marine OM at the site (C702) impacted by frequent algal blooms has not apparently increased the burial of labile OM, as has been demonstrated earlier. Consequently, a scant fraction of labile OM which has escaped decomposition and eventually becomes buried has been largely diluted by quickly deposited terrestrial material, rendering the bulk OM generally low in decomposability. Quick oxidation regeneration of reduced Fe species at the redox interface due to dynamic resuspension ensures high availability of reactive Fe oxides. Dissimilatory Fe reduction has been speculated to be playing a role in OM oxidation in the sediment (Bao 1989; Lü et al. 2011; Zhu et al. 2012b), which is probably one of the reasons for generally low AVS contents in the sediments in comparison with many other costal sediments in the world (Chambers et al. 2000; Goldhaber 2004). Because of the suppression of sulfide formation to some extent, a portion of Fe(II) produced via dissimilatory reduction can remain un-sulfidized, and is poised to quickly buffer upward-diffusing sulfide.
If dissolved sulfide reacts only with LFe pool [i.e., ∑(LFe(II) + LFe(III)], the LFe pool would have decreased because a fraction of LFe has been converted to HCl nonextractable pyrite (Fig. 4b). On the contrary, the LFe pool in the three cores increased with depth in the upper 15 cm (Fig. 3a), which strongly indicates that parts of less reactive Fe oxides have also been reduced to LFe(II). Given the undetectable free sulfide along with much higher LFe than AVS contents, we conclude that the reduction of less reactive Fe oxides is not through chemical reduction by dissolved sulfide but through dissimilatory reduction when dissolved sulfide has become low or exhausted. This argument is further supported by the observation that although LFe(III) is available for reaction with dissolved sulfide within the upper 20 cm of C802 and 30 cm of C702, LFe increase was still seen, suggesting that the LFe pool is continuously replenished from Fe sources other than LFe(III). Studies have suggested that some crystalline Fe oxides even sheet silicate-Fe(III) are susceptible to microbial reduction (Roden and Zachara 1996; Dong et al. 2009). Note that simultaneous chemical and dissimilatory reductions of LFe(III) cannot be excluded when dissolved sulfide is at its low, though chemical reduction is more favored while dissolved sulfide is present at a high level on basis of both the thermodynamic and kinetic viewpoints.
To summarize, continuous formation of un-sulfidized LFe(II) via dissimilatory reduction of less reactive Fe oxides may have played an important role in quick sulfide buffering in the ECS inner shelf sediments, in spite of a low level of LFe(III) at depth. Dissimilatory Fe reduction is believed to play a dual role in controlling free sulfide level in the sediments: (1) suppressing sulfate reduction and in turn sulfide production and (2) replenishing un-sulfidized Fe(II) for quick buffering of upward-diffusing sulfide. This study implies that a low LFe(III) or its exhaustion in marine sediments where dissimilatory Fe reduction is prevalent may not necessarily mean a low quick sulfide-buffering capacity, and low availability of LFe(III) cannot reliably suggest an depletion of the quick sulfide-buffering capacity.
Quick sulfide-buffering capacity
Quick sulfide-buffering capacities (β L) for C702, C802, and C803, calculated using Eq. 6, are 117–167, 73–117, and 107–130 μmol/g, respectively (Fig. 5). The values of β L are roughly within (t test, p < 0.05) β L range for eutrophic Jiaozhou Bay sediment (China) (LFe 71–148 μmol/g, β L 75–142 μmol/g) (Zhu et al. 2012a) and Fe-rich sediments of eutrophic Sacca di Goro lagoon (Italy) (LFe 95–285 μmol/g, β L 79–244 μmol/g) (Azzoni et al. 2005; Giordani et al. 2008), but much higher (t test, p < 0.05) than for Fe-poor sediments of this lagoon (LFe 32.6 μmol/g, β L < 20 μmol/g) (Giordani et al. 2008) and also much higher than for Fe-poor, sulfide-rich sediments of Arcachon Bay (France) (LFe 3.4–20 μmol/g, β L < 60 μmol/g) (Heijs et al. 1999). The above comparison reconfirms the critical role of high LFe in maintaining high β L. As shown in Fig. 5, the β L for the three cores displayed somewhat downcore increase although the LFe(II) pool has been consumed by pyrite formation and the LFe(III) decreased quickly with depth and maintained very low at depth (Fig. 3c). The increase in the β L can be attributable to continuous replenishment of un-sulfidized LFe(II) from dissimilatory reduction of less reactive Fe oxides, as has been documented earlier.

Depth profiles of quick sulfide-buffering capacity, β L, for cores C702, C802, and C803
Ratio of AVS/LFe can be used as a measure of buffering saturation of Fe towards free sulfide (i.e., fraction of LFe that has been sulfidized), with the AVS/LFe ratio less than 0.25 indicating a state of high residual buffering capacity and larger than 0.75 indicating a symptom of depletion of buffering capacity (Azzoni et al. 2005). AVS/LFe ratios for the ECS inner shelf sediments are 0–0.04, roughly similar to the ratios for Jiaozhou Bay sediments (Zhu et al. 2012a), but much lower than 0.25, suggesting a very low buffering saturation. This implies that β L for the sediments of the three cores has not become exhausted in spite of the impacts of seasonal eutrophication and frequent algal blooms in the past. It is predictable that eutrophication-induced pulsed inputs of labile OM in the near future may enhance sulfate reduction, but dissolved sulfide can be efficiently sequestrated in situ in the sediments given the high availability of un-sulfidized LFe pool and the prevalence of dissimilatory Fe reduction. Therefore, free sulfide is difficult to accumulate in porewater and to cause detrimental effects on the benthic ecosystem, like in Jiaozhou Bay (Zhu et al. 2012a). The three sites investigated in this study cannot be used to describe spatial variations of the geochemical parameters discussed above, particularly the spatial patterns from the inner shelf to the mid and outer shelves. However, the parameters for the three sites may have revealed a general feature of the whole inner shelf since the inner shelf has many things in common including hydrodynamic conditions and main sediment sources. This argument is seemingly supported by generally similar diagenesis of Fe and sulfur of this study and previous studies in the northern and southern inner shelves (Aller et al. 1985; Lin et al. 2000). Further work is needed to reveal the whole picture of spatial variability of the geochemical parameters.
Biogeochemical behavior of coastal marine sediment may vary nonlinearly in response to ever-increasing climate change or/and anthropogenic perturbations at an alarming rate (Middelburg 2009; Lehtoranta et al. 2009). The ECS shelf sediments, if subject to persisting high eutrophication and/or hypoxia, may have a quite different scenarios of S and Fe cycling in comparison with the current picture. Lasting reductive consumption of sedimentary Fe oxides via sulfide buffering due to persisting eutrophication and/or hypoxia may result in not only depletion of highly reactive Fe, but also enhanced release of phosphate into the overlaying water, which will exacerbate the eutrophication and sulfide production, and eventually establish a positive feedback between the eutrophication and phosphate and sulfide release (Rozan et al. 2002). Under this circumstance, high availability of LFe(III) is critically important for quick titration of dissolved sulfide because dissimilatory Fe reduction to replenish un-sulfidized LFe will be diminished to a great extent, even completely inhibited. Therefore, further work is needed to elucidate the long-term impacts of persisting hypoxia and eutrophication on Fe and S diagenetic cycling and quick-sulfide buffering capacity in the coastal ECS, given the ever-increasing climate change or/and anthropogenic perturbations.
Conclusions
High fluxes of terrestrial OM and Fe together with dilution of marine labile OM in the ECS inner shelf sediments have encouraged dissimilatory Fe reduction but suppressed sulfate reduction, which has resulted in low production of sulfide. Free sulfide in the sediments can be quickly buffered by LFe pool and therefore cannot accumulate in the porewater. Dissimilatory reduction of less reactive Fe oxides in the sediments is an important process to supply un-sulfidized LFe(II) for quick sulfide buffering. Quick sulfide-buffering capacity of the sediments has not become exhausted in spite of the impacts of seasonal eutrophication and frequent algal blooms. It is predictable that dissolved sulfide is difficult to accumulate in the porewater and to cause detrimental effect on the benthic ecosystem in the near future, given the availability of un-sulfidized LFe pool and the prevalence of dissimilatory Fe reduction. However, further work is needed to predict the long-term impacts of persisting hypoxia and eutrophication on diagenetic behavior of Fe and S, and the quick sulfide-buffering capacity, given the ever-increasing climate change and anthropogenic perturbations.
References
Aller RC (1998) Mobile deltaic and continental shelf muds as suboxic, fluidized bed reactors. Mar Chem 61:143–155
Aller RC (2004) Conceptual models of early diagenetic processes: the muddy seafloor as an unsteady, batch reactor. J Mar Res 62:815–835
Aller RC, Mackin JE, Ullman WJ, Wang C-H, Tsai S-M, Jin J-C, Sui Y-N, Hong J-Z (1985) Early chemical diagenesis, sediment-water solute exchange, and storage of reactive organic matter near the mouth of the Changjiang, East China Sea. Cont Shelf Res 4:227–251
Azzoni R, Giordani G, Viaroli P (2005) Iron–sulphur–phosphorus interactions: implications for sediment buffering capacity in a Mediterranean eutrophic lagoon (Sacca di Goro, Italy). Hydrobiologia 550:131–148
Bao G-D (1989) Separation of iron and manganese in the early diagenetic processes and its mechanism of biogeochemistry. Sci China (Ser B) 19:93–102 (in Chinese)
Burdige DJ (2006) Geochemistry of marine sediments. Princeton Univ Press, Princeton
Burton ED, Sullivan LA, Bush RT, Johnston SG, Keene AF (2008) A simple and inexpensive chromium-reducible sulfur method for acid-sulfate soils. Appl Geochem 23:2759–2766
Canfield DE (1989) Reactive iron in marine sediments. Geochim Cosmochim Acta 53:619–632
Canfield DE, Raiswell R, Bottrell S (1992) The reactivity of sedimentary iron minerals toward sulfide. Am J Sci 292:659–683
Canfield DE, Kristensensen E, Thamdrup B (2005) Aquatic geomicrobiology. Elsevier, Amsterdam
Chai C, Yu Z, Song X, Cao X (2006) The status and characteristics of eutrophication in the Yangtze River (Changjiang) Estuary and the adjacent East China Sea, China. Hydrobiologia 563:313–328
Chambers RM, Hollibaugh JT, Snively CS, Plant JN (2000) Iron, sulfur, and carbon diagenesis in sediments of Tomales Bay, California. Estuaries 23:1–9
Chen C-C, Gong G-C, Shiah F-K (2007) Hypoxia in the East China Sea: one of the largest coastal low-oxygen areas in the world. Mar Environ Res 64:399–408
Cline JD (1969) Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol Oceanogr 14:454–458
Cooper SR, Brush GS (1991) Long-term history of Chesapeake Bay anoxia. Science 254:992–996
DeMaster DJ, McKee BA, Nittrouer CA, Jiangchu Q, Guodong C (1985) Rates of sediment accumulation and particle reworking based on radiochemical measurements from continental shelf deposits in the East China Sea. Cont Shelf Res 4:143–158
Dong H, Jaisi DP, Kim J, Zhang G (2009) Microbe-clay mineral interactions. Am Miner 94:1505–1519
Fang T-H, Chen J-L, Huh C-A (2007) Sedimentary phosphorus species and sedimentation flux in the East China Sea. Cont Shelf Res 27:1465–1476
Fang T-H, Li J-Y, Feng H-M, Chen H-Y (2009) Distribution and contamination of trace metals in surface sediments of the East China Sea. Mar Environ Res 68:178–187
Giordani G, Azzoni R, Viaroli P (2008) A rapid assessment of the sedimentary buffering capacity towards free sulphides. Hydrobiologia 611:55–66
Goldhaber MB (2004) Sulfur-rich sediment. In: Mackenzie FT (ed) Sediments, diagenesis, and sedimentary rocks. Treatise on geochemistry, vol 7. Elsevier, Amsterdam, pp 257–288
Heijs SK, van Gemerden H (2000) Microbiological and environmental variables involved in the sulfide buffering capacity along an eutrophication gradient in a coastal lagoon (Bassin d’Arcachon, France. Hydrobiologia 437:121–131
Heijs SK, Jonkers HM, van Gemerden H, Schaub BEM, Stal LJ (1999) The buffering capacity towards free sulphide in sediments of a coastal lagoon (Bassin d’Arcachon, France)—the relative importance of chemical and biological processes. Estuar Coast Shelf Sci 49:21–35
Hsieh YP, Shieh YN (1997) Analysis of reduced inorganic sulfur by diffusion methods: improved apparatus and evaluation for sulfur isotopic studies. Chem Geol 137:255–261
Huang K-M, Lin S (1995) The carbon-sulfur-iron relationship and sulfate reduction rate in the East China Sea continental shelf sediments. Geochem J 29:301–315
Huh C-A, Su C-C (1999) Sedimentation dynamics in the East China Sea elucidated from 210Pb, 137Cs and 239,240Pu. Mar Geol 160:183–196
Jensen MM, Thamdrup B, Rysgaard S, Holmer M, Fossing H (2003) Rates and regulation of microbial iron reduction in sediments of the Baltic-North Sea transition. Biogeochemistry 65:295–317
Jørgensen BB (1982) Mineralization of organic matter in the sea bed—the role of sulfate reduction. Nature 296:643–645
Kallmeyer J, Ferdelman TG, Weber A, Fossing H, Jørgensen B (2004) A cold chromium distillation procedure for radiolabeled sulfide applied to sulfate reduction measurements. Limnol Oceanogr: Method 2:171–180
Kanaya G, Kikuchi E (2004) Relationships between sediment chemical buffering capacity and H2S accumulation: comparative study in two temperate estuarine brackish lagoons. Hydrobiologia 528:187–199
Kao SJ, Lin FJ, Liu KK (2003) Organic carbon and nitrogen contents and their isotopic compositions in surficial sediments from the East China Sea shelf and the southern Okinawa Trough. Deep-Sea Res Part II 50:1203–1217
Lehtoranta J, Ekholm P, Heikki P (2009) Coastal eutrophication thresholds: a matter of sediment microbial processes. Ambio 38:303–308
Lin S, Huang K-M, Chen S-K (2000) Organic carbon deposition and its control on iron sulfide formation of the southern East China Sea continental shelf sediments. Cont Shelf Res 20:619–635
Lin S, Hsieh I-J, Huang K-M, Wang C-H (2002a) Influence of the Yangtze River and grain size on the spatial variations of heavy metals and organic carbon in the East China Sea continental shelf sediments. Chem Geol 182:377–394
Lin S, Huang K-M, Chen S-K (2002b) Sulfate reduction and iron sulfide mineral formation in the southern East China Sea continental slope sediment. Deep-Sea Res Part I 49:1837–1852
Liu JP, Xu KH, Li AC, Milliman JD, Velozzi DM, Xiao SB, Yang ZS (2007) Flux and fate of Yangtze River sediment delivered to the East China Sea. Geomorphology 85:208–224
Liu S, Shi X, Liu Y, Zhu Z, Yang G, Zhu A, Gao J (2011) Concentration distribution and assessment of heavy metals in sediments of mud area from inner continental shelf of the East China Sea. Environ Earth Sci 64:567–579
Lü R-Y, Zhu M-X, Li T, Yang G-P, Deng F-F (2011) Speciation of solid-phase iron in mud sediments collected from the shelf of the East China Sea: constraints on diagenetic pathways of organic matter, iron, and sulfur. Geochimica 40:363–371 (in Chinese with English abstract)
Luther GW (1991) Pyrite synthesis via polysulfide compounds. Geochim Cosmochim Acta 55:2839–2849
McKee BA, Nittrouer CA, DeMaster DJ (1983) Concepts of sediment deposition and accumulation applied to the continental shelf near the mouth of the Yangtze River. Geology 11:631–633
Middelburg JJ (2009) Coastal hypoxia and sediment biogeochemistry. Biogeosciences 6:1273–1293
Nickel M, Vandieken V, Bruchert V, Jørgensen BB (2008) Microbial Mn(IV) and Fe(III) reduction in northern Barents Sea sediments under different conditions of ice cover and organic carbon deposition. Deep-Sea Res Part II 55:2390–2398
Otero XL, Calvo de Anta RM, Macias F (2006) Sulphur partitioning in sediments and biodeposits below mussel rafts in the Ria de Arousa (Galicia, NW Spain). Mar Environ Res 61:305–325
Poulton SW, Krom MD, Raiswell R (2004) A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide. Geochim Cosmochim Acta 68:3703–3715
Raiswell R, Canfield DE (1998) Sources of iron for pyrite formation in marine sediments. Am J Sci 298:219–245
Rickard DT (1975) Kinetics and mechanism of pyrite formation at low temperatures. Am J Sci 275:636–652
Roden EE, Zachara JM (1996) Microbial reduction of crystalline iron(III) oxides: influence of oxide surface area and potential for cell growth. Environ Sci Technol 30:1618–1628
Rozan TF, Taillefert M, Trouwborst RE, Glazer BT, Ma S, Herszage J, Valdes LM, Price KS, Luther GW (2002) Iron–sulfur–phosphorus cycling in the sediments of a shallow coastal bay: implications for sediment nutrient release and benthic macroalgal blooms. Limnol Oceanogr 47:1346–1354
Sell KS, Morse JW (2006) Dissolved Fe2+ and ΣH2S behavior in sediments seasonally overlain by hypoxic-to-anoxic waters as determined by CSV microelectrodes. Aquat Geochem 12:179–198
Sorokin YI, Zakuskina OY (2012) Acid-labile sulfides in shallow marine bottom sediments: a review of the impact on ecosystems in the Azov Sea, the NE Black Sea Shelf and NW Adriatic lagoons. Estuar Coast Shelf Sci 98:42–48
Stookey LL (1970) Ferrozine—a new spectrophotometric reagent for iron. Anal Chem 42:779–781
Stumm W, Morgan JJ (1996) Aquatic chemistry: chemical equilibrium and rates in natural waters, 3rd edn. Wiley, New York
Thamdrup B (2000) Bacterial manganese and iron reduction in aquatic sediments. Adv Microbiol Ecol 16:41–84
Valdemarsen T, Kristensen E, Holmer M (2009) Metabolic threshold and sulfide-buffering in diffusion controlled marine sediments impacted by continuous organic enrichment. Biogeochemistry 95:335–353
Valdemarsen T, Kristensen E, Holmer M (2010) Sulfur, carbon, and nitrogen cycling in faunated marine sediments impacted by repeated organic enrichment. Mar Ecol Prog Ser 400:37–53
Wang X-L, Sun X, Han X-R, Zhu C-J, Zhang C-S, Xin Y, Shi X-Y (2004) Comparison in macronutrient distributions and composition for high frequency HAB occurrence areas in East China Sea between summer and spring 2002. Oceanol Limn Sin 35:323–331 (in Chinese with English abstract)
Wang X-C, Sun M-Y, Li A-C (2008) Contrasting chemical and isotopic compositions of organic matter in Changjiang (Yangtze River) estuarine and East China Sea shelf sediments. J Oceanogr 64:311–321
Wu Y, Zhang J, Liu S, Zhang Z, Yao Q, Hong G, Cooper L (2007) Sources and distribution of carbon within the Yangtze River system. Estuar Coast Shelf Sci 71:13–25
Zhu M-X, Liu J, Yang G-P, Li T, Yang R-J (2012a) Reactive iron and its buffering capacity towards dissolved sulfide in sediments of Jiaozhou Bay, China. Mar Environ Res 80:46–55
Zhu M-X, Hao X-C, Shi X-N, Yang G-P, Li T (2012b) Speciation and spatial distribution of solid-phase iron in surface sediments of the East China Sea continental shelf. Appl Geochem 27:892–905
Zhu M-X, Shi X-N, Yang G-P, Hao X-C (2013) Formation and burial of pyrite and organic sulfur in mud sediments of the East China Sea inner shelf: constraints from solid-phase sulfur speciation and stable sulfur isotope. Cont Shelf Res 54:24–36
Acknowledgments
This research was jointly supported by the NSF of China (Grants 41076045 to M.X.Z.), the Shandong Province Natural Science Foundation (Grants ZR2011DM003 to M.X.Z.), and the Taishan Scholars Programme of Shandong Province (to G.P.Y.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, J., Zhu, MX., Yang, GP. et al. Quick sulfide buffering in inner shelf sediments of the East China Sea impacted by eutrophication. Environ Earth Sci 71, 465–473 (2014). https://doi.org/10.1007/s12665-013-2454-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12665-013-2454-4