
Abstract
Despite the fact that the nitrogen (N) fixers act as the key regulator of ecosystem process, a detailed study of their abundance, diversity, and dynamics in arsenic (As)-contaminated rice fields is missing so far. DNA extracted from soil followed by 16S rRNA and nifH gene-based real-time qPCR, clone library analysis, and DNA sequencing were used to examine the status of the total and diazotrophic communities in two agricultural fields with and without arsenic contamination history during one rice cultivation season. In general, higher nifH and 16S rRNA gene copy numbers were observed in rice growing soils with lesser As than that with higher As. Elevated levels of 16S rRNA and nifH genes in soil is directly associated with total and nitrogen fixers abundance in the agricultural land without As contamination history through the cultivation period, but the copy number of 16S rRNA gene was decreased, and the nifH gene remained unchanged in the As-contaminated land. Additionally, Canonical Correspondence Analysis (CCA) indicated the possible suppression of nifH gene abundance by soil pH, phosphate, and As content. Increased abundance of total and Acidobacterial lineages in low As-containing soil and the detection of several uncultured groups among nifH gene sequence in higher frequency indicated the presence of novel nifH bearing bacterial groups. Conversely, the abundance of copiotrophic Proteobacterial lineages gradually increased in soil with higher As. Herein, our study demonstrated that the dynamics of free-living nitrogen-fixing bacterial communities were perturbed due to As contamination in agricultural land.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic (As) contamination of soil and water is a serious issue due to its toxicity toward many organisms of the biome including humans; thus, it poses a severe threat to biodiversity. Higher concentration of As in underground aquifers is a serious concern, as most, if not all people are dependent on the underground water for drinking and irrigation purposes (Li et al. 2015; Wang et al. 2016a). Irrigation with As-laden groundwater is the major source of arsenic contamination in rice growing soil (Christopher and Haque, 2012; Polizzotto et al. 2013). Accumulation of As in agricultural soil enhances its translocation into rice plant and also in the rice grain in greater amounts (Khan et al. 2010). Consequently, people of rice-consuming countries like Bangladesh and India where As-laden groundwater is used for irrigation, in particular, were severely affected by As (Meharg and Rahman 2003; Mondal and Polya 2008; Karagas et al. 2016). Furthermore, several investigations noticed the negative correlation between crop yield and As contamination in soil (Brammer and Ravenscroft 2009; Srivastava et al. 2013). Therefore, elevated accumulation of As in rice field reduces soil fertility, and simultaneously its translocation to rice plant increases human health hazard through food chain contamination, thereby indirectly obstructing the sustainable agriculture (Brammer and Ravenscroft 2009; Bhattacharya et al. 2016).
The fertile quality of soil in particular zone is largely determined by the composition and diversity of the inhabitant microbial communities which influence nutrient transformation and edaphic factors (Reed et al. 2011; Luo et al. 2016). Nitrogen-fixing bacteria, the diazotrophs, are the major drivers of soil nitrogen input in the terrestrial ecosystem, reduce atmospheric nitrogen into ammonia which in turn gets converted to nitrate by nitrifying bacteria (Herridge et al. 2008; Vitousek et al. 2013; Vicente and Dean 2017). Biologically fixed ammonia and nitrate are used by all plants as their sole source of nitrogen (Santi et al. 2013). The conversion of free N2 into ammonia is exclusively carried out by prokaryotic nitrogenase (Yang et al. 2017, Vicente and Dean 2017). Among several subtypes, the molybdenum-dependent nitrogenase NifH encoded by the highly conserved nifH gene is widely distributed among all diazotrophs and well studied (Gaby and Buckley 2012; Santos et al. 2012; Kaschuk and Hungria 2017). Therefore, the soil fertility, as well as the optimum growth of plant directly or indirectly depends on the expression of nifH gene or in other words diversity of diazotrophic microorganisms (Kaschuk and Hungria 2017). Previous studies showed the effects of agricultural irrigation on in situ microbial community in high As aquifer but the impact of aquifer microbes in the irrigated water on the surface sediment geochemistry is less studied and should be explored in much detail (Li et al. 2015; Wang et al. 2016a). Considering these facts, present agricultural practice emphasizes predominantly on microbial community management, among many other factors to sustain the soil fertility (Das et al. 2016). Several investigations targeted As abatement into the plant by managing the agricultural practice and also by on focussing soil microbial communities (Somenahally et al. 2011; Das et al. 2016). Also, the rhizosphere microbial community thought to play a pivotal role during enhanced phytoextraction of As from soil (Sessitsch et al. 2013). All these studies revolved around the interaction between plant, microbes, and arsenic. However, not a single study reported so far on the impact of As exposure on soil microbial community during crop cultivation and therefore information on their abundance, diversity, dynamics, and function in the arsenic contaminated rice field has been carried out in this study to help ameliorate the problem.
Consequently, the abundance, composition, and distribution of microbes are site-specific and influenced by the chemistry of soil microhabitats (Wang et al. 2007; Islam et al. 2011; Gittel et al. 2014; Wang et al. 2017). Any perturbation to the soil composition and microenvironment may lead to restructuring the inhabitant microbial community composition, diversity, and function (Griffiths and Philippot 2013; Huhe et al. 2014; Wang et al. 2016b). For this reason, these microbial community parameters are generally used to assess the environmental changes influenced by varied factors (Islam and Sar 2011; Silva et al. 2013). Microorganisms capable of assimilating and fixing atmospheric N2, are considered as one of such important bio-indicators of soil fertility of several agricultural fields including paddy field (Hsu and Buckley 2009; Huhe et al. 2014). Changes in diazotroph composition implicating As contamination might be resulting in the decrease of rice production.
In this study, dynamics of total and free-living diazotrophic population was assessed in the soil of two agricultural fields with and without the history of irrigation with As-laden groundwater, throughout the rice cultivation period. Free-living diazotrophs are recalcitrant to laboratory cultivation; thus, little is explored about them; the nifH gene was used to study their diversity using culture-independent methods (Hsu and Buckley 2009; Gaby and Buckley 2012; Huhe et al. 2014). The abundance, distribution, and diversity of the total and nitrogen-fixing bacterial community were monitored by quantitative real-time PCR (qPCR), clone library analysis, and DNA sequencing.
Materials and methods
Site description and sample collection
Samples were collected from agricultural fields cultivating rice rotationally with other crops. Two agricultural fields with and without As contamination history in two different locations of West Bengal, India, were selected. One agricultural field was located in Arazi Ramakantapur village (24° 34′ 15′′ N, 88° 3′ 59′′ E) of Murshidabad district (MBD) with a history of As contamination, and the other was in Baruipara village (22° 45′ 33′′ N, 88° 14′ 26′′ E) of Hooghly district (BP). Arsenic-laden groundwater is used in Arazi Ramakantapur village for rice cultivation but there was no history of As contamination in groundwater of Baruipara village. Alternate wetting and drying irrigation strategy was followed in both places, where the field was re-flooded when saturated water of the soil reduced. Soil samples were collected during drying cycle from three randomly selected points (~ 5 cm deep), composited, and kept on ice. Samples collected from Murshidabad (MBD) and Hooghly (BP) district were numbered according to the place of collection followed by numerals showing collection sequence. Samples from both the agricultural lands were collected in three phases, during tillage (BP1 and MBD1), late tillering stage (BP2 and MBD2), and after harvesting of rice (BP3 and MBD3). Collected samples were transported in the ice-cooled chest to the laboratory and stored at − 20 °C until used for DNA extraction. One aliquot of the composite sample was analyzed for soil physicochemical properties and the other was used for culture independent microbiological analysis.
Chemical analysis of soil samples
Aliquots of soil samples were oven dried and grinded to obtain powder samples. For estimation of arsenic and other metal content, aliquot of the powdered soil samples was acid-digested with the digestion mixture (nitric, sulfuric, and perchloric acid in a volume ratio of 3:1:1). Arsenic and other metal concentration were estimated by Atomic Absorption Spectrophotometer (Thermo Scientific™ iCE™ 3000 Series) attached with vapor generator (VP100). Total organic carbon was estimated using standard Walkley-Black method (Walkley and Black 1934). Nitrate (NO3-N) and total phosphorus were measured following standard spectrophotometric methods (Jackson 1958). Briefly, nitrate was estimated from soil extract by the development of yellow color with nitrophenoldisulfonic acid and measuring absorbance at 420 nm wavelength light. Total phosphorus was measured from the acid digested soil sample by the development of 1, 2, 4-aminonaphtholsulfonic acid-reduced molybdophosphoric blue color in the perchloric acid system. Exchangeable ammonium (NH4 +-N) from soil was estimated by Kjeldahl distillation followed by titration with H2SO4 (Jackson 1958). Soil pH was measured by pH meters following resuspending the powdered soil in deionized water (10:1). Sand, silt, and clay content in the collected samples were determined by standard procedure (Lu 1999).
DNA extraction and PCR amplification
Total DNA from the six soil samples was extracted in triplicate taking 0.25 g subsample using MoBio PowerSoil DNA kit (MoBio, Carlsbad, CA). All three extractions from each sample were pooled and quantified by NanoDrop spectrophotometer (Thermo Scientific) and stored at − 20 °C until use. Diluted DNA (15 ng/μl) was used to amplify the nifH and 16S rRNA genes. Each PCR reaction mixture contained the following (final concentration): 2 mM MgCl2, 240 μM of each dNTPs, 10 pmol (16S rRNA gene) or 40 pmol (nifH gene) of each primer, 10× PCR buffer, and 1.5 U Taq DNA polymerase in 25 μl reaction. The bacterial 16S rRNA genes were amplified with the primer set 27F and 1492R using previously describe touchdown PCR across a broad range of annealing temperatures (55–60 °C) (Islam et al. 2014). The primer pair IGK3/DVV was used to amplify nifH gene according to Gaby and Buckley (2012).
Construction of 16S rRNA and nifH gene clone libraries and OTU assignment
Amplified PCR products were pooled, gel purified, ligated into the pGEM-T vector (Promega, USA), and cloned into E. coli JM109. Positive transformants with proper insert size were considered for clone library construction with nearly 100 (16S rRNA) and 30 (nifH gene) randomly chosen positive colonies per samples. The cloned gene fragments from each positive colony were re-amplified using vector specific primer SP6 and T7 by whole-cell PCR. The amplified products were digested with restriction endonuclease (MspI) in separate reactions and analyzed by 2.5% agarose gel electrophoresis. The restriction patterns of amplified cloned genes were grouped visually, and each group was considered as an Operational Taxonomic Unit (OTU). Representative clones from each OTU were identified through plasmid extraction and DNA sequencing with ABI PRISM BigDye terminator ready reaction mix (Life Technologies, USA) and Applied Biosystems ABI 3730xl DNA analyzer. Aliquots of each clone were preserved at − 80 °C until further analysis.
DNA translation and operational protein unit (OPU) assignment
For better taxonomic resolution of the nifH sequences and to associate their presence with the specific soil sample, DNA sequences were translated into amino acid sequences by translation tools of ExPASy. Respective protein sequence obtained for each RFLP group was referred as OPU. These protein sequences were used to find and retrieved similar putative bacterial groups in the NCBI database through protein blast.
Quantification of 16S rRNA and nifH gene copies by qPCR
The abundance of total bacteria and nitrogen fixers were quantified by qPCR targeting the 16S rRNA gene (primer 341F and 518R) (Muyzer et al. 1993) and nifH gene (primer polF and polR) (Poly et al. 2001). All real-time PCR runs were performed in duplicate, and each reaction mixture was prepared using the Quantifast SYBR Green master mix (Qiagen) in a total volume of 20 μl: 10.0 μl Master mix, 1.25 μl of each primer (5 pmol), 4.0 μl template DNA, and 3.5 μl PCR-grade water. The thermal cycling protocol was as follows: initial denaturation for 3 min at 95 °C followed by 40 cycles of 10 s at 95 °C, 30 s at 55 °C, and 5 s at 72 °C. The fluorescence signal was measured at the end of each extension step at 72 °C.
Standards for the qPCR assays were prepared by PCR amplification of each gene of interest (16S rRNA and nifH genes) from the genomic DNA of pure cultures using the universal bacteria specific primers (16S rRNA gene) (Islam and Sar 2011) and polF and polR (nifH gene) (Poly et al. 2001) followed by cloning into a pGEM-T Easy vector according to Islam and Sar (2011). Plasmid DNA concentrations ranging from 1.0 to 0.0001 ng/μl were prepared to generate the qPCR standard curves. Copy numbers were calculated from the concentration of DNA added to the reaction according to Lee et al. (2008).
Phylogenetic and statistical analyses
Based on number and frequency of OTU identified in each library, Shannon diversity index (H) and evenness (E) were calculated as described previously (Islam et al. 2014). Phylogenetic analysis of the DNA sequence data was performed using MEGA 4.1 software. The relationship between the samples in respect to their geochemical properties (Supplementary Table S1) and bacterial assemblage (Supplementary Table S2) was ascertained using Unweighted Pair Group Method with Arithmetic Mean analysis (UPGMA) by building the resemblance dendrogram using MVSP 3.1 software. The differences in soil chemical parameters and nifH gene abundance in the different soils over time were estimated with independent -tests. To test the influence of soil physicochemical factors on the community, canonical correspondence analysis (CCA) was performed using XLSTAT 2014.5.03 software.
Nucleotide accession number
DNA sequences retrieved in this study were submitted to the GenBank under the following accession number, 16S rRNA genes: KU603353-KU603417; nifH genes: KX273131-KX273174.
Results
Comparison of soil parameters
The physicochemical data are presented in Table 1. Relatively higher concentration of As was observed in MBD soils (~ 11 mg/kg) compared to BP (~ 7 mg/kg). Through the cultivation period in BP, the As concentrations in soil remain same but in MBD, it gradually declined from 12.8 to 10 mg/kg. The pH of the BP soils was nearly 5.0, whereas ~ 6.5 for MBD. The moisture content of the samples was nearly the same for both the fields but relatively increased humidity was noticed during tillering. Compared to NH4-N, relatively higher level of NO3-N was observed in both the rice fields. NH4-N level declined with the progression of cultivation in both rice fields. Reduction of NH4-N level in MBD during tillering and fall of NO3-N level in BP through the cultivation period, in particular, was noticeable. BP soil was characterized by higher sulfate and lower phosphate content than MBD soil. In addition, nearly similar level of total organic carbon was noticed in both the fields and remains unchanged throughout the cultivation period.
Abundance of nifH and 16S rRNA gene in the soils
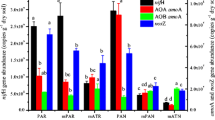
Real-time qPCR was used to compare the fluctuation of the nifH and 16S rRNA gene copy numbers in two rice fields across one cultivation cycle. Irrespective of rice field, the abundance of nifH gene copy numbers varied from (1.40 ± 0.21) × 106 to (2.49 ± 0.24) × 107 g−1 dry soil during the cultivation period (Fig. 1a). In general, significantly higher (p < 0.05) nifH gene copy numbers in BP, particularly during tillering and post-harvesting [(2.23 ± 0.36) × 107 and (2.49 ± 0.24) × 107 g−1dry soil)] was observed to that in MBD (~ 1.45 × 106 g−1dry soil) in all sampling time. In BP, nifH gene copy numbers gradually increased from tillage to late tillering (13-fold) and post-harvesting (15-fold) while no significant (p > 0.05) change in nifH gene copy number was observed in MBD.

Copies of the target gene per gram of soil. a Changes in abundance of bacterial 16S rRNA and nifH genes during the cultivation period.b Relative abundance of nifH gene in respect to abundance of 16S rRNA gene copy
Abundance of total bacterial community in terms of 16S rRNA gene copy number was assessed to draw conclusions on the fluctuations of the diazotrophic population using nifH gene copy number. During the cultivation period, the 16S rRNA gene copy numbers fluctuated between (3.88 ± 1.03) × 108 to (6.72 ± 2.09) × 108 g−1 dry soil in BP and (2.98 ± 0.71) × 108 to (5.15 ± 0.74) × 108 g−1 dry soil in MBD (Fig. 1a). During tillage, higher level of 16S rRNA gene abundance was observed in MBD than BP. However, during tillering and post-harvesting abundance of 16S rRNA gene gradually declined in MBD but enhanced in BP.
To compare the fluctuation of the diazotrophic bacterial population in respect to the fluctuation of total bacterial population in each sampling period, the abundance of nifH gene copy was expressed as per copy of 16S rRNA gene (Fig. 1b). In BP, relative abundance of nifH gene showed, although, diazotroph population first increased ninefold during tillering, however, decreased subsequently during post-harvesting. On the contrary, the relative abundance of nifH gene copy in MBD insignificantly increased (p > 0.05) round the cultivation period. Noticeably, the relative abundance of diazotrophs in MBD in all sampling period was similar to that observed in BP during tillage.
Fluctuation of total bacterial communities
To address how the total and diazotrophic bacterial diversity fluctuated, we focused on ARDRA richness, Shannon diversity index, and evenness of six 16S rRNA gene clone libraries (BP1, BP2, BP3, MBD1, MBD2, and MBD3) presented in Table 1. During tillage, both the agricultural fields showed the similar diversity of total bacteria with nearly equal OTU richness and evenness. During tillering, total bacterial diversity in both agricultural fields decreased in terms of OTU richness, Shannon index, and evenness (Table 1). In post-harvesting time, OTU richness again grew up with increasing diversity compared to tillering in BP. On the contrary, in the same period, MBD had less OTU richness with constant diversity.
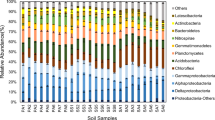
DNA sequencing of representative 16S rRNA gene clones followed by phylogenetic analysis; we tried to find out the taxonomic groups that were really fluctuated (Fig. 2a). During tillage, more than eight bacterial groups were observed in two rice fields at the phylum level. Proteobacteria, Acidobacteria, and Verruccomicrobia were found as predominant bacterial groups in both the rice field but with different proportionate (Fig. 2a). Apart from these, Cyanobacteria, Gematimonadetes, and Candidates saccharibacteria were found in BP1, whereas, Bacteroidetes, Actinobacteria, and Chloroflexi were detected in MBD1. During tillering, four and five different bacterial groups were found in BP2 and MBD2, respectively. Compared to tillage, declined abundance of Proteobacteria, as well as the proliferation of Acidobacteria, was noticed in BP2. In the contrary, very lesser increased in abundance of these bacterial groups along with the decrease in Bacteroidetes and Actinobacteria was observed in MBD2. During post-harvesting, in BP3 and MBD3, eight and six, respectively, bacterial groups were observed. In this period, abundance of both Acidobacteria and Proteobacteria was reduced to that of tillering.

Frequencies of bacterial phylogenetic lineages detected in a 16S rRNA and b nifH gene clone libraries derived from six soil samples collected from two rice fields in different cultivation period. Calculations were made based on the total number of clones associated with operational taxonomic units (OTUs) and sequenced representatives of each OTUs
During tillage, α-, ß-, γ- and δ-Proteobacteria were distributed equally in BP1, but during tillering, α-Proteobacteria flourished and other classes were depleted. β-Proteobacteria became abundant population during post-harvesting. In contrast α-, γ- and δ-Proteobacteria were detected during tillage at MBD1 with the abundance of α-Proteobacteria population, which became more abundant during tillering and declined during post-harvesting. Emergence of β-Proteobacteria, Bacilli, Cyanobacteria, and Ktedonobacteria as dominant population during post-harvesting in MBD3 was noteworthy although they were not detected in tillage and tillering period.
Fluctuation of nitrogen-fixing bacterial communities
An attempt had been made to find out the diazotroph that proliferated or inhibited during cultivation cycle through the analysis of six nifH gene clone libraries (Table 1). Sequencing of representative nifH gene clones and BLAST analyses in NCBI database revealed putative nitrogen fixer were mostly affiliated to uncultured microorganisms (Fig. 2b), although blast search with translated putative amino acid sequence indicated their relatedness to dinitrogenase protein. Phylogenetic analysis of nifH gene sequence revealed the presence of 11 bacterial taxa (C1 to C11) throughout the cultivation period in two rice fields of which eight (C2 and C5 to C11) remained unclassified (Fig. 3). The three known taxonomic groups with which some sequences showed relatedness were α-Proteobacteria (C1), γ-Proteobacteria (C3), and Cyanobacteria (C4). Sequences affiliated with α-Proteobacterial group were more abundantly detected in all MBD samples. Although the putative nifH gene sequences of Cyanobacterial origin were more frequent in BP1 and remain undetected in BP2 and BP3, they found to be flourished during tillering in MBD. Sequences-related group C7 and C11 were frequently observed in all sampling period in BP but the latter remain undetected in MBD. During the assessed period, in BP and MBD, the following numbers of taxonomic groups were observed: tillage, 4 and 6; tillering, 5 and 4; and post-harvesting, 6 and 4, respectively (Fig. 3). Nitrogen-fixing gene sequences retrieved from one library were used to find similar sequences in other libraries based on identity matrix. Both in BP and MBD, except few (α-Proteobacteria and Cyanobacteria in MBD), those bacterial population detected during tillage were not observed in late tillering or in post-harvesting.

Bootstrap neighbors joining phylogenetic tree based on DNA sequences of the nifH gene from six different soil samples collected from two rice fields in different cultivation period. Each taxonomic group (C) include representative sequences with closer identity query coverage. Numbers at the nodes reflect bootstrap support values with branches within clusters being collapsed. Frequency of each taxonomic group in each soil sample is shown in percentage and closer putative taxonomic affiliation nifH gene sequences are provided
Through the maximum-likelihood phylogenetic analysis, we addressed, whether the above differences were maintained at the protein level or not. Based on NCBI protein database, total 11 OPU (OPU1 to OPU11) were found across all samples that were distributed to two distinct NifH clusters (groups I and III) (Fig. 4). The OPUs with more than 50% identity was mentioned. Although putative protein sequences showed considerable identity with the sequences in the database with very lower query coverage, five OPU were affiliated with Proteobacteria, one with Cyanobacteria, four with Firmicutes, and rest with unidentified organisms. OPU1 affiliated with α-Proteobacteria (94–100% sequence identity) noticed only in all samples of MBD. Although OPU4 affiliated to Cyanobacteria was observed only during tillage in BP at higher abundance, it was ubiquitously noticed in all MBD samples. OPU7 and OPU11, affiliated with δ-Proteobacteria and Firmicutes were frequently observed in BP.

Maximum likelihood phylogenetic tree based on translated amino acid sequences of the nifH gene from six different soil samples collected from two rice fields in different cultivation period. Each operational protein unit (OPU) include sequences with range identity shown in percentage and 90% query coverage. Numbers at the nodes reflect bootstrap support values with branches within clusters being collapsed. Frequency of each OPU in each soil sample is shown in percentage, and closer putative taxonomic affiliation of translated NifH protein sequences is provided
Relation of soil variables to nifH gene carrying communities
To understand the potential effect of soil variables on the dynamics nifH gene carrying communities and total bacterial communities, we performed canonical correspondence analysis (CCA) on the abundance of nifH and 16S rRNA genes copy number, diversity indices, and physicochemical data (Fig. 5). CCA revealed that arsenic, pH, phosphate, and sulfate were the main factors affecting both absolute and relative abundance of nifH genes. The nitrogen-fixing bacterial abundance was negatively correlated with As, pH, and phosphate but positively correlated with TOC and sulfate. Total bacterial diversity remained unchanged in respect to these soil variables.

Canonical correspondence analysis (CCA) revealed the changes in the abundance 16S rRNA (16SA) and nifH (NFA) gene copies, relative abundance of nifH (nifH RA) and diversity of total bacteria (H), and the influence of environmental parameters. The number in each axis shows the percentage of total variation explained
UPGMA clustering based on abundance of different bacterial groups in each sample showed similar bacterial community composition during tillage and tillering at MBD, but at BP, similarity was observed during tillering and post-harvesting (Fig. 6a). A closer relation between bacterial composition during tillage at BP and during post-harvesting of MBD was observed.

Comparative analysis of bacterial community composition and geochemical properties of the samples. UPGMA cluster analysis among the samples based on abundance of different bacterial groups as detected in each samples (a), heat map of bacterial abundance observed in different samples (b), and UPGMA cluster analysis among the samples based on geochemical data (c) and neighbors joining phylogenetic relationship based on 16S rRNA gene sequences from six different soil samples. Each taxonomic group includes representative sequences with closer identity and query coverage (d). Samples detail is given in the text (Materials and methods)
Heat map (Fig. 6b) based on relative abundance of individual phyla/class detected within the samples showed Acidobacteria Gp6 Cyanobacteria 1, α-Proteobacteria 2, and ß-Proteobacteria were prevalent in BP1 and MBD3. Similarly, Bacteroidetes, α-Proteobacteria 2, α-Proteobacteria 3, δ-Proteobacteria 2, and Acidobacteria Gp3 were predominant in MBD1 and MBD2; and Acidobacteria Gp1 and ß- Proteobacteria were in BP2 and BP3. But UPGMA based on geochemical data showed in Table 1, closer relationship between BP1, BP2, and MBD3 and close clustering between MBD1 and MBD2 (Fig. 6c). Most of the retrieved 16S rRNA genes did not show high sequence identity at lower taxonomic level (the known genera). However, phylogenetic analysis (Fig. 6d) indicated that the 16S rRNA gene sequences from different samples altogether represented 25 bacterial taxa (at higher taxonomic level) distributed among more than six prominent clusters. Clustering pattern indicated, except few, the types of Acidobacteria and Proteobacteria sequences retrieved from BP were different than those from MBD in their respective counterpart.
Discussion
Total and free-living N2-fixing bacterial community structure and diversity is associated with the change in various physical, chemical, and biological factors such as pH, chemical fertilizer, and crop rotation. (Chunleuchanon et al. 2003; Orr et al. 2011; Silva et al. 2013; Wang et al. 2016b). Herein, we tried to explore the effects of As contamination on the microbial community in soil by comparing the dynamics of abundance and diversity of total and free-living N2-fixing bacteria during rice cultivation in two lands, with and without any history of As contamination. The observed abundance of total and N2-fixing bacteria in our studied rice fields as indicated by qPCR (based on nifH gene), corroborated with the previous reports from agricultural field, cropland, managed agricultural system, and rainforest (Orr et al. 2011; Silva et al. 2013; Mirza et al. 2014; Huhe et al. 2014). Although the two studied rice field exhibited almost identical levels of total bacterial abundance as revealed by 16S rRNA gene-based qPCR, a marked difference was noticed in abundance of N2-fixers. The lower absolute and relative abundance and insignificant proliferation of N2-fixers during the progression of cultivation in MBD soil compared to BP indicate the existence of factors that did not allow N2-fixers to flourish in MBD soil. We tried to correlate this difference of shifting of N2-fixing communities with various physical and chemical parameters to find out the causative factors. The CCA revealed quite dissimilar distribution of abundance of free-living N2-fixing bacteria to that of total bacteria in respect to distribution of probable influencing factors. This suggests that the factors that affect total bacterial community did not likely influence the free-living N2-fixers in studied agricultural soils. CCA also revealed pH, PO4 −, and As as the major factors that negatively influence the nifH gene abundance.
We observed elevated level of As in rice field of MBD while in BP As level remains below the global average (10.0 mg/kg) set for As in soil environment, across the cultivation period (Das et al. 2013a). This difference might be corroborated with the level of As in groundwater of respective villages as the rice fields were irrigated with groundwater (Christopher and Haque 2012; Santra et al. 2013). It is well reported that groundwater of MBD is significantly contaminated with As (> 0.05 mg/L) while there is no history of As contamination of groundwater in Baruipara village (Das et al. 1996; Roychowdhury et al. 2002). Therefore, whatever the As concentration detected in the BP soils might be the background As naturally present in this region. Investigation showed > 10 mg/kg As in soil significantly decrease the growth of crop plant (Yan-Chu 1994; Smith et al. 1998; Abedin et al. 2002; Khan et al. 2010; Das et al. 2013b). Consequently, it could be assumed that compared to MBD, the level of As in BP did not impart any negative influence on microbial community. Therefore, the observed lower level of diazotrophic bacterial population in MBD suggests a possible detrimental effect of As contamination. Although, very little information is available regarding the impact of As contamination on soil microbial diversity, previous research based on culture-dependent approaches hypothesized that As might decrease the nitrogenase activity in cyanobacteria and leguminous plant by interacting with iron, an important cofactor of dinitrogenase reductase (Ahmad et al. 2000; Mandal et al. 2011, Singh et al. 2014). In this regard, the pattern of fluctuation of bacterial community composition in both the rice fields could be elaborated in much detail. Here, we noticed the proliferation of Acidobacteria in BP but in MBD, the abundance of this group remained same with the progression of cultivation. This observation is well supported by the findings of Luo et al. (2014), who noticed negative correlation between abundance of Acidobacterial population and As concentration in the soil. Sheik et al. (2012) also noticed the dominance of Proteobacteria with minor representation of Acidobacteria in As-contaminated soil. Study also showed the prevalence of Acidobacterial communities in soil with lower pH (Jones et al. 2009). We also observed lower pH in BP soil compared to MBD which showed insignificant change during the cultivation. Therefore, this lower Acidobacterial abundance that remained unchanged in MBD soil might be due to As toxicity rather than pH.
The effort was also made to trace the bacterial groups that were fluctuated and contributed in increased nifH gene abundance. Our data suggested that the increased nifH gene abundance might be due to the proliferation of Acidobacteria in BP and Proteobacteria in MBD soil. Interestingly, based on nifH gene sequence, we could not found Acidobacterial lineage in the database; instead, we noticed the prevalence of unidentified lineages based on the nifH gene sequences. Although Acidobacteria group is prevalent in the soil environment, this might be due to unculturability of Acidobacteria and lack of nifH sequence of Acidobacterial origin in the database (Liu et al. 2016). In recent years, Santos et al. (2012) however, noticed few Acidobacterial nifH sequences through in silico analysis. Soil pH was also considered as an important driver controlling nifH gen-bearing bacterial population. Previous study noticed the positive correlation between soil pH and clay content but these two showed a negative correlation with nifH gene abundance (Silva et al. 2013). Similar clay content in both agricultural lands suggests increasing abundance of nifH gene in BP likely due to low pH rather than soil texture. Sulfur is another important component of dinitrogenase reductase enzyme used to fix N2 by microorganisms (Speelman and Holland 2016). Therefore, it could be assumed that at certain extends, the more available sulfur to the microorganisms, the more formation of dinitrogenase reductase and hence require more NifH (Rao and Rao 1986; Yuvaniyama et al. 2000; Cheng et al. 2016). In the present study, the observed higher level of sulfur in the form of sulfate in BP soil than MBD could be the reason of increased level of nifH in BP. However, increased level of sulfate does not guarantee its translocation into the cell as it depends on the highly regulated sulfate transporter (Aguilar-Barajas et al. 2011; Cheng et al. 2016). Previous investigations showed that available inorganic N in the form of NO3 in environment inhibits the function of di-nitrogenase reductase of N2 fixers (Knowles and Denike 1974; Yin et al. 2015). Therefore, removal of NO3-N from the environment might allow the expression of nifH gene (Yin et al. 2015). Gradual decrease in NO3-N level in BP soil is corroborated well with the increased level of nifH gene in this region. In this respect, although the level of NO3-N decreased in tillering stage and increased in post-harvesting period of MBD, the level of nifH did not align with the fluctuation of NO3-N, indicating the involvement of other factors in soil that reduce N2-fixing bacterial community.
Previous study showed soil microbial community shift is principally driven by the growing plant within a specific soil (Cline and Zak 2015). In this regard, however, Huhe et al. (2014) found a negative correlation between plant diversity and nifH copy number. The observed gradual decrease in As level might also be due to the uptake of arsenic by rice plants after converting it to available forms or volatilizing it by plant or microbes (Abedin et al. 2002; Meng et al. 2011).
The observed high soil phosphate, increased As, and decreased nifH gene abundance in MBD compared to BP soil also indicated the negative influence of As on nifH gene abundance in MBD. Although the As and inorganic phosphate ratio in soil sap and their retention in soil system determine the As uptake, however, the phosphate fertilization or increased phosphate availability in soil decrease the acquisition of As uptake by rice plant (Pigna et al. 2010). Conversely, previous investigation also showed a positive influence of available phosphorous on nitrogen fixation as well as abundance of nifH gene (Reed et al. 2007).
Conclusion
In summary, using a molecular method and statistical analysis with physicochemical data, this study revealed that free-living N-fixer could not flourish in As-contaminated soil. Our findings also indicated Proteobacterial dominance in As-contaminated soil while Acidobacteria were prevalent in the soil without As contamination. This study also indicated diverse nifH gene bearing unidentified bacteria in arsenic-rich soil, but this does not imply all nifH genes bearing bacteria actually express dinitrogenase. However, our study on microbial community structure involved in nitrogen fixation might be useful to elucidate the functional significance of diazotrophs for sustainable agriculture in As-contaminated soil and opens up an avenue to investigate the relationships between microorganisms and the environment.
References
Abedin MJ, Cresser MS, Meharg AA, Feldmann J, Cotter-Howells J (2002) Arsenic accumulation and metabolism in rice (Oryza sativa L). Environ Sci Technol 36(5):962–968. https://doi.org/10.1021/es0101678
Aguilar-Barajas E, Díaz-Pérez C, Ramírez-Díaz MI, Riveros-Rosas H, Cervantes C (2011) Bacterial transport of sulfate, molybdate, and related oxyanions. Biometals 24(4):687–707. https://doi.org/10.1007/s10534-011-9421-x
Ahmad S, Kitchin KT, Cullen WR (2000) Arsenic species that cause release of iron from ferritin and generation of activated oxygen. Arch Biochem Biophys 382(2):195–202. https://doi.org/10.1006/abbi.2000.2023
Bhattacharya P, Vahter M, Jarsjö J, Kumpiene J, Ahmad A, Sparrenbom C, Jacks G, Donselaar ME, Bundschuh J (2016) Arsenic research and global sustainability: proceedings of the sixth international congress on arsenic in the environment. CRC Press Stockholm Sweden
Brammer H, Ravenscroft P (2009) Arsenic in groundwater: a threat to sustainable agriculture in south and south-east Asia. Environ Int 35(3):647–654. https://doi.org/10.1016/j.envint.2008.10.004
Cheng G, Karunakaran R, East AK, Poole PS (2016) Multiplicity of sulfate and molybdate transporters and their role in nitrogen fixation in Rhizobium leguminosarum bv. Viciae Rlv3841. Mol Plant-Microbe Interact 29(2):143–152. https://doi.org/10.1094/MPMI-09-15-0215-R
Christopher OA, Haque AMM (2012) Arsenic contamination in irrigation water for rice production in Bangladesh: a review. Trends Appl Sci Res 7:331–349
Chunleuchanon S, Sooksawang A, Teaumroong N, Boonkerd N (2003) Diversity of nitrogen-fixing cyanobacteria under various ecosystems of Thailand: population dynamics as affected by environmental factors. World J Microbiol Biotechnol 19(2):167–173. https://doi.org/10.1023/A:1023286823958
Cline LC, Zak DR (2015) Soil microbial communities are shaped by plant-driven changes in resource availability during secondary succession. Ecology 96(12):3374–3385. https://doi.org/10.1890/15-0184.1
Das D, Samanta G, Mandal BK, Chowdhury TR, Chanda CR, Chowdhury PP, Basu GK, Chakraborti D (1996) Arsenic in ground water in six districts of West Bengal India. Environ Geochem Hlth 18(1):5–15. https://doi.org/10.1007/BF01757214
Das I, Ghosh K, Das DK, Sanyal SK (2013a) Assessment of arsenic toxicity in rice plants in areas of West Bengal. Chem Spec Bioavailab 25(3):201–208. https://doi.org/10.3184/095422913X13785717162124
Das S, Chou ML, Jean JS, Liu CC, Yang HJ (2016) Water management impacts on arsenic behavior and rhizosphere bacterial communities and activities in a rice agro-ecosystem. Sci Total Environ 542(Pt A):642–652. https://doi.org/10.1016/j.scitotenv.2015.10.122
Das S, Jean JS, Kar S, Liu CC (2013b) Changes in bacterial community structure and abundance in agricultural soils under varying levels of arsenic contamination. Geomicrobiol J 30(7):635–644. https://doi.org/10.1080/01490451.2012.746407
Gaby JC, Buckley DH (2012) A comprehensive evaluation of pcr primers to amplify the nifH gene of nitrogenase. PLoS One 7(7):e42149. https://doi.org/10.1371/journal.pone.0042149
Gittel A, Bárta J, Kohoutová I, Schnecker J, Wild B, Čapek P, Kaiser C, Torsvik VL, Richter A, Schleper C, Urich T (2014) Site- and horizon-specific patterns of microbial community structure and enzyme activities in permafrost-affected soils of Greenland. Front Microbiol 5:541. https://doi.org/10.3389/fmicb.2014.00541
Griffiths BS, Philippot L (2013) Insights into the resistance and resilience of the soil microbial community. FEMS Microbiol Rev 37(2):112–129. https://doi.org/10.1111/j.1574-6976.2012.00343.x
Herridge DF, Peoples MB, Boddey RM (2008) Marschner review: global inputs of biological nitrogen fixation in agricultural systems. Plant Soil 311(1-2):1–18. https://doi.org/10.1007/s11104-008-9668-3
Hsu SF, Buckley DH (2009) Evidence for the functional significance of diazotroph community structure in soil. ISME J 3(1):124–136. https://doi.org/10.1038/ismej.2008.82
Huhe BS, Cheng Y, Nomura N, Nakajima T, Nakamura T, Uchiyama H (2014) Effect of abandonment on diversity and abundance of free-living nitrogen-fixing bacteria and total bacteria in the cropland soils of Hulun Buir Inner Mongolia. PLoS One 9(9):e106714. https://doi.org/10.1371/journal.pone.0106714
Islam E, Dhal P, Kazy SK, Sar P (2011) Molecular analysis of bacterial communities in uranium ores and surrounding soils from Banduhurang open cast uranium mine India: a comparative study. J Environ Sci Health Part A 46(3):271–280. https://doi.org/10.1080/10934529.2011.535433
Islam E, Paul D, Sar P (2014) Microbial diversity in uranium deposits from Jaduguda and Bagjata uranium mines India as revealed by clone library and denaturing gradient gel electrophoresis analyses. Geomicrobiol J 31(10):862–887. https://doi.org/10.1080/01490451.2014.907375
Islam E, Sar P (2011) Molecular assessment on impact of uranium ore contamination in soil bacterial diversity. Int Biodeter Biodegr 65(7):1043–1051. https://doi.org/10.1016/j.ibiod.2011.08.005
Jackson ML (1958) Soil chemical analysis. Prentice-Hall, Inc Englewood Cliffs, p 498
Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N (2009) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 4:442–453
Karagas MR, Punshon T, Sayarath V, Jackson BP, Folt CL, Cottingham KL (2016) Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr 170(6):609–616. https://doi.org/10.1001/jamapediatrics.2016.0120
Kaschuk G, Hungria M (2017) Diversity and importance of diazotrophic bacteria to agricultural sustainability in the tropics. In: de Azevedo J, Quecine M (eds) Diversity and benefits of microorganisms from the tropics. Springer, Cham, pp 269–292. https://doi.org/10.1007/978-3-319-55804-2_12
Khan MA, Stroud JL, Zhu YG, McGrath SP, Zhao FJ (2010) Arsenic bioavailability to rice is elevated in Bangladeshi paddy soils. Environ Sci Technol 44(22):8515–8521. https://doi.org/10.1021/es101952f
Knowles R, Denike D (1974) Effect of ammonium, nitrate, and nitrate-nitrogen on anaerobic nitrogenase activity in soil. Soil Biol Biochem 6(6):353–358. https://doi.org/10.1016/0038-0717(74)90043-1
Lee C, Lee S, Shin S, Hwang S (2008) Real-time PCR determination of rRNA gene copy number: absolute and relative quantification assays with Escherichia coli. Appl Microbiol Biotechnol 78(2):371–376. https://doi.org/10.1007/s00253-007-1300-6
Li P, Wang Y, Dai X, Zhang R, Jiang Z, Jiang D, Wang S, Jiang H, Wang Y, Dong H (2015) Microbial community in high arsenic shallow groundwater aquifers in Hetao Basin of Inner Mongolia, China. PLoS One 10(5):e0125844. https://doi.org/10.1371/journal.pone.0125844
Liu J, Sui Y, Yu Z, Yao Q, Shi Y, Chu H, Jin J, Liu X, Wang G (2016) Diversity and distribution patterns of acidobacterial communities in the black soil zone of northeast China. Soil Biol Biochem 95:212–222. https://doi.org/10.1016/j.soilbio.2015.12.021
Lu R (1999) Analytical methods of soil agrochemistry. Beijing Science and Technology Press
Luo J, Bai Y, Liang J, Qu J (2014) Metagenomic approach reveals variation of microbes with arsenic and antimony metabolism genes from highly contaminated soil. PLoS One 9(10):e108185. https://doi.org/10.1371/journal.pone.0108185
Luo X, Fu X, Yang Y, Cai P, Peng S, Chen W, Huang Q (2016) Microbial communities play important roles in modulating paddy soil fertility. Sci Rep 6(1):20326. https://doi.org/10.1038/srep20326
Mandal SM, Gouri SS, De D, Das BK, Mondal KC, Pati BR (2011) Effect of arsenic on nodulation and nitrogen fixation of black gram (Vigna mungo). Indian J Microbiol 51(1):44–47. https://doi.org/10.1007/s12088-011-0080-y
Meharg AA, Rahman M (2003) Arsenic contamination of Bangladesh paddy field soils: implications for rice contribution to arsenic consumption. Environ Sci Technol 37(2):229–234. https://doi.org/10.1021/es0259842
Meng XY, Qin J, Wang LH, Duan GL, Sun G-X, HL W, Chu CC, Ling H-Q, Rosen BP, Zhu YG (2011) Arsenic biotransformation and volatilization in transgenic rice. New Phytol 191(1):49–56. https://doi.org/10.1111/j.1469-8137.2011.03743.x
Mirza BS, Potisap C, Nüsslein K, Bohannan BJM, Rodrigues JLM (2014) Response of free-living nitrogen-fixing microorganisms to land use change in the amazon rainforest. Appl Environ Microbiol 80(1):281–288. https://doi.org/10.1128/AEM.02362-13
Mondal D, Polya DA (2008) Rice is a major exposure route for arsenic in Chakdaha block Nadia district West Bengal India: a probabilistic risk assessment. Appl Geochem 23(11):2987–2998. https://doi.org/10.1016/j.apgeochem.2008.06.025
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59(3):695–700
Orr CH, James A, Leifert C, Cooper JM, Cummings SP (2011) Diversity and activity of free-living nitrogen-fixing bacteria and total bacteria in organic and conventionally managed soils. Appl Environ Microbiol 77(3):911–919. https://doi.org/10.1128/AEM.01250-10
Pigna M, Cozzolino V, Caporale GA, Mora ML, Meo VD, Jara AA, Violante A (2010) Effects of phosphorus fertilization on arsenic uptake by wheat grown in polluted soils. J Soil Sci Plant Nutr 10(4):428–442. https://doi.org/10.4067/S0718-95162010000200004
Polizzotto ML, Lineberger EM, Matteson AR, Neumann RB, Badruzzaman ABM, Ali M (2013) Arsenic transport in irrigation water across rice-field soils in Bangladesh. Environ Pollut 179:210–217. https://doi.org/10.1016/j.envpol.2013.04.025
Poly F, Ranjard L, Nazaret S, Gourbière F, Monrozier LJ (2001) Comparison of nifH gene pools between soils and between soil microenvironments of contrasting properties. Appl Environ Microbiol 67(5):2255–2267. https://doi.org/10.1128/AEM.67.5.2255-2262.2001
Rao J, Rao V (1986) Nitrogen fixation (C2H2 reduction) as influenced by sulfate in paddy soils. J Agric Sci 106(02):331–336. https://doi.org/10.1017/S0021859600063929
Reed SC, Cleveland CC, Townsend AR (2011) Functional ecology of free-living nitrogen fixation: a contemporary perspective. Annu Rev Ecol Evol Syst 42(1):489–512. https://doi.org/10.1146/annurev-ecolsys-102710-145034
Reed SC, Seastedt TR, Mann CM, Suding KN, Townsend AR, Cherwin KL (2007) Phosphorus fertilization stimulates nitrogen fixation and increases inorganic nitrogen concentrations in a restored prairie. Appl Soil Ecol 36(2-3):238–242. https://doi.org/10.1016/j.apsoil.2007.02.002
Roychowdhury T, Uchino T, Tokunaga H, Ando M (2002) Survey of arsenic in food composites from arsenic affected area of West Bengal India. Food Chem Toxicol 40(11):1611–1621. https://doi.org/10.1016/S0278-6915(02)00104-7
Santi C, Bogusz D, Franche C (2013) Nitrogen fixation in non legumes. Ann Bot 111(5):743–767. https://doi.org/10.1093/aob/mct048
Santos PCD, Fang Z, Mason SW, Setubal JC, Ray Dixon R (2012) Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes. BMC Genomics 13(1):162. https://doi.org/10.1186/1471-2164-13-162
Santra SC, Samal AC, Bhattacharya P, Banerjee S, Biswas A, Majumdar J (2013) Arsenic in food chain and community health risk: a study in Gangetic West Bengal. Procedia Environ Sci 18:2–13. https://doi.org/10.1016/j.proenv.2013.04.002
Sessitsch A, Kuffner M, Kidd P, Vangronsveld J, Wenzel WW, Fallmann K, Puschenreiter M (2013) The role of plant-associated bacteria in the mobilization and phytoextraction of trace elements in contaminated soils. Soil Biol Biochem 60(100):182–194. https://doi.org/10.1016/j.soilbio.2013.01.012
Sheik CS, Mitchell TW, Rizvi FZ, Rehman Y, Faisal M, Hasnain S, McInerney MJ, Krumholz LR (2012) Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS One 7(6):e40059. https://doi.org/10.1371/journal.pone.0040059
Silva MCP, Hai BS, Schloter M, Elsas JDV, Salles JF (2013) Temporal dynamics of abundance and composition of nitrogen-fixing communities across agricultural soils. PLoS One 8(9):e74500. https://doi.org/10.1371/journal.pone.0074500
Singh S, Shrivastava AK, Singh VK (2014) Arsenic and cadmium are inhibitors of cyanobacterial dinitrogenase reductase (nifH1) gene. Funct Integr Genomics 14(3):571–580. https://doi.org/10.1007/s10142-014-0375-2
Smith E, Naidu R, Alston AM (1998) Arsenic in the soil environment: a review. Adv Agron 64:149–195. https://doi.org/10.1016/S0065-2113(08)60504-0
Somenahally AC, Hollister EB, Loeppert RH, Yan W, Gentry TJ (2011) Microbial communities in rice rhizosphere altered by intermittent and continuous flooding in fields with long-term arsenic application. Soil Biol Biochem 43(6):1220–1228. https://doi.org/10.1016/j.soilbio.2011.02.011
Speelman AL, Holland PL (2016) Sulfur-supported iron complexes for understanding N2 reduction. In: Nishibayashi Y (ed) Nitrogen fixation, Topics in organometallic chemistry, vol 60. Springer, Cham, pp 197–213. https://doi.org/10.1007/3418_2016_4
Srivastava PK, Singh M, Singh N, Tripathi RD (2013) Soil arsenic pollution: a threat to crops. J Biorem Biodegrad 4:7
Vicente EJ, Dean DR (2017) Keeping the nitrogen-fixation dream alive. Proc Natl Acad Sci 114(12):3009–3011. https://doi.org/10.1073/pnas.1701560114
Vitousek PM, Menge DNL, Reed SC, Cleveland CC (2013) Biological nitrogen fixation: rates, patterns and ecological controls in terrestrial ecosystems. Philos Trans R Soc B 368(1621):20130119. https://doi.org/10.1098/rstb.2013.0119
Walkley A, Black IA (1934) An examination of Degtjareff method for determining soil organic matter and a proposed modification of the chromic acid titration method. Soil Sci 37(1):29–37. https://doi.org/10.1097/00010694-193401000-00003
Wang J, Zhang D, Zhang L, Li J, Raza W, Huang Q, Shen Q (2016b) Temporal variation of diazotrophic community abundance and structure in surface and subsoil under four fertilization regimes during a wheat growing season. Agric Ecosyst Environ 216:116–124. https://doi.org/10.1016/j.agee.2015.09.039
Wang R, Zhang H, Sun L, Qi G, Chen S, Zhao X (2017) Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Sci Rep 7(1):343. https://doi.org/10.1038/s41598-017-00472-6
Wang Y, Li P, Jiang Z, Sinkkonen A, Wang S, Tu J, Wei D, Dong H, Wang Y (2016a) Microbial community of high arsenic groundwater in agricultural irrigation area of Hetao plain, Inner Mongolia. Front Microbiol 7:1917. https://doi.org/10.3389/fmicb.2016.01917
Wang Y, Shi J, Wang H, Lin Q, Chen X, Chen Y (2007) The influence of soil heavy metals pollution on soil microbial biomass enzyme activity and community composition near a copper smelter. Ecotoxicol Environ Saf 67(1):75–81. https://doi.org/10.1016/j.ecoenv.2006.03.007
Yan-Chu H (1994) Arsenic distribution in soils. In: Nriagu JO (ed) Arsenic in the environment, Part I: cycling and characterization. John Wiley & Sons Inc, New York, pp 17–51
Yang J, Xie X, Yang M, Dixon R, Wang YP (2017) Modular electron-transport chains from eukaryotic organelles function to support nitrogenase activity. Proc Natl Acad Sci U S A 114(12):E2460–E2465. https://doi.org/10.1073/pnas.1620058114
Yin TT, Pin UL, Ghazali AHA (2015) Influence of external nitrogen on nitrogenase enzyme activity and auxin production in Herbaspirillum seropedicae (Z78). Trop Life Sci Res 26(1):101–110
Yuvaniyama P, Agar JN, Cash VL, Johnson MK, Dean DR (2000) NifS-directed assembly of a transient [2Fe-2S] cluster within the NifU protein. Proc Natl Acad Sci U S A 97(2):599–604. https://doi.org/10.1073/pnas.97.2.599
Acknowledgements
Arindam Chakraborty acknowledges the financial assistance from the University of Kalyani under URS fellowship scheme. We thankfully acknowledge Kayce Booth, Department of Cell Biology and Anatomy, University of South Carolina School of Medicine, Columbia, USA for language editing of the script. We are also thankful to Md Nur Alam, Ph.D. for improving the English language of the manuscript.
Funding
This work was carried out with the support of BSR start-up grants from University Grants Commission, Extra Mural Research grant from DST-SERB, Government of India, and DST-PURSE Programme, University of Kalyani, India.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Diane Purchase
Electronic supplementary material
Supplementary Table S1
(XLSX 10 kb)
Supplementary Table S2
(XLSX 10 kb)
Rights and permissions
About this article

Cite this article
Chakraborty, A., Islam, E. Temporal dynamics of total and free-living nitrogen-fixing bacterial community abundance and structure in soil with and without history of arsenic contamination during a rice growing season. Environ Sci Pollut Res 25, 4951–4962 (2018). https://doi.org/10.1007/s11356-017-0858-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-017-0858-5