
Abstract
Microorganisms attached on the surfaces of substrate materials in constructed wetland play crucial roles in the removal of organic and inorganic pollutants. However, the impact of substrate material on wetland microbial community structure remains unclear. Moreover, little is known about microbial community in constructed wetland purifying polluted surface water. In this study, Illumina high-throughput sequencing was applied to profile the spatial variation of microbial communities in three pilot-scale surface water constructed wetlands with different substrate materials (sand, zeolite, and gravel). Bacterial community diversity and structure showed remarkable spatial variation in both sand and zeolite wetland systems, but changed slightly in gravel wetland system. Bacterial community was found to be significantly influenced by wetland substrate type. A number of bacterial groups were detected in wetland systems, including Proteobacteria, Chloroflexi, Bacteroidetes, Acidobacteria, Cyanobacteria, Nitrospirae, Planctomycetes, Actinobacteria, Firmicutes, Chlorobi, Spirochaetae, Gemmatimonadetes, Deferribacteres, OP8, WS3, TA06, and OP3, while Proteobacteria (accounting for 29.1–62.3 %), mainly composed of Alpha-, Beta-, Gamma-, and Deltaproteobacteria, showed the dominance and might contribute to the effective reduction of organic pollutants. In addition, Nitrospira-like microorganisms were abundant in surface water constructed wetlands.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Due to its advantages of low cost, easy maintenance, and environmental friendliness, constructed wetland (CW) has been widely used in treatment of industrial, agricultural, and municipal wastewater (Chang et al. 2015; Ji et al. 2012; Mulling et al. 2014; Xiong et al. 2015; Zhi and Ji 2014) and polluted surface water (Dzakpasu et al. 2015; Tu et al. 2014). Biofilm microbial community, attached to the surfaces of substrate materials (or filter media) in CW, plays crucial roles in the reduction of degradable organic pollutants and transformation of inorganic pollutants including nitrogen, sulfate, and phosphate (Iasur-Kruh et al. 2010; Liu et al. 2013; Ramond et al. 2012). In addition, microbial assemblage is sensitive to operational and environmental conditions, and its structure is a good indicator for the status of CW ecosystem (Chang et al. 2015). Previous studies using traditional molecular biology approaches [e.g., denaturing gradient gel electrophoresis (DGGE), terminal restriction fragment length polymorphism (TRFLP), and clone library analysis] have greatly aided in our understanding of microbial community structure in CW ecosystem (Arroyo et al. 2013; Elsayed et al. 2014; Iasur-Kruh et al. 2010; Morato et al. 2014). A variety of factors have also been found to influence the structure of CW microbial community, such as layer depth (Bouali et al. 2014; Iasur-Kruh et al. 2010), operational time (Bouali et al. 2014), plant type (Menon et al. 2013), plant species richness (Liu et al. 2013), plant development (Zhao et al. 2012), wastewater quality characteristics (Chang et al. 2015; Zhao et al. 2012), flow (Arroyo et al. 2013), and water depth (Morato et al. 2014). These previous studies mainly focused on CW systems treating industrial and municipal wastewater, yet microbial community in CW systems purifying polluted surface water has received little attention. Moreover, the impact of substrate material on CW microbial community structure remains unclear.
High-throughput sequencing technology is a highly efficient tool for identifying the profile of complicated microbial community (Liao et al. 2015; Wang et al. 2015a, b; Yang et al. 2015), and can provide a new opportunity to resolve the microbial assemblage of CW biofilm and systematically investigate its links with operational and environmental conditions (Ansola et al. 2014; Arroyo et al. 2015; Zhong et al. 2015). Due to its relatively low costs and great throughput, Illumina sequencing has been applied to profile microbial communities in various natural and man-made ecosystems (Kim et al. 2014; Shi et al. 2014; Sun et al. 2015; Wu et al. 2015; Yang et al. 2014; Zhang et al. 2015). However, information on high-throughput sequencing of microbial assemblage in CW treating surface water is still very limited (Ligi et al. 2014). Therefore, the main objective of the current study was to use Illumina high-throughput sequencing to investigate the impact of substrate type on the structure of microbial community in CW treating surface water.
Materials and methods
Wetland description
Three vertical-flow pilot-scale wetland systems (length 1.2 m, width 1 m, height 1 m) were constructed to treat the water of a heavily polluted river located in Guangdong Province (southern China). Each wetland system was filled with coarse gravel in the bottom layer (diameter 70–120 mm, height 20 cm). Wetlands WA, WB, and WC were filled with an 80-cm upper layer of sand (1–2 mm), natural zeolite (5–8 mm), and fine gravel (6–12 mm), respectively. The top of each wetland system was planted with Pennisetum purpureum Schum. at a density of nearly 30 plants/m2. The retention time of river water in each wetland was set to 5 days. Before the beginning of experiments, these three wetland systems had been under continuous operation for nearly 1 year. During this period, the dissolved organic carbon (DOC) and ammonia nitrogen for the influents of each wetland system were 8–12 and 2–4 mg/L, respectively. The average DOC removal rates of wetlands WA, WB, and WC were 60, 79, and 49 %, respectively, while the average ammonia removal rate of each wetland system was above 95 %.
Molecular analyses
In the current study, substrate particle samples WA1–WA4, WB1–WB4, and WC1–WC4 were collected from 0.2, 0.4, 0.6, and 0.8 m below the surface of wetlands WA, WB, and WC, respectively. Total genomic DNA of each sample was extracted using Powersoil DNA extraction kit (Mobio Laboratories) according to the protocol recommended by the manufacturer. PCR amplicon libraries were constructed for Illumina MiSeq sequencing with the primer set 515F (5′-GTGCCAGCMGCCGCGG-3′)/R907 (5′-CCGTCAATTCMTTTRAGTTT-3′) that targets V4–V5 hypervariable regions of bacterial 16S rRNA genes (Wang et al. 2015b). The reads from raw DNA fragments were merged using FLASH and quality filtering of sequences was carried out according to the literature (Caporaso et al. 2011). Chimeric reads were checked and filtered out using UCHIME (Edgar et al. 2011), and the high-quality sequences were clustered into the operational taxonomic units (OTUs) by setting a 0.03 distance. Bacterial community richness (Chao 1 estimator) and diversity (Shannon index) were obtained using the UPARSE pipeline (Edgar 2013). The Classifier program of the RDP-II was used to assign taxonomic identity of the representative sequence from each OTU (Wang et al. 2007). The OTU-based beta diversity was calculated using UniFrac analysis to compare similarity among wetland microbial communities. Weighted UniFrac using Quantitative Insights into Microbial Ecology (QIIME) program was applied for weighted pair group method with arithmetic mean (WPGMA) clustering. The reads obtained from Illumina high-throughput analysis in this study were deposited in the NCBI short-read archive under accession number SRP059159.
Results and discussion
Bacterial community diversity
In this study, a total of 367,800 valid reads for 12 wetland samples were retrieved from Illumina MiSeq sequencing platform. Each library contained 11,466 to 28,561 reads, normalized to 11,460 for comparison of bacterial community diversity. Good’s coverage estimator indicated that 96–99 % of the OTUs were retrieved in all wetland samples, using a 0.03 distance (Table 1), suggesting that the OTU diversity has been well captured. The OTU number ranged from 1203 to 2144 for each wetland sample. The Chao 1 estimator showed that the bacterial community richness ranged from 1622 to 2629 taxa in the three CW systems used to treat polluted river water. The bacterial community richness illustrated a remarkable spatial variation in each CW system. The samples from wetland A showed lower richness than those from wetlands B and C. Moreover, the value of Shannon’s diversity index of bacterial communities in wetland samples fell between 5.42 and 6.76. A remarkable spatial variation of Shannon’s diversity was found in wetlands A and B, while Shannon’s diversity only slightly increased with increasing wetland layer depth. The samples from wetland C showed higher Shannon’s diversity than those from wetlands A and B.
In a large-scale CW system purifying polluted river water, DGGE analysis indicated that Shannon’s diversity of wetland sediment bacterial community decreased from inlet zone (3.471) to outlet zone (2.566) (Zhi et al. 2015). In this study, high-throughput sequencing was used to profile the microbial communities in surface water CW systems. The observed Shannon’s diversity (5.42–6.76) in this study was much higher than that reported in a previous study using DGGE analysis (Zhi et al. 2015). In addition, a remarkable spatial variation of bacterial community diversity was observed in both sand and zeolite CW systems, instead of gravel CW system. The gravel CW system harbored higher bacterial community diversity than the sand and zeolite CW systems. These results suggested the strong impact of substrate type on bacterial community diversity in surface water CW system. In contrast, Huang et al. (2013) reported a weak impact of substrate type on bacterial community diversity in CW treating piggery wastewater.
Bacterial community structure
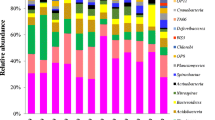
In this study, only a small proportion of sequences retrieved from the three surface water CW systems could not be affiliated with known bacterial phylum or candidate division (accounting for 0.17–6.35 %) (Fig. 1). A total of 13 bacterial phyla and four candidate divisions were frequently identified among 12 wetland samples, including Proteobacteria, Chloroflexi, Bacteroidetes, Acidobacteria, Cyanobacteria, Nitrospirae, Planctomycetes, Actinobacteria, Firmicutes, Chlorobi, Spirochaetae, Gemmatimonadetes, Deferribacteres, OP8, WS3, TA06, and OP3. Among these bacterial phyla, Proteobacteria (accounting for 29.1–62.3 %) was the largest bacterial group in all the wetland samples. Phylum Chloroflexi (4–19.4 %) was the second largest bacterial group in samples WA2, WA3, WA4, WC2, and WC4, and was also dominant in other wetland samples. The relative abundance of Bacteroidetes and Acidobacteria in the wetland samples was 5.5–14.5 % and 2.8–13.3 %, respectively. Cyanobacteria showed high proportion in samples WB3 (18 %) and WB4 (19 %), but became much less abundant in other wetland samples (0.4–6.4 %). The organisms belonging to phyla Nitrospirae, Planctomycetes, Actinobacteria, Firmicutes, and Chlorobi were also abundant in all the wetland samples (>1 %). In addition, the relative abundance of these major bacterial groups usually illustrated a remarkable spatial variation in each CW system.

Comparison of the quantitative contribution of the sequences affiliated with different phyla to the total number of sequences from CW samples. The rare species with relative abundance of less than 0.5 % in each sample are included as others
Figure 2 illustrates the composition of proteobacterial community in each wetland sample. Epsilonproteobacterial organisms only showed very low proportion in wetland samples (below 0.3 %). The wetland proteobacterial communities were mainly composed of Alpha-, Beta-, Gamma-, and Deltaproteobacteria. The samples from wetland WA (1–7.7 %) had much lower proportion of alphaproteobacterial microorganisms than those from the other two wetlands (12–28.2 %). Except for sample WA1 (35.1 %), the samples from wetland WA (10.7–18.6 %) also showed lower proportion of betaproteobacterial species than those from the other two wetlands (26.3–38.6 %). Moreover, betaproteobacterial proportion was generally much higher than alphaproteobacterial proportion in the studied wetland samples. Gammaproteobacterial proportion (10.2–45.5 %) was also usually lower than betaproteobacterial proportion in wetland samples. The samples from wetland WA (32.2–77.6 %) had much higher deltaproteobacterial proportion than those from the other two wetlands (13.1–31.4 %). In addition, the relative abundance of these major proteobacterial classes also illustrated a large spatial variation in each CW system.

Comparison of the quantitative contribution of the sequences affiliated with different proteobacterial classes to the total number of proteobacterial sequences from CW samples. Proteobacterial sequences not related to any known proteobacterial class are included as others
The result of WPGMA clustering based on OTU level illustrated that all of the studied wetland samples fell into three distinctive clusters (Fig. 3). Except for sample WB1, samples from the same wetland were grouped together. This suggested that substrate type could have a profound impact on microbial community structure. However, for the four samples in each wetland, the ones from 0.6 and 0.8 m below the wetland surface were more closely clustered, which showed the strong impact of layer depth on microbial community structure.

WPGMA clustering of CW samples
Microorganisms from Proteobacteria might be involved in the biodegradation or biotransformation of numerous organic compounds in natural or manmade ecosystems (Cheng et al. 2014; Liao et al. 2013a, 2015; Liu et al. 2014a; Zhang et al. 2014). The dominance of Proteobacteria has been found in natural wetlands (Ansola et al. 2014; Liu et al. 2014b), wastewater CW systems (Ansola et al. 2014; Bouali et al. 2014; Huang et al. 2013), and biofilters treating surface water (Feng et al. 2013a; Liao et al. 2013b). Moreover, a previous study using DGGE analysis also suggested the dominance of proteobacterial organisms in a large-scale CW system purifying polluted river water (Zhi et al. 2015). To date, the influential factors governing the structure of wetland proteobacterial community remain unclear. The spatial variation of proteobacterial proportion in surface water CW has never been addressed. In this study, Illumina MiSeq sequencing analysis also showed the dominance of Proteobacteria in surface water CW. However, a remarkable spatial variation of proteobacterial proportion was observed in the sand and zeolite CW systems, while the gravel CW system showed a relatively slight change. In addition, the proportion of major proteobacterial classes also experienced a more profound spatial shift in the sand and zeolite CW systems, compared with the gravel CW system. These results suggested the strong impact of substrate type on proteobacterial community structure in surface water CW system.
So far, the impact of substrate material on CW microbial community structure remains in debate. Silyn-Roberts and Lewis (2004) revealed a very slight difference of CW microbial population structure between biofilms on slag and on greywacke, while Huang et al. (2013) showed the significant impact of substrate type on CW microbial community structure. In this study, the result of WPGMA clustering further confirmed the profound impact of substrate type on the structure of total bacterial community in surface water CW system.
Bacterial genera
There were a total of 33 frequently detected genera in the 12 studied wetland samples. The difference of bacterial community structure among these wetland samples was also evident at genus level (Table 2). For an example, most of the frequently detected genera only showed dominance (with relative abundance of 1 %) in a sole wetland sample. Microorganisms from genera Arenimonas and Thermomonas showed relatively high proportion in samples WB2, WB3, and WB4 (>1 %). Moreover, Planktothrix species dominated in sample WB4 (11.55 %), but became less abundant in other wetland samples. Although Nitrospira was dominant in each wetland sample, its proportion illustrated a large shift (1.89–6.12 %).
Microorganisms from proteobacterial genera Burkholderia, Pseudoxanthomonas, Smithella, Syntrophobacter, Syntrophorhabdus, and Syntrophus are able to aerobically or anaerobically degrade various organic compounds (Arora et al. 2014; Chen et al. 2005, 2013; Gan et al. 2014; Liu et al. 1999; Mouttaki et al. 2008; Nopcharoenkul et al. 2013; Patel et al. 2012; Qiu et al. 2008). Members of genus Flavobacterium (Bacteroidetes) have been linked to biodegradation of a variety of organic pollutants (Nedashkovskaya et al. 2014; Sack et al. 2011; Sun et al. 2011). In addition, microorganisms within genus Bacillus (Firmicutes) can biodegrade a number of organic compounds (Chebbi et al. 2014; Nakkabi et al. 2015; Patowary et al. 2015; Reddy et al. 2014; Xiao et al. 2015; Zhao et al. 2014). Therefore, the dominance of microorganisms from a variety of bacterial genera might play important roles in the removal of organic compounds in the three CV systems and thus contribute to the effective reduction of DOC in polluted river water. Moreover, Nitrospira species (Nitrospirae) are known for their role in nitrite oxidation (Feng et al. 2013b; Liao et al. 2013b). In this study, Nitrospira-like organisms showed high relative abundance in each wetland sample, suggesting the presence of strong nitrifying activity in the three CV systems. This might indirectly explain the effective removal of ammonia in polluted river water.
Conclusions
Illumina high-throughput sequencing indicated that bacterial community diversity and structure varied remarkably in sand and zeolite CW systems treating polluted river water, but slightly in gravel CW system. Substrate type could have a strong impact on bacterial community in surface water CW system. Proteobacteria dominated in all the three CW systems. Microorganisms from a variety of bacterial genera might contribute to the effective reduction of DOC. Moreover, Nitrospira illustrated high relative abundance in each CW system.
References
Ansola G, Arroyo P, de Miera LES (2014) Characterisation of the soil bacterial community structure and composition of natural and constructed wetlands. Sci Total Environ 473:63–71
Arora PK, Srivastava A, Singh VP (2014) Novel degradation pathway of 4-chloro-2-aminophenol via 4-chlorocatechol in Burkholderia sp. RKJ 800. Environ Sci Pollut Res 21:2298–2304
Arroyo P, Ansola G, de Miera LES (2013) Effects of substrate, vegetation and flow on arsenic and zinc removal efficiency and microbial diversity in constructed wetlands. Ecol Eng 51:95–103
Arroyo P, de Miera LES, Ansola G (2015) Influence of environmental variables on the structure and composition of soil bacterial communities in natural and constructed wetlands. Sci Total Environ 506:380–390
Bouali M, Zrafi I, Bakhrouf A, Chaussonnerie S, Sghir A (2014) Bacterial structure and spatiotemporal distribution in a horizontal subsurface flow constructed wetland. Appl Microbiol Biotechnol 98:3191–3203
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108:4516–4522
Chang JJ, Wu SQ, Liang K, Wu ZB, Liang W (2015) Comparative study of microbial community structure in integrated vertical-flow constructed wetlands for treatment of domestic and nitrified wastewaters. Environ Sci Pollut Res 22:3518–3527
Chebbi A, Mnif S, Mhiri N, Jlaiel L, Sayadi S, Chamkha M (2014) A moderately thermophilic and mercaptan-degrading Bacillus licheniformis strain CAN55 isolated from gas-washing wastewaters of the phosphate industry, Tunisia. Int Biodeterior Biodegrad 94:207–213
Chen SY, Liu XL, Dong XZ (2005) Syntrophobacter sulfatireducens sp. nov., a novel syntrophic, propionate-oxidizing bacterium isolated from UASB reactors. Int J Syst Evol Microbiol 55:1319–1324
Chen K, Zhu Q, Qian YG, Song Y, Yao J, Choi MMF (2013) Microcalorimetric investigation of the effect of non-ionic surfactant on biodegradation of pyrene by PAH-degrading bacteria Burkholderia cepacia. Ecotox Environ Safe 98:361–367
Cheng W, Zhang JX, Wang Z, Wang M, Xie SG (2014) Bacterial communities in sediments of a drinking water reservoir. Ann Microbiol 64:875–878
Dzakpasu M, Wang XC, Zheng YC, Ge Y, Xiong JQ, Zhao YQ (2015) Characteristics of nitrogen and phosphorus removal by a surface-flow constructed wetland for polluted river water treatment. Water Sci Technol 71:904–912
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996
Elsayed OF, Maillard E, Vuilleumier S, Imfeld G (2014) Bacterial communities in batch and continuous-flow wetlands treating the herbicide S-metolachlor. Sci Total Environ 499:327–335
Feng S, Chen C, Wang QF, Zhang XJ, Yang ZY, Xie SG (2013a) Characterization of microbial communities in a granular activated carbon-sand dual media filter for drinking water treatment. Int J Environ Sci Technol 10:917–922
Feng S, Chen C, Wang QF, Yang ZY, Zhang XJ, Xie SG (2013b) Microbial community in a full-scale drinking water biosand filter. J Environ Biol 34:321–324
Gan L, Cheng Y, Palanisami T, Chen ZL, Megharaj M, Naidu R (2014) Pathways of reductive degradation of crystal violet in wastewater using free-strain Burkholderia vietnamiensis C09V. Environ Sci Pollut Res 21:10339–10348
Huang X, Liu CX, Gao CF, Wang Z, Zhu GF, Liu L, Lin GX (2013) Comparison of nutrient removal and bacterial communities between natural zeolite-based and volcanic rock-based vertical flow constructed wetlands treating piggery wastewater. Desalin Water Treat 51:4379–4389
Iasur-Kruh L, Hadar Y, Milstein D, Gasith A, Minz D (2010) Microbial population and activity in wetland microcosms constructed for improving treated municipal wastewater. Microb Ecol 59:700–709
Ji GD, Wang RJ, Zhi W, Liu XX, Kong YP, Tan YF (2012) Distribution patterns of denitrification functional genes and microbial floras in multimedia constructed wetlands. Ecol Eng 44:179–188
Kim M, Kim WS, Tripathi BM, Adams J (2014) Distinct bacterial communities dominate tropical and temperate zone leaf litter. Microb Ecol 67:837–848
Liao XB, Chen C, Wang Z, Wan R, Chang CH, Zhang XJ, Xie SG (2013a) Changes of biomass and bacterial communities in biological activated carbon filters for drinking water treatment. Process Biochem 48:312–316
Liao XB, Chen C, Wang Z, Wan R, Chang CH, Zhang XJ, Xie SG (2013b) Pyrosequencing analysis of bacterial communities in drinking water biofilters receiving influents of different types. Process Biochem 48:703–707
Liao XB, Chen C, Zhang JX, Dai Y, Zhang XJ, Xie SG (2015) Operational performance, biomass and microbial community structure: impacts of backwashing on drinking water biofilter. Environ Sci Pollut Res 22:546–554
Ligi T, Oopkaup K, Truu M, Preem JK, Nolvak H, Mitsch WJ, Mander U, Truu J (2014) Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing. Ecol Eng 72:56–66
Liu YT, Balkwill DL, Aldrich HC, Drake GR, Boone DR (1999) Characterization of the anaerobic propioate-degrading syntrophs Smithella propionica gen. nov., sp. nov. and Syntrophobacter wolinii. Int J Syst Bacteriol 49:545–556
Liu WL, Pan XC, Zhang CB, Wang J (2013) Characterization of substrate microbial communities in vertical flow mesocosms as impacted by both planting pattern and species richness. Res Microbiol 164:941–948
Liu Y, Zhang JX, Zhao L, Zhang XL, Xie SG (2014a) Spatial distribution of bacterial communities in high-altitude freshwater wetland sediment. Limnology 15:249–256
Liu JJ, Zheng CY, Song CC, Guo SD, Liu XB, Wang GH (2014b) Conversion from natural wetlands to paddy field alters the composition of soil bacterial communities in Sanjiang Plain, Northeast China. Ann Microbiol 64:1395–1403
Menon R, Jackson CR, Holland MM (2013) The influence of vegetation on microbial enzyme activity and bacterial community structure in freshwater constructed wetland sediments. Wetlands 33:365–378
Morato J, Codony F, Sanchez O, Perez LM, Garcia J, Mas J (2014) Key design factors affecting microbial community composition and pathogenic organism removal in horizontal subsurface flow constructed wetlands. Sci Total Environ 481:81–89
Mouttaki H, Nanny MA, McInerney MJ (2008) Use of benzoate as an electron acceptor by Syntrophus aciditrophicus grown in pure culture with crotonate. Environ Microbiol 10:3265–3274
Mulling BTM, Soeter AM, van der Geest HG, Admiraal W (2014) Changes in the planktonic microbial community during residence in a surface flow constructed wetland used for tertiary wastewater treatment. Sci Total Environ 466:881–887
Nakkabi A, Sadiki M, Fahim M, Ittobane N, Koraichi SI, Barkai H, El Abed S (2015) Biodegradation of poly(ester urethane)s by Bacillus subtilis. Int J Environ Res 9:157–162
Nedashkovskaya OI, Balabanova LA, Zhukova NV, Kim SJ, Bakunina IY, Rhee SK (2014) Flavobacterium ahnfeltiae sp. nov., a new marine polysaccharide-degrading bacterium isolated from a Pacific red alga. Arch Microbiol 196:745–752
Nopcharoenkul W, Netsakulnee P, Pinyakong O (2013) Diesel oil removal by immobilized Pseudoxanthomonas sp. RN402. Biodegradation 24:387–397
Patel V, Cheturvedula S, Madamwar D (2012) Phenanthrene degradation by Pseudoxanthomonas sp. DMVP2 isolated from hydrocarbon contaminated sediment of Amlakhadi canal, Gujarat, India. J Hazard Mater 201:43–51
Patowary K, Saikia RR, Kalita MC, Deka S (2015) Degradation of polyaromatic hydrocarbons employing biosurfactant-producing Bacillus pumilus KS2. Ann Microbiol 65:225–234
Qiu YL, Hanada S, Ohashi A, Harada H, Kamagata Y, Sekiguchi Y (2008) Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl Environ Microbiol 74:2051–2058
Ramond JB, Welz PJ, Cowan DA, Burton SG (2012) Microbial community structure stability, a key parameter in monitoring the development of constructed wetland mesocosms during start-up. Res Microbiol 163:28–35
Reddy GVS, Reddy BR, Tlou MG (2014) Biodegradation of 2-hydroxyquinoxaline (2-HQ) by Bacillus sp. J Hazard Mater 278:100–107
Sack ELW, van der Wielen PWJJ, van der Kooij D (2011) Flavobacterium johnsoniae as a model organism for characterizing biopolymer utilization in oligotrophic freshwater environments. Appl Environ Microbiol 77:6931–6938
Shi YW, Yang HM, Zhang T, Sun J, Lou K (2014) Illumina-based analysis of endophytic bacterial diversity and space-time dynamics in sugar beet on the north slope of Tianshan mountain. Appl Microbiol Biotechnol 98:6375–6385
Silyn-Roberts G, Lewis G (2004) Substrata effects on bacterial biofilm development in a subsurface flow dairy waste treatment wetland. Water Sci Technol 48:261–269
Sun BZ, Ko K, Ramsay JA (2011) Biodegradation of 1,4-dioxane by a Flavobacterium. Biodegradation 22:651–659
Sun M, Xiao TF, Ning ZP, Xiao EZ, Sun WM (2015) Microbial community analysis in rice paddy soils irrigated by acid mine drainage contaminated water. Appl Microbiol Biotechnol 99:2911–2922
Tu YT, Chiang PC, Yang J, Chen SH, Kao CM (2014) Application of a constructed wetland system for polluted stream remediation. J Hydrol 510:70–78
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Wang Z, Yang YY, Dai Y, Xie SG (2015a) Anaerobic biodegradation of nonylphenol in river sediment under nitrate- or sulfate-reducing conditions and associated bacterial community. J Hazard Mater 286:306–314
Wang Z, Yang YY, He T, Xie SG (2015b) Change of microbial community structure and functional gene abundance in nonylphenol-degrading sediment. Appl Microbiol Biotechnol 99:3259–3268
Wu HT, Zhang JX, Mi ZL, Xie SG, Chen C, Zhang XJ (2015) Biofilm bacterial communities in urban drinking water distribution systems transporting waters with different purification strategies. Appl Microbiol Biotechnol 99:1947–1955
Xiao Y, Chen SH, Gao YQ, Hu W, Hu MY, Zhong GH (2015) Isolation of a novel beta-cypermethrin degrading strain Bacillus subtilis BSF01 and its biodegradation pathway. Appl Microbiol Biotechnol 99:2849–2859
Xiong YJ, Peng SZ, Luo YF, Xu JZ, Yang SH (2015) A paddy eco-ditch and wetland system to reduce non-point source pollution from rice-based production system while maintaining water use efficiency. Environ Sci Pollut Res 22:4406–4417
Yang Y, Yu K, Xia Y, Lau FTK, Tang DTW, Fung WC, Fang HHP, Zhang T (2014) Metagenomic analysis of sludge from full-scale anaerobic digesters operated in municipal wastewater treatment plants. Appl Microbiol Biotechnol 98:5709–5718
Yang YY, Wang Z, He T, Dai Y, Xie SG (2015) Sediment bacterial communities associated with anaerobic biodegradation of bisphenol A. Microb Ecol 70:97–104
Zhang JX, Zhang XL, Liu Y, Xie SG, Liu YG (2014) Bacterioplankton communities in a high-altitude freshwater wetland. Ann Microbiol 64:1405–1411
Zhang JX, Yang YY, Zhao L, Li YZ, Xie SG, Liu Y (2015) Distribution of sediment bacterial and archaeal communities in plateau freshwater lakes. Appl Microbiol Biotechnol 99:3291–3302
Zhao YJ, Li JH, Wang ZF, Yan C, Wang SB, Zhang JB (2012) Influence of the plant development on microbial diversity of vertical-flow constructed wetlands. Biochem Syst Ecol 44:4–12
Zhao M, Sun PF, Du LN, Wang G, Jia XM, Zhao YH (2014) Biodegradation of methyl red by Bacillus sp. strain UN2: decolorization capacity, metabolites characterization, and enzyme analysis. Environ Sci Pollut Res 21:6136–6145
Zhi EQ, Song YH, Duan L, Yu HB, Peng JF (2015) Spatial distribution and diversity of microbial community in large-scale constructed wetland of the Liao River Conservation Area. Environ Earth Sci 73:5085–5094
Zhi W, Ji GD (2014) Quantitative response relationships between nitrogen transformation rates and nitrogen functional genes in a tidal flow constructed wetland under C/N ratio constraints. Water Res 64:32–41
Zhong F, Wu J, Dai YR, Yang LH, Zhang ZH, Cheng SP, Zhang Q (2015) Bacterial community analysis by PCR-DGGE and 454-pyrosequencing of horizontal subsurface flow constructed wetlands with front aeration. Appl Microbiol Biotechnol 99:1499–1512
Ethical statement
No conflict of interest exists in this manuscript. The work has not been published previously and not under consideration for publication elsewhere.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article

Cite this article
Guan, W., Yin, M., He, T. et al. Influence of substrate type on microbial community structure in vertical-flow constructed wetland treating polluted river water. Environ Sci Pollut Res 22, 16202–16209 (2015). https://doi.org/10.1007/s11356-015-5160-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5160-9