
Abstract
Introduction
Although it is still at a very early stage compared to its mass spectrometry (MS) counterpart, proton nuclear magnetic resonance (NMR) lipidomics is worth being investigated as an original and complementary solution for lipidomics. Dedicated sample preparation protocols and adapted data acquisition methods have to be developed to set up an NMR lipidomics workflow; in particular, the considerable overlap observed for lipid signals on 1D spectra may hamper its applicability.
Objectives
The study describes the development of a complete proton NMR lipidomics workflow for application to serum fingerprinting. It includes the assessment of fast 2D NMR strategies, which, besides reducing signal overlap by spreading the signals along a second dimension, offer compatibility with the high-throughput requirements of food quality characterization.
Method
The robustness of the developed sample preparation protocol is assessed in terms of repeatability and ability to provide informative fingerprints; further, different NMR acquisition schemes—including classical 1D, fast 2D based on non-uniform sampling or ultrafast schemes—are evaluated and compared. Finally, as a proof of concept, the developed workflow is applied to characterize lipid profiles disruption in serum from β-agonists diet fed pigs.
Results
Our results show the ability of the workflow to discriminate efficiently sample groups based on their lipidic profile, while using fast 2D NMR methods in an automated acquisition framework.
Conclusion
This work demonstrates the potential of fast multidimensional 1H NMR—suited with an appropriate sample preparation—for lipidomics fingerprinting as well as its applicability to address chemical food safety issues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Over the last decade, the growth of lipidomics, defined as “the full characterization of lipid molecular species and of their biological roles with respect to expression of proteins involved in lipid metabolism and function, including gene regulation” (Spener et al. 2003), has been noteworthy. It encountered a revival of research works thanks to recent advances in both analytical chemistry and data analysis, which greatly accelerated progresses in the field. Lipidomics is now considered a relevant approach for addressing a broad range of research questions, as attested by the large number of review articles dedicated to the topic (Khalil et al. 2010; Cajka and Fiehn 2014; Hyotylainen et al. 2017; Li et al. 2014; Wenk 2005, 2010; Yang and Han 2016). The applications of this field of research are diverse and include disease biomarker discovery, drug development, drug safety assessment, nutrition or plant research (Zhao et al. 2014). Some studies also showed its potential as a tool for the assessment of food quality; authenticity applications in particular have been widely reported (Li et al. 2017). In chemical food safety, lipidomics-based strategies are also relevant, both at the risk analysis and management steps, for instance for the control of forbidden growth promoting agents in livestock (Nzoughet et al. 2015). It is worth noting that most of the reported lipidomics studies use mass spectrometry (MS) as a fingerprinting analytical strategy, often hyphenated to liquid chromatography. Although MS allows sensitive measurement of hundreds of species in a single scan (Veenstra 2012), it is a destructive technique, which suffers from the various capabilities of ionization between lipid species. Nuclear magnetic resonance (NMR), in contrast, does not induce degradation of the sample upon analysis and is a highly reproducible and directly quantitative technique in spite of its lower sensitivity. NMR is therefore a very promising tool for lipidomics, as illustrated by a recent perspective on the subject (Li et al. 2017). Moreover, covering the wide range of lipid species with one single untargeted MS method is difficult (Lee and Yokomizo 2018) and NMR lipidomics could be a useful complementary strategy for identifying the discriminant lipid classes.
However, even though a few studies reported the use of 1H NMR for lipidomics (Beger et al. 2006; Ekman et al. 2009; Fernando et al. 2011), this technique is still minor in the field (Cajka and Fiehn 2014; Li et al. 2014). The reasons for this lack of popularity are diverse. First of all, 1D 1H NMR spectra suffer from severe peak overlaps, which hinder the accurate determination of peak areas (Giraudeau 2017). Heteronuclear NMR such as 13C NMR is considered an interesting alternative as it offers a better resolution of the NMR signals, thanks to the large bandwidth involved. Indeed, 13C NMR has been used for decades in the analysis of lipids, for instance to determine the composition of fatty acids in food samples (Mavromoustakos et al. 1997; Vlahov 1997). Nevertheless, such method suffers from a poor sensitivity compared to 1H, due to the low abundance of 13C (1.1% at natural abundance) and to the low gyromagnetic ratio of this nucleus—although the latter can be circumvented by indirect detection methods (Merchak et al. 2017). Interestingly, 31P NMR has also been reported for lipidomics (Li et al. 2017) but does not allow the analysis of global lipid fingerprint, as it focuses on the determination of phospholipids.
Alternatively, 2D 1H NMR is an appealing solution in order to obtain a better resolution of the lipid signals without sacrificing the sensitivity, as it allows to spread the signals on a 2D plane. Such experiments are however difficult to apply to lipidomics, as they involve long experiments −up to several hours of acquisition− which do not meet the high-throughput requirements associated with omics approaches. Moreover, the absolute quantification is not straightforward and thus requires specific and time-consuming acquisition schemes or calibration procedures (Giraudeau 2014).
Fortunately, fast 2D NMR approaches have emerged to tackle the issue of acquisition time (Rouger et al. 2017). Two of these approaches, ultrafast (UF) NMR and non-uniform sampling (NUS) have already shown great potential and usefulness in the field of metabolomics (Marchand et al. 2017), either used for fingerprinting (Le Guennec et al. 2014) or associated with a calibration procedure for targeted analyses (Jézéquel et al. 2015) when absolute quantification is necessary.
Based on such developments, fast 2D NMR could also be relevant within NMR-based lipidomics workflows. However, in order to use such strategies on a routine basis, there is a need for the development and testing of a suitable and comprehensive workflow, including a repeatable sample preparation protocol and an efficient fingerprinting method. In this perspective, we have set up a complete strategy for serum lipidomics. First a sample preparation procedure was developed and assessed in terms of repeatability, a critical characteristic for the ultimate comparison of the samples. Then two different fast 2D 1H NMR techniques for untargeted lipidomics fingerprinting were experimented and their performances were compared to 1D 1H NMR. Finally, to evaluate the capacity of the developed approach to address key challenges in NMR lipidomics—namely exploitability, wealth of information and relevance with regard to research question raised—we applied the protocol to a current chemical food safety issue: the detection of forbidden vet drugs administration in livestock. In that context and as a proof of concept, serum samples from ractopamine diet fed pigs have been characterized and their lipids fingerprints compared to control ones with the objective of highlighting specific patterns. Such a set of serum samples (n > 40) was obtained in a well-controlled animal experiment which already enabled reporting specific patterns when investigating the polar fraction of the metabolome by MS-metabolomics (Peng et al. 2017). Subsequent data processing and analysis enabled describing specific lipidomics patterns upon NMR fingerprinting associated to ractopamine treated animals, as could be expected upon the use of such growth promoter (Guitton et al. 2017).
2 Materials and methods
The detailed experimental protocol (sample preparation, NMR data acquisition and processing, data analysis) is provided as Supplementary Material. We will focus here on the most critical points of the workflow.
2.1 Animal experiment/samples
Blood samples were collected from an ethically approved experiment described elsewhere (Peng et al. 2017) and involving ten four-month-old female pigs randomly divided in control and treated groups, the latter one being daily exposed to ractopamine hydrochloride (Sigma Aldrich) through feed (10 ppm). Four QC samples consisting of all the collected samples pooled in identical quantities and mixed together, were prepared. All serum samples were aliquoted and stored at − 20 °C before analysis. A schematic of this design can be found in Supplementary Fig. 3.
For all preliminary sample preparation optimization steps, a serum mix constituted from routine pig blood tests was used as matrix reference.
2.2 Solvents, chemicals
Details on the solvent and chemicals used for extraction/analysis can be found in Supplementary Material.
2.3 Sample preparation
The lipidic fraction was obtained from serum (sample size 300 µL) according to a modified Bligh and Dyer extraction, inspired from Nzoughet (Nzoughet et al. 2015). The final extracts were suspended in 700 µL of CDCl3 containing 0.3 mmol/L of dimethylsulfone (DMSO2) as an internal standard.
2.4 NMR analysis
All the spectra were automatically recorded using IconNMR (Bruker Biospin) on a 16.4 T Bruker Avance-III HD spectrometer operating at a 1H frequency of 700.13 MHz, equipped with an inverse 1H/13C/15N/2H cryogenically cooled probe. The sample temperature was set at 298 K and a SampleJet auto-sampler set at 277 K, requiring 4 inches long NMR tubes in 96-well plates was used. 1D 1H, 2D 1H NUS ZQF-TOCSY and 2D 1H UF COSY spectra were separately acquired. Acquisition and processing parameters are detailed in Supplementary Material.
3 Results and discussion
3.1 Sample preparation
As described in Fig. 1, the first step of the lipidomics workflow aims at extracting the lipidic fraction of the samples in a repeatable way so that they can eventually be compared, while eliminating interfering signals such as those from proteins. Therefore, it is crucial to check the repeatability of this step and the apparent composition of the resulting fraction, via NMR spectra observation. To this aim, the same reference serum mix was extracted four different times, using the modified Bligh and Dyer protocol described in Supplementary Material. The four NMR tubes were thus submitted to 1D 1H analysis (see Supplementary Material for the detailed parameters of acquisition, processing and integration) in identical conditions. Since 1D NMR is considered repeatable at ca. 1% for signal-to-noise ratio (SNR) values higher than 50 (Barding et al. 2012; Malz 2008), any variation observed across the four resulting NMR spectra above this value is mainly expected to reflect the repeatability of the preparation step, provided that the observed signals are intense enough. The spectra (Supplementary Fig. 1, top) obtained for these samples showed a rich fingerprint with characteristic signals mainly originating from cholesterol, glycerol backbone and fatty acyl chains, therefore attesting for the lipidic rich fraction obtained. The assignment of the 1D NMR signals, achieved both from comparison with literature data (Ekman et al. 2009; Fernando et al. 2011; Jayalakshmi et al. 2011) and the analysis of various lipid standards, is available in Supplementary Material (Supplementary Fig. 2). The repeatability of the extraction protocol was evaluated through the integration of the main observable NMR signals, by drawing large buckets in order to avoid “cutting” overlapping signals which could eventually account for the observed variability. This was followed by normalization on the total sum for each spectrum and calculation of the coefficients of variation (CVs) across the different replicates for each of these normalized areas (Supplementary Fig. 1, bottom). All resulting CV values were observed below 9%, with only two regions respectively around 3.5 and 1.9 ppm presenting variations above 4%. The value observed for the 3.5 ppm (labeled “k” in Supplementary Material) region is probably due to the low SNR of the associated signal, which accentuates the measurement error. For the 1.9 ppm region (labeled “f”), the high CV can be attributed to the broad water signal around 1.6 ppm, caused by an unfortunate water residue in the CDCl3 bottle used for this experiment. Even if this particular signal was voluntarily discarded for the bucketing, its broad base overlaps the signal integrated in the 1.9 ppm region and therefore influences the CV of this bucketing region. Consequently, a new CDCl3 bottle was used for all subsequent experiments of the study resulting in no observable water signal thereafter. Apart from those particular cases, the CV values can be deemed as satisfying as they are consistent with repeatability values from the literature. For instance, Pellegrino et al. (2014) tested various methods for serum lipids by LC–MS and obtained optimal values (ca. 3% relative standard deviation). Therefore, the proposed extraction method ensures a sufficient repeatability to compare NMR signals, it is also reasonably simple and can be used for high-throughput purposes. However, as both the serum and solvent volumes involved are relatively high compared with an extraction dedicated to MS, particular care is necessary to take the organic phase, without drawing part of the thicker protein layer. Moreover, the stability of the extracts has been verified and resulted in no change in the apparent lipidic fingerprint (superimposed 1D NMR spectra at 128 scans) neither when stored at 277 K for one week or stored at 253 K for up to 3 weeks; thus validating the use of a 277 K autosampler and the storage conditions (253 K) of the NMR tubes used between analysis batches. Consequently, we further implemented this protocol to a set of samples selected for its relevance in the present lipidomics context.

Schematic of the developed workflow for 1H NMR lipidomics
3.2 NMR fingerprinting
For NMR lipidomics, a broad range of 1D and 2D pulse sequences are of potential relevance (Barding et al. 2012; Marchand et al. 2017). 1D 13C NMR was initially considered, either through direct detection or polarization transfer experiments, because of the better natural spectral resolution of this nucleus compared to 1H. Unfortunately, because of limited sample availability, 13C NMR spectra of the serum lipid extract at natural abundance could not be recorded with enough sensitivity in a reasonable time. For similar reasons, the use of 2D heteronuclear pulse sequences such as heteronuclear single-quantum correlation (HSQC) was also discarded.
Therefore, we focused on 1H NMR spectroscopy for optimal sensitivity, testing three different possibilities. Moreover, in order to keep the workflow compatible with high-throughput expectations, an auto-sampler was employed, which can automatically transfer the NMR tubes into the magnet and allows an automatic setting of the shims as described in Supplementary Material. Such procedure allows optimal shim adjustment while keeping limited the human intervention throughout analysis.
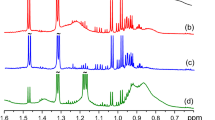
Firstly, the classical 1H 1D pulse-acquire sequence was selected because of its recognized high repeatability and reproducibility, as detailed above. However, when lipid extracts are analyzed with this method, significant overlaps are observed between the signals, as can be observed in Fig. 2a.

1H NMR sequences used for lipidomics fingerprinting and associated spectra from pig serum extract. a 1D pulse-acquire. b 2D ZQF-TOCSY. c Interleaved multi-scan UF COSY. Spectra recorded at 700 MHz with a cryogenically cooled probe
To increase the resolving power without sacrificing the analysis duration, the use of fast 2D NMR approaches was tested. While many fast 2D NMR methods have been described in the literature (Rouger et al. 2017), here this paper focuses on those whose repeatability has already been studied (Le Guennec et al. 2012; Martineau et al. 2013) and whose potential for metabolomics has already been demonstrated, e.g. NUS and ultrafast (Jézéquel et al. 2015; Le Guennec et al. 2014). The objective of the present work was not to propose new method optimizations, but rather to evaluate the potential of recently published approaches in the conditions that were previously optimized and published in methodological papers. In 2D NMR, as opposed to 1D, the coefficient of proportionality between the NMR signal (peak volumes) and the analyte concentration depends on numerous parameters such as coupling constants, relaxation times or pulse sequence delays, arising from the multi-pulse nature of 2D NMR experiments. Nevertheless, each individual signal intensity remains directly proportional to the analyte concentration (Giraudeau 2014). In an untargeted lipidomics framework, this feature ensures the validity of the comparison of a signal from samples of a similar nature. In addition, it has been shown that such 2D spectra used for relative quantification do not need to be recorded with full relaxation between scans, since longitudinal relaxation is only one among numerous factors impacting the peak volume (Giraudeau 2014). This choice relies on the assumption that relaxation times do not vary significantly between samples of the same nature with small concentration variations. This explains the choice of a short recovery time (4.9 s) to remain compatible with rapid analysis.
The first fast 2D NMR approach used in the proposed workflow is the ZQF-TOCSY, acquired with a non-uniform sampling (NUS) scheme. The resulting lipid extract spectra (see Fig. 2b), with 50% NUS, show an important number of signals while keeping a reasonable analysis time of 1 h 47 min (A fully annotated spectrum can be found in Supplementary Fig. 2c). NUS, depending on the way it is used, either allows saving time for a 2D experiment or multiplying the number of points in the indirect dimension, thus enhancing spectral resolution. Le Guennec et al. demonstrated the potential of this approach, in the case of the ZQF-TOCSY pulse sequence, for homonuclear 2D NMR metabolomics (Le Guennec et al. 2014). The choice of the ZQF-TOCSY pulse sequence was motivated by the clean in-phase resulting lineshapes and the high number of observable correlations, as illustrated in Fig. 2b. As shown by Le Guennec et al., a 50% level of NUS (resulting in an overall experiment time divided by two) is the optimal choice for ZQF-TOCSY on complex mixtures; higher levels of NUS would result in reconstruction artefacts that could alter the subsequent data extraction and analysis (Le Guennec et al. 2015).
The second fast 2D approach evaluated in this paper is ultrafast 2D NMR, with a hybrid multi-scan experiment based on the UF COSY sequence. While such experiment has already been used in a metabolomics context (Jézéquel et al. 2015; Le Guennec et al. 2014), this is, to our knowledge, the first time that such an approach is used for lipidomics purposes and the first time it is applied in an automatic way, using an auto-sampler. While UF 2D NMR allows the acquisition of a complete 2D NMR spectrum within a single scan, the sensitivity of sub-second experiments is not suitable for complex samples with realistic concentrations. However, it has been shown that a multi-scan experiment based on UF 2D NMR offers an appealing alternative to conventional NMR for typical experiment durations below 30 min (Le Guennec et al. 2012). In these conditions, even if they suffer from the need to compromise between resolution, spectral width and sensitivity (Akoka and Giraudeau 2015), such hybrid experiments can offer a much higher repeatability than conventional 2D NMR due to their better immunity towards spectrometer instabilities. Indeed, UF spectra are not affected by t1 noise contrary to their conventional counterparts, and this is a significant advantage for samples with large dynamic ranges (Pathan et al. 2011). Here, we used the UF version of the COSY pulse sequence. Although less information is obtained with COSY sequence compared to ZQF-TOCSY, UF COSY has a much higher sensitivity compared to UF TOCSY—the latter being hampered by the effect of molecular diffusion during the spin-lock period. Indeed, hybrid COSY spectra based on UF spectroscopy yield clean and rich spectra (see Fig. 2c) free of t1 noise, which facilitates the bucketing step (A fully annotated spectrum can be found in Supplementary Fig. 2d). Moreover, the analysis duration (26 min in the present work, corresponding to the maximum allowed for UF experiments regarding hardware considerations) is much reduced compared to conventional 2D NMR, making such sequence precious for high-throughput applications.
3.3 Application of the workflow to food safety issues
Once this robust workflow was set up, it was applied to study the effect of ractopamine in pigs, and in particular to assess lipids profile disruption in serum upon such treatment. Ractopamine is a synthetic drug belonging to the β-agonist family that may be used as a growth promoter in finishing pigs to promote leaner meat (Ricks et al. 1984). While being authorized in a number of countries worldwide (n > 25), it has been banned within the EU since the late 80s (Council Directive 88/146/EEC; Council Directive 96/22/EC). To comply efficiently with such ban, new screening techniques, including untargeted approaches, are expected, to detect any potential abuse (Dervilly-Pinel et al. 2012; Pinel et al. 2010). Untargeted approaches have already proved their efficiency in the bovine species for the detection of urinary specific signatures upon β-agonists treatment (Dervilly-Pinel et al. 2015). Regarding the porcine species, untargeted approaches have not been reported so far in that context, except for a study dealing with the investigation of polar metabolome modifications in the serum as a consequence of ractopamine treatment (Peng et al. 2017). However, as the effect of growth promoters, particularly βagonists, on the expression of lipids has been studied and reported for a long time (Dunshea 1993; Dunshea et al. 1998; Soares da Silva Ferreira et al. 2013), it appears that globally investigating lipid profiles through untargeted approach such as lipidomics would be a relevant strategy to generate new knowledge about biological pathways involved. It could also potentially highlight candidate biomarkers that may be further used in a screening context for classification purposes. In this part, results obtained with the three analysis methods (1D, 2D NUS ZQF-TOCSY, 2D UF COSY) are discussed and compared.
3.3.1 1D NMR
After application of the workflow, each 1D spectrum was subjected to manual bucketing. As can be seen in Fig. 2a, much overlap is observed on the 1D lipid spectra, in particular in the 0.8–2.4 ppm region. Therefore, manual bucketing was optimized in order to prevent, as much as possible, splitting a signal in two different buckets, while drawing a large number of buckets in order to get enough variables for the statistical analysis and subsequent data interpretation. Buckets were first drawn on a superposition of all the spectra, to take potential chemical shift variations into account. These buckets were then applied individually to each spectrum and normalized on the total sum, to ensure comparability of the samples, including QC. Here, the QC are used for setting up shim file as well as assessment of the quality of the analysis. Thus, the data quality was checked by calculating the CVs for each variables across all QC samples. As no bucket presented a CVQC > 10%, all the variables (35 in total) were kept for subsequent statistical analysis. Firstly, a principal component analysis (PCA) on all the samples was performed in order to check the quality of the data, paying particular attention to the QC samples. The PCA score plot showed clustered QCs, illustrating the reproducibility of the analysis during the entire experiment (Supplementary Fig. 4). QCs were then removed for subsequent analyses. As can be expected, samples from days 3 and 9 did not appear different on the PCA score plot between control and treated groups (Supplementary Fig. 5), in accordance with previous findings in bovine reporting a slower response of the lipidome to β-agonist actions compared to metabolome one (Nzoughet et al. 2015). Those time points were then discarded for the rest of the analyses; corresponding PCA is shown in Supplementary Fig. 6.
The data were then analyzed with a two-component partial least squares-discriminant analysis (PLS-DA), whose score plot is illustrated in Fig. 3a. The first principal component (PC) accounts for 35.8% whereas the second PC accounts for 13.8%. As can be seen in Fig. 3a, a discrimination is observed along the PC1 between the control group (in red) and the treated group (in green). A very good discrimination between sample groups is obtained (R2 = 0.84 and Q2 = 0.69). The robustness of the model for discriminating sample classes was further validated by a permutation test (n = 100)—available in Supplementary Fig. 7a—and a CV-ANOVA. The latter resulted in a p value of 2.6 × 10−5, thus denoting significance of the model (Eriksson et al. 2008) and the ability of the NMR lipidomics workflow to discriminate sample classes.

PLS-DA score plot of the lipidomics study of ractopamine in pigs from: a 1D data b 2D NUS ZQF-TOCSY data c 2D UF COSY data. Associated R2 and Q2 values are specified within each box. Each dot represents an individual (i.e. a sample) and is labeled as PXX/D_YY corresponding to the pig number (P) and the sampling day (D). Green dots correspond to the Treated group whereas red dots correspond to the control group
3.3.2 2D NUS ZQF-TOCSY
The 2D 1H-1H ZQF-TOCSY experiment could potentially improve the quality of the results by spreading the signals into an additional dimension. After processing the spectra, manual bucketing was operated on the 2D peak volumes, by manually drawing rectangular buckets on a 2D contour plot of the spectra. Correlation and diagonal signals were integrated as both contain valuable information; in particular, the information from singlets is only observable on the diagonal. Note that peak overlap was still present in the CH2 region, albeit to a lesser extent than for 1D spectra. Moreover, t1 noise, mainly originating from the very intense CH2 signal of the fatty acyl chains, complicated the bucketing of neighboring signals. Fortunately, as TOCSY provides symmetrical spectra, bucketing could be performed by individually selecting, for each pair of symmetric correlation signals, the one less disturbed by surrounding noise or peak overlap. After normalization on the total sum, a cleaning step similar to the one performed on 1D data was carried out. This step led to the suppression of only 13 variables from the dataset which originally contained 153 variables, thus highlighting the good quality of the original data. After checking the quality of the fingerprints by PCA, a two component PLS-DA was performed, from which the score plot is illustrated in Fig. 3b. The first PC explains 38.1% of the variance whereas the second PC explains 11.0%. Samples from both classes could be separated efficiently by the model, as attested by the associated performances (R2 = 0.84 and Q2 = 0.71), permutation tests (Supplementary Fig. 7b) and the p value (3.8 × 10−4) from the CV-ANOVA. The discrimination performance achieved with this model from 2D spectra is similar to the one achieved with 1D. However, the added value of such 2D fingerprint lies in the additional dimension that reduces signal overlap, leading to a facilitated bucketing step and to the generation of a higher number of variables, which are less affected by peak overlap. This was further confirmed by orthogonal PLS-DA (OPLS-DA), that allow for an easier interpretation of the variable involvement in the discrimination between classes, through loading plot examination. In the associated loading plot from the 1D data, some neighboring overlapping buckets appear close to each other, suggesting similar apparent pattern towards the control/treated status. Figure 4a illustrates such an example where the bucket attributed to the –CH 2 –CH=CH signal from fatty acyls (FA) in both buckets n°14 and 15 in the 1D dataset, is merged with the CH (C12) signal from Cholesterol/Esterified cholesterols (Chol/CholE) in bucket n°14 and overlaps with the neighboring CH (C7) from Chol/CholE (bucket n°13). These three buckets are located close to each other in the associated loading plot from OPLS-DA (see Supplementary Fig. 8a). In such cases, doubt still remains about the real relevance of all variables. It is difficult to assess if their relative positions on the loading plot arise from a genuine similar biological behavior regarding the control/treated status or are the result of overlapping signals. On the contrary, with 2D data, the corresponding correlation signals can resolve such ambiguity, as the peaks are spread along a second dimension. In Fig. 4b from a ZQF-TOCSY spectrum, signals can be separated and integrated according to associated lipid classes. As a consequence, the signal –CH 2 –CH=CH from FA (bucket n°27 in the ZQF-TOCSY dataset) can be integrated in a separated bucket from the signals CH (C7) and CH (C12) from Chol/Chol (bucket n°26). Such integration shows that these two variables actually present opposite behavior towards the control/treated status and are located at opposite sides of the associated OPLS-DA loading plot (Supplementary Fig. 8b). Similar occurrences could also be observed, which can be more generally objectivized through examination of the variables loading values according to their positions on the spectra (Supplementary Fig. 9). The enhanced and better resolved fingerprint provided by 2D NMR therefore offers an increased confidence in the identification of potential biomarkers and hence in the investigation of lipid metabolism pathways. A similar conclusion was reached in the field of 2D NMR metabolomics (Le Guennec et al. 2014). This result therefore confirms the potential of fast 2D 1H NMR for lipidomics applications.

Zooms on specific bucketing regions in spectra from pig serum lipid extracts. a Bucketing from a 1D spectrum. The bucket n°15 contains the –CH 2 –CH=CH signal from FA; the bucket n°14 contains both the –CH 2 –CH=CH signal from FA and CH (C12) from Chol/CholE; the bucket n°13 contains the CH (C7) from Chol/CholE. b Bucketing from a ZQF-TOCSY spectrum. The bucket n°27 contains the –CH 2 –CH=CH signal from FA whereas the bucket n°26 contains the CH (C12) and CH (C7) from Chol/CholE
3.3.3 2D UF COSY
While the ZQF-TOCSY experiment still suffers from the time penalty inherent to conventional 2D NMR acquisitions, the UF COSY allows to record a 2D spectrum in a time comparable to the one used for 1D spectra. Fourty-eight buckets could originally be drawn, resulting in a dataset containing 39 variables after normalization and suppression of buckets with CVQC > 10%. This number is comparable to the 35 variables of the 1D dataset, but in this case 2D buckets suffer from less signal overlap than in 1D; this is mainly due to the spreading of the signals along a second dimension. Again, the 2D UF COSY dataset was submitted to PLS-DA, after data processing. The resulting PLS-DA score plot (Fig. 3c) shows a similar distribution of the samples compared to 1D and 2D NUS ZQF-TOCSY data, with explained variances of 30.1% for PC 1 and 9.1% for PC 2. This model achieved an efficient discrimination of the two sample classes with R2 and Q2 values of 0.82 and 0.60 and with confidence parameters associated to the model (p value from the CV-ANOVA (5.1 × 10−4), permutation tests (Supplementary Fig. 7c). Such performance is similar to the one obtained from 1D and ZQF-TOCSY, while the analysis duration remains as low as 26 min. This result can probably be explained by the high repeatability of UF COSY experiments, as previously described in the literature (Akoka and Giraudeau 2015; Le Guennec et al. 2012). The main advantage related to UF COSY is the resolving of ambiguities in the identification of putative biomarkers thanks to the contribution of the second dimension, as explained above for ZQF-TOCSY, with as an acquisition time comparable to the presented 1D experiment and four times faster than the ZQF-TOCSY NUS approach. These results confirm the potential of this technique for lipidomics applications, considering both the relevance of the information provided as well as rapidity of the process. However, it is important to note that the resolution of the observed signals with UF spectra is lower than the NUS spectra and that the COSY sequence only allows to observe correlations between spins which are coupled to each other, as opposed to ZQF-TOCSY, resulting in fewer variables than the latest. Therefore, when one uses the herein presented workflow for lipidomics applications where sample availability is limited, the choice of the selected acquisition approach strongly depends on the desired requirements. If many variables are needed and the number of samples is limited, ZQF-TOCSY NUS appears as a reasonable option. Alternatively, in the case of large-scale applications, where high-throughput is most important, UF COSY would appear as the most appropriate choice.
3.3.4 Biological effects of ractopamine
Although the full biological interpretation of our data was not the primary aim of this work, a preliminary interpretation of the effect of Ractopamine on serum lipid profiles in pigs has been carried out, based on the results obtained with the developed workflow. The link between ractopamine and lipid metabolism has been investigated decades ago in farm animals (Dunshea 1993) and a reduction in the deposition of adipose tissue in the carcass of pigs fed diets containing ractopamine is commonly reported. Such an effect is hypothesized to occur through either reduction in lipogenesis and/or increase in lipolysis and is expected to be reflected in blood through characterization of the lipid profiles disruption, as targeted in the present work.
As similar lipids exhibit the same signals on various zones of 1H NMR spectra, the variables obtained after the bucketing step are rarely unique to one particular lipid. Therefore, this technique does not allow the identification of specific lipid species but rather generally informs on chemical groups or lipid classes, from which qualitative and semi-quantitative variations in observed signals may be the basis for biological interpretation. Consequently, the results from the NUS ZQF-TOCSY dataset were explored, as it contains the highest amount of variables and spectral resolution, facilitating subsequent interpretation. From the PLS-DA described above, the main variables responsible for class separation were extracted using the associated loading plot. Each of these variables was submitted to a Wilcoxon test in order to confirm its discriminative ability, for each time point of this dataset i.e. for days 16, 18, 23 and 29. Of particular interest for class separation were the variables related to Chol/CholE, phospholipids (PL) and triacylglycerols (TAG) signals. Boxplots for each of these lipid classes can be found in Supplementary Fig. 10. Chol/CholE were found to be significant for discrimination between the control and the treated group for day-18, day-29 and marginally significant (p < 0.07) for day-23. PL were significant for day-29 and marginally significant for day-23 whereas TAG were significant for day-18 and marginally significant for day-23. PL and TAG both presented higher levels in samples from treated population, associated to lower Chol/CholE levels compared to controlled population. In a recent review, da Sylva Ferreira et al. (Soares da Silva Ferreira et al. 2013) went through published studies to understand the in vivo mechanism behind the reduction of adipose tissue in carcass of ractopamine-treated animals. In their work, they discussed the two hypothesized pathways in respect of the literature: the reduction in lipogenesis and/or the increase in lipolysis; considering the evaluation of different parameters such as enzymatic activities or quantification of non-esterified fatty acids (NEFA) in porcine blood samples. They concluded on a predominant inhibition of lipogenesis to explain the reduction of lipid deposition on the carcass, rather than a positive effect on lipolysis. Indeed, ractopamine administration is generally not associated with an increase of serum NEFA.
Our results suggest lower concentrations of free and esterified cholesterol in pig serum upon the use of ractopamine. The TAG seem to be affected with higher levels, yet this was only observed as significant on a short time window (day 18, day 23). The PL were affected in the same way as TAG, albeit at a later stage. The disrupted PL profiles observed in the present study are in accordance with previous observations on muscle where diacylglycerophosphoethanolamine, phosphatidylinositol and sphingomyelin have been associated with ractopamine administration to pigs (Guitton et al. 2017). These observations provide complementary and yet undescribed pieces of information that may contribute to a better understanding of lipid metabolism modifications as a consequence of β-agonist exposure. The full biological investigation of the metabolic and lipidic consequences of β-agonist exposure will be the purpose of future research.
4 Conclusion
In this paper, the development of a robust NMR workflow for untargeted lipidomics, using serum as a matrix, is proposed. First, the repeatability of the sample preparation, using only 300 µL as sample size, was assessed. Our results demonstrate the ability of the protocol to answer lipidomics requirements in terms of reduced analytical variability. Further, the developed approach proposes, for high-throughput purposes, the innovative combination of automated analysis together with fast 1H 2D NMR acquisition schemes while keeping a satisfying resolution of the NMR signals. Afterwards, the whole workflow was successfully applied on serum samples collected in the frame of an animal experiment in which disruption of the seric lipid profile was expected within the treated group of animals involved. The observed results confirmed the impact of β-agonists on lipids as already suggested in previous studies in blood (Dunshea and King 1994), adipose or muscle tissues (Guitton et al. 2017; Reiter et al. 2007). Further biological work is currently ongoing to identify the lipids involved and investigate the biological pathways impacted. Despite these encouraging results, further improvements of the approach are still necessary since several steps of the workflow are still performed manually (sample extraction, data integration). The next step will be dedicated at testing enhanced bucketing approaches, in order to make the best of our data while limiting human intervention, as 2D automatic processing for semi-quantitative approaches emerge in the literature (Puig-Castellvi et al. 2018). Concerning the ractopamine application, our results also showed that further investigations are still necessary for a complete biological understanding. Still, this study highlights the potential of advanced NMR methods for high-throughput lipidomics, thus paving the way towards a better complementarity of NMR and MS in the field. For the analysis of the results and the comparison of the different NMR datasets, the standard PLS-DA was used although the experimental design includes repeated measures on the same animals. For thorough analysis and complete understanding of the biological effects explaining our results, advanced statistical methods suitable for this particular design could be used, such as multilevel methods (Liquet et al. 2012; Westerhuis et al. 2010). Moreover, an appealing perspective is the combination of the described NMR data with MS data, as performed by Marshall et al. for metabolomics (Marshall et al. 2015) in order to optimize the understanding of the effect of Ractopamine.
References
Akoka, S., & Giraudeau, P. (2015). Fast hybrid multi-dimensional NMR methods based on ultrafast 2D NMR. Magnetic Resonance in Chemistry, 53, 986–994.
Barding, G. A. Jr., Salditos, R., & Larive, C. K. (2012). Quantitative NMR for bioanalysis and metabolomics. Analytical and Bioanalytical Chemistry, 404, 1165–1179.
Beger, R. D., Schnackenberg, L. K., Holland, R. D., Li, D., & Dragan, Y. (2006). Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma. Metabolomics, 2, 125–134.
Cajka, T., & Fiehn, O. (2014). Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC Trends in Analytical Chemistry, 61, 192–206.
Council Directive 88/146/EEC Council Directive 88/146/EEC prohibiting the use livestock farming of certain substances having a hormonal action, pp. 16–18.
Council Directive 96/22/EC concerning the prohibition on the use in stockfarming of certain substances having a hormonal or thyrostatic action and of beta-agonists, and repealing directives 81/602/EEC, 88/146/EEC and 88/299/EEC, pp. 3–9.
Dervilly-Pinel, G., Chereau, S., Cesbron, N., Monteau, F., & Le Bizec, B. (2015). LC-HRMS based metabolomics screening model to detect various β-agonists treatments in bovines. Metabolomics, 11, 403–411.
Dervilly-Pinel, G., Courant, F., Chereau, S., Royer, A., Boyard-Kieken, F., Antignac, J., & Le Bizec, B. (2012). Metabolomics in food analysis: Application to the control of forbidden substances. Drug Testing and Analysis, 4, 59–69.
Dunshea, F. R. (1993). Effect of metabolism modifiers on lipid metabolism in the pig. Journal of Animal Science, 71, 1966–1977.
Dunshea, F. R., & King, R. H. (1994). Temporal response of plasma metabolites to ractopamine treatment in the growing pig. Australian Journal of Agricultural Research, 45, 1683–1692.
Dunshea, F. R., Leur, B. J., Tilbrook, A. J., & King, R. H. (1998). Ractopamine increases glucose turnover without affecting lipogenesis in the pig. Australian Journal of Agricultural Research, 49, 1147–1152.
Ekman, D. R., Teng, Q., Villeneuve, D. L., Kahl, M. D., Jensen, K. M., Durhan, E. J., Ankley, G. T., & Collette, T. W. (2009). Profiling lipid metabolites yields unique information on sex- and time-dependent responses of fathead minnows (Pimephales promelas) exposed to 17α-ethynylestradiol. Metabolomics, 5, 22–32.
Eriksson, L., Trygg, J., & Wold, S. (2008). CV-ANOVA for significance testing of PLS and OPLS® models. Journal of Chemometrics, 22, 594–600.
Fernando, H., Bhopale, K. K., Kondraganti, S., Kaphalia, B. S., & Ansari, S., G.A (2011). Lipidomic changes in rat liver after long-term exposure to ethanol. Toxicology and Applied Pharmacology, 255, 127–137.
Giraudeau, P. (2014). Quantitative 2D liquid-state NMR. Magnetic Resonance in Chemistry, 52, 259–272.
Giraudeau, P. (2017). Challenges and perspectives in quantitative NMR. Magnetic Resonance in Chemistry, 55, 61–69.
Guitton, Y., Dervilly-Pinel, G., Jandova, R., Stead, S., Takats, Z., & Le Bizec, B. (2017). Rapid evaporative ionisation mass spectrometry and chemometrics for high-throughput screening of growth promoters in meat producing animals. Food Additives & Contaminants. https://doi.org/10.1080/19440049.2017.1421778.
Hyotylainen, T., Ahonen, L., Poho, P., & Oresic, M. (2017). Lipidomics in biomedical research-practical considerations. Biochimica et Biophysica Acta, 1862, 800–803.
Jayalakshmi, K., Sonkar, K., Behari, A., Kapoor, V. K., & Sinha, N. (2011). Lipid profiling of cancerous and benign gallbladder tissues by 1H NMR spectroscopy. NMR in Biomedicine, 24, 335–342.
Jézéquel, T., Deborde, C., Maucourt, M., Zhendre, V., Moing, A., & Giraudeau, P. (2015). Absolute quantification of metabolites in tomato fruit extracts by fast 2D NMR. Metabolomics, 11, 1231–1242.
Khalil, M. B., Hou, W., Zhou, H., Elisma, F., Swayne, L. A., Blanchard, A. P., Yao, Z., Bennett, S. A., & Figeys, D. (2010). Lipidomics era: Accomplishments and challenges. Mass Spectrometry Reviews, 29, 877–929.
Le Guennec, A., Dumez, J. N., Giraudeau, P., & Caldarelli, S. (2015). Resolution-enhanced 2D NMR of complex mixtures by non-uniform sampling. Magnetic Resonance in Chemistry, 53, 913–920.
Le Guennec, A., Giraudeau, P., & Caldarelli, S. (2014). Evaluation of fast 2D NMR for metabolomics. Analytical Chemistry, 86, 5946–5954.
Le Guennec, A., Tea, I., Antheaume, I., Martineau, E., Charrier, B., Pathan, M., Akoka, S., & Giraudeau, P. (2012). Fast determination of absolute metabolite concentrations by spatially encoded 2D NMR: Application to breast cancer cell extracts. Analytical Chemistry, 84, 10831–10837.
Lee, H.-C., & Yokomizo, T. (2018). Applications of mass spectrometry-based targeted and non-targeted lipidomics. Biochemical and Biophysical Research Communications. https://doi.org/10.1016/j.bbrc.2018.03.081.
Li, J., Vosegaard, T., & Guo, Z. (2017). Applications of nuclear magnetic resonance in lipid analyses: An emerging powerful tool for lipidomics studies. Progress in Lipid Research, 68, 37–56.
Li, M., Yang, L., Bai, Y., & Liu, H. (2014). Analytical methods in lipidomics and their applications. Analytical Chemistry, 86, 161–175.
Liquet, B., Le Cao, K. A., Hocini, H., & Thiebaut, R. (2012). A novel approach for biomarker selection and the integration of repeated measures experiments from two assays. BMC Bioinformatics, 13, 325.
Malz, F. (2008). Quantitative NMR in the solution state NMR A2. In U. Holzgrabe, I. Wawer, B. Diehl (Eds.), NMR spectroscopy in pharmaceutical analysis (pp. 43–62). Amsterdam: Elsevier.
Marchand, J., Martineau, E., Guitton, Y., Dervilly-Pinel, G., & Giraudeau, P. (2017). Multidimensional NMR approaches towards highly resolved, sensitive and high-throughput quantitative metabolomics. Current Opinion in Biotechnology, 43, 49–55.
Marshall, D. D., Lei, S., Worley, B., Huang, Y., Garcia-Garcia, A., Franco, R., Dodds, E. D., & Powers, R. (2015). Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics, 11, 391–402.
Martineau, E., Akoka, S., Boisseau, R., Delanoue, B., & Giraudeau, P. (2013). Fast quantitative 1H–13C two-dimensional NMR with very high precision. Analytical Chemistry, 85, 4777–4783.
Mavromoustakos, T., Zervou, M., Theodoropoulou, E., Panagiotopoulos, D., Bonas, G., Day, M., & Helmis, A. (1997). 13C NMR analysis of the triacylglycerol composition of Greek virgin olive oils. Magnetic Resonance in Chemistry, 35, S3-S7.
Merchak, N., Silvestre, V., Loquet, D., Rizk, T., Akoka, S., & Bejjani, J. (2017). A strategy for simultaneous determination of fatty acid composition, fatty acid position, and position-specific isotope contents in triacylglycerol matrices by (13)C-NMR. Analytical and Bioanalytical Chemistry, 409, 307–315.
Nzoughet, J. K., Gallart-Ayala, H., Biancotto, G., Hennig, K., Dervilly-Pinel, G., & Le Bizec, B. (2015). Hydrophilic interaction (HILIC) and reverse phase liquid chromatography (RPLC)–high resolution MS for characterizing lipids profile disruption in serum of anabolic implanted bovines. Metabolomics, 11, 1884–1895.
Pathan, M., Akoka, S., Tea, I., Charrier, B., & Giraudeau, P. (2011). “Multi-scan single shot” quantitative 2D NMR: A valuable alternative to fast conventional quantitative 2D NMR. Analyst, 136, 3157–3163.
Pellegrino, R. M., Di Veroli, A., Valeri, A., Goracci, L., & Cruciani, G. (2014). LC/MS lipid profiling from human serum: A new method for global lipid extraction. Analytical and Bioanalytical Chemistry, 406, 7937–7948.
Peng, T., Royer, A.-L., Guitton, Y., Le Bizec, B., & Dervilly-Pinel, G. (2017). Serum-based metabolomics characterization of pigs treated with ractopamine. Metabolomics, 13, 77.
Pinel, G., Weigel, S., Antignac, J. P., Mooney, M. H., Elliott, C., Nielen, M. W. F., & Le Bizec, B. (2010). Targeted and untargeted profiling of biological fluids to screen for anabolic practices in cattle. TrAC Trends in Analytical Chemistry, 29, 1269–1280.
Puig-Castellvi, F., Perez, Y., Pina, B., Tauler, R., & Alfonso, I. (2018). Compression of multidimensional NMR spectra allows a faster and more accurate analysis of complex samples. Chemical Communications, 54, 3090–3093.
Reiter, S. S., Halsey, C. H., Stronach, B. M., Bartosh, J. L., Owsley, W. F., & Bergen, W. G. (2007). Lipid metabolism related gene-expression profiling in liver, skeletal muscle and adipose tissue in crossbred Duroc and Pietrain Pigs. Comparative Biochemistry and Physiology Part D: Genomics and Proteomics, 2, 200–206.
Ricks, C. A., Dalrymple, R. H., Baker, P. K., & Ingle, D. L. (1984). Use of a β-agonist to alter fat and muscle deposition in steers1, 2. Journal of Animal Science, 59, 1247–1255.
Rouger, L., Gouilleux, B., & Giraudeau, P. (2017). Fast n-dimensional data acquisition methods A2. In J.C. Lindon, G. E. Tranter & D. W. Koppenaal (Eds.), Encyclopedia of spectroscopy and spectrometry (third edition) (pp. 588–596). Oxford: Academic Press.
Soares da Silva Ferreira, M., Garbossa, P., Oberlender, C. A., Pereira, G., Zangeronimo, L., Vicente, M., de Sousa, R., & Cantarelli, V. (2013). Effect of ractopamine on lipid metabolism in vivo: A systematic review. Brazilian Archives of Biology and Technology, 56, 35–43.
Spener, F., Lagarde, M., Géloên, A., & Record, M. (2003). Editorial: What is lipidomics? European Journal of Lipid Science and Technology, 105, 481–482.
Veenstra, T. D. (2012). Metabolomics: The final frontier? Genome Medicine, 4, 40.
Vlahov, G. (1997). Quantitative 13C NMR method using the DEPT pulse sequence for the detection of olive oil adulteration with soybean oil. Magnetic Resonance in Chemistry, 35, S8–S12.
Wenk, M. R. (2005). The emerging field of lipidomics. Nature Reviews Drug Discovery, 4, 594–610.
Wenk, M. R. (2010). Lipidomics: New tools and applications. Cell, 143, 888–895.
Westerhuis, J. A., van Velzen, E. J., Hoefsloot, H. C., & Smilde, A. K. (2010). Multivariate paired data analysis: Multilevel PLSDA versus OPLSDA. Metabolomics, 6, 119–128.
Yang, K., & Han, X. (2016). Lipidomics: Techniques, applications, and outcomes related to biomedical sciences. Trends in Biochemical Sciences, 41, 954–969.
Zhao, Y. Y., Wu, S. P., Liu, S., Zhang, Y., & Lin, R. C. (2014). Ultra-performance liquid chromatography-mass spectrometry as a sensitive and powerful technology in lipidomic applications. Chemico-Biological Interactions, 220, 181–192.
Acknowledgements
The authors would like to thank the Région Pays de la Loire for funding through the “Recherche-Formation-Innovation: Food 4.2” program (Grant LipidoTool) and the technical platform CORSAIRE.
Author information
Authors and Affiliations
Contributions
GDP, BLB and PG designed research; JM performed research; EM contributed analytic tools; JM, EM, YG, GDP and PG analyzed data; JM, PG and GDP wrote the paper. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research Involving Animal Participants
The animal study was approved by the national Ethical Committee n°6 (Comité n°6 - Ministère de l’Enseignement Supérieur et de la Recherche – Direction Générale pour la Recherche et l’Innovation – Secrétariat « Autorisation de projet » - 1, rue Descartes, 75231 PARIS cedex 5) under agreement 2,015,092,516,084,715/APAFIS 1914 (Protocol Number CRIP-2015-054). The study was implemented at Centre de Recherche et d’Investigation Préclinique-CRIP-ONIRIS- Plate-forme de chirurgie et animaleries expérimentales, Oniris, Nantes, France, under agreement number F.44-271.
Institutional and national guidelines were followed for the animal experimentation, as mentioned in Cerfa N° 51706#02 and N° 14906*02; in particular ARTICLES R. 214-87 to 214-137 from CODE RURAL ET DE LA PÊCHE MARITIME (French Regulation).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Marchand, J., Martineau, E., Guitton, Y. et al. A multidimensional 1H NMR lipidomics workflow to address chemical food safety issues. Metabolomics 14, 60 (2018). https://doi.org/10.1007/s11306-018-1360-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11306-018-1360-x