
Abstract
Metabolomics datasets are commonly acquired by either mass spectrometry (MS) or nuclear magnetic resonance spectroscopy (NMR), despite their fundamental complementarity. In fact, combining MS and NMR datasets greatly improves the coverage of the metabolome and enhances the accuracy of metabolite identification, providing a detailed and high-throughput analysis of metabolic changes due to disease, drug treatment, or a variety of other environmental stimuli. Ideally, a single metabolomics sample would be simultaneously used for both MS and NMR analyses, minimizing the potential for variability between the two datasets. This necessitates the optimization of sample preparation, data collection and data handling protocols to effectively integrate direct-infusion MS data with one-dimensional (1D) 1H NMR spectra. To achieve this goal, we report for the first time the optimization of (i) metabolomics sample preparation for dual analysis by NMR and MS, (ii) high throughput, positive-ion direct infusion electrospray ionization mass spectrometry (DI-ESI–MS) for the analysis of complex metabolite mixtures, and (iii) data handling protocols to simultaneously analyze DI-ESI–MS and 1D 1H NMR spectral data using multiblock bilinear factorizations, namely multiblock principal component analysis (MB-PCA) and multiblock partial least squares (MB-PLS). Finally, we demonstrate the combined use of backscaled loadings, accurate mass measurements and tandem MS experiments to identify metabolites significantly contributing to class separation in MB-PLS-DA scores. We show that integration of NMR and DI-ESI–MS datasets yields a substantial improvement in the analysis of metabolome alterations induced by neurotoxin treatment.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The analysis of metabolomics samples is routinely carried out using either mass spectrometry (MS) (Dettmer et al. 2007) or nuclear magnetic resonance (NMR) spectroscopy (Nicholson et al. 1999). However, NMR and MS have distinct, complementary sets of strengths and limitations. The advantages of NMR for metabolomics include relatively high-throughput, non-destructive data acquisition, minimal sample handling, simple methods for quantitation of metabolite alterations, and redundant spectral information to improve the accuracy of metabolite identification (Pan and Raftery 2007; t’Kindt et al. 2010; Zhang et al. 2013). However, one-dimensional (1D) 1H NMR is limited by low sensitivity (≥1 µM), low information content (~0.02 ppm resolution over a ~10 ppm spectral width) and low dynamic range, all of which reduce the observable set of metabolites. These deficiencies of NMR are strengths of MS (Lenz and Wilson 2007; Pan and Raftery 2007). For instance, MS has a much higher sensitivity compared to NMR, readily measuring concentrations in the nanomolar (nM) range. MS also boasts higher resolution (~103–104) and dynamic range (~103–104). As a result, MS-based metabolomics studies can potentially detect a much greater subset of the metabolome than NMR.
More often than not, MS metabolomics relies on hyphenated analytical platforms, such as GC–MS (Kuehnbaum and Britz-McKibbin 2013) or LC–MS (Crockford et al. 2006), to reduce peak overlap and improve coverage of the metabolome. Peak overlap in the mass spectrum occurs because of the relatively narrow molecular-weight distribution of the metabolome (Kell 2004). Ion suppression is also a significant concern given the complexity and heterogeneity of metabolomics samples. The competition for charge between co-eluting analytes may lead to altered or missing metabolite signals (Metz et al. 2008). While the coupling of a chromatographic separation to MS potentially alleviates these issues, it also increases analysis time and requires additional sample preparation in comparison with NMR.
The extra sample processing required by chromatography may lead to variations in the observed metabolome not relevant to the biological system under study (Canelas et al. 2009). As an example, the chemical derivatization step required by GC–MS may individually bias metabolite concentrations. The derivatization yields may differ for each metabolite, and no derivatizing agent exists that will universally and efficiently label all metabolites in any given biological sample (Kanani et al. 2008). Compound decomposition during derivatization or separation may also contribute to this bias (Xu et al. 2009), and co-eluting matrix compounds may further suppress the ionization of true analytes in LC–MS (Taylor 2005). In fact, there is now a growing body of evidence suggesting that, with a judicious choice of instrumental conditions, direct infusion electrospray MS (DI-ESI–MS) may achieve equal or greater ion transmission efficiency in metabolic fingerprinting relative to LC–MS (Draper et al. 2014). DI-ESI–MS requires less sample pre-treatment and allows for shorter instrument cycle times than LC–MS and GC–MS, and does not require post-acquisition alignment of retention times (Kopka 2006; Lange et al. 2008). Thus, DI-ESI–MS is an attractive choice of analytical platform to complement NMR for high-throughput metabolic fingerprinting and profiling.
Ion sources such as direct analysis in real time (DART) and desorption electrospray ionization (DESI) have also been utilized for MS analysis of metabolomics samples (Chen et al. 2006; Gu et al. 2011). DESI and DART are ambient ionization methods and provide an abundance of reproducible data (Chen et al. 2006; Gu et al. 2011). However, DESI and DART are not nearly as widely accessible as DI-ESI, which is nearly universally available in modern instrumentation facilities. DI-ESI–MS is also easily automated and can be performed with minimal sample preparation, which is indispensable to high-throughput studies (Draper et al. 2014).
The combination of NMR and MS techniques for metabolic fingerprinting and profiling is a growing trend (Pan and Raftery 2007) and has been shown to improve metabolome coverage (Barding et al. 2013). A number of metabolomics studies combine 1D 1H NMR experiments with LC–MS (Atherton et al. 2006; Jung et al. 2013) or GC–MS (Barding et al. 2013). In these cases, samples and data for each instrumental platform are handled in effective isolation. Most importantly, such studies require the preparation of separate sets of metabolite samples that meet the specific needs of NMR and MS instrumentation (Beltran et al. 2012). Nevertheless, such parallel approaches greatly enhance the structural characterization and quantitation of metabolites (Dai et al. 2010). An alternative approach is to use MS as the primary analytical tool, relying on NMR to validate the results or confirm the identification of key metabolites (Mullen et al. 2012). To date, a limited number of metabolomics studies actually integrate NMR spectral data with information obtained from direct infusion ion sources.
The combined multivariate statistical analysis of data from multiple instrumental platforms is a nascent and underutilized practice in the metabolomics field. Most studies that integrate NMR and DI-ESI–MS data still perform separate analyses of their respective data matrices and combine the results in an attempt to enhance the total information content. For example, Chen et al. performed independent principal component analyses (PCA) on NMR and MS datasets and combined the scores from each analysis into a three dimensional (3D) scores plot (Chen et al. 2006). While the resulting combined scores yielded greater between-class separations than the original NMR or MS scores, such an analysis completely ignores the highly informative correlations that exist between the two datasets. Similarly, Gu et al. (2011) replaced binary class designations in an orthogonal projections to latent structures (OPLS) analysis of MS data with scores from a PCA of the corresponding NMR spectra. While the resulting OPLS-R class separations were greater than the original OPLS-DA separations, such an analysis carries no statistical guarantee of success for any other dataset.
Multiblock bilinear factorizations such as Consensus PCA (CPCA), Hierarchical PCA, Hierarchical PLS and Multiblock PLS provide a powerful framework for analyzing a set of multivariate observations from multiple analytical measurements containing potentially correlated variables (Smilde et al. 2003; Westerhuis et al. 1998; Wold 1987). Such algorithms provide analogous information to classical PCA and PLS in situations where extra knowledge is available to subdivide the measured variables into multiple “blocks”. As a result, the correlation structures of each block and the between-block correlations may be simultaneously utilized. Due to the existence of trends common to each block, this use of between-block correlations during modeling will ideally bring the model loadings (latent variables) into better agreement with the true underlying biology (hidden variables). In short, multiblock algorithms provide an ideal means of integrating 1D 1H NMR and DI-ESI–MS datasets for metabolic fingerprinting studies (Xu et al. 2013).
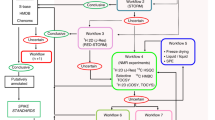
The successful integration of DI-ESI–MS data with 1D 1H NMR data for metabolic fingerprinting and profiling necessitates improving sample preparation, data collection and data processing protocols. Our described optimization of sample preparation protocols enabled the utilization of a single sample for both NMR and MS analysis. To further diminish the impact of sample handling, samples were infused directly into the mass spectrometer without pre-source separation. Electrospray source conditions were then optimized in order to maximize the performance of DI-ESI–MS and minimize ion suppression and/or enhancement (matrix effects). Multiblock PCA (MB-PCA) and multiblock PLS (MB-PLS) were used to analyze the collected NMR and mass spectral data, allowing the identification of key metabolites that significantly contributed to class separation from the resulting scores and loadings. Finally, NMR, accurate mass and MS/MS data were collected to enhance the accuracy and efficiency of metabolite identification. Our resulting protocol for combining DI-ESI–MS with 1D 1H NMR for metabolic fingerprinting and profiling is summarized in Fig. 1.

A flow chart illustrating our protocol for combining NMR and MS datasets for metabolomics. 2.0 mL of a single metabolite extract was split into 1.8 and 0.2 mL for NMR and MS analysis, respectively. Spectral binning of the NMR data used adaptive intelligent binning. First, the background is subtracted from the MS spectrum followed by spectral binning. Spectral binning of the MS data used fixed binning with a set bin width of 0.5 m/z. Baseline noise removal and normalization separately applied to the NMR and MS data sets. The NMR and MS datasets were modeled by MB-PCA and MB-PLS. The resulting block scores and loadings were analyzed for significantly contributing metabolites
2 Materials and methods
2.1 Samples and reagents
All standard reagents and isotopically labeled chemicals were obtained from Sigma Aldrich (St. Louis, MO), Fischer Scientific (Fair Lawn, NJ) and Cambridge Isotopes (Andover, MA). A standard metabolite mixture was prepared by mixing six compounds together: caffeine, l-histidine, β-alanine, l-glutamine, (S)-(+)-ibuprofen, and l-asparagine at concentrations of 10 mM in double distilled water (ddH2O)/methanol/FA (49.75:49.75:0.5). The solution was diluted by a factor of 1000 for MS analysis. Metabolite extracts from Escherichia coli Mach1 were prepared as previously described in detail (Zhang et al. 2013). To generate analytical replicates, each of the metabolite extracts were separated into three aliquots of 100 μL and then diluted ten-fold with ddH2O/methanol/FA (49.75:49.75:0.5) containing 10 μM caffeine as an internal mass reference. A complete description of the preparation of standard samples is available in the supporting information.
2.2 Preparation of metabolomics samples from human dopaminergic neuroblastoma cells
Human dopaminergic neuroblastoma cells (SK-N-SH) with different neurotoxin treatments were used as a metabolomics test system for developing the methodology for integrating NMR and MS data. SK-N-SH cell lines were cultured in DMEM/F12 medium consisting of 100 units/mL penicillin–streptomycin and 10 % fetal bovine serum. 1.5 × 106 SK-N-SH cells were plated on a 10 cm dish for overnight incubation with 5 % CO2 at 37 °C. The cells were then treated with 2.5 mM 1-methyl-4-phenylpyridinium (MPP+), 50 µM 6-hydroxydopamine (6-OHDA), 0.5 mM paraquat or 4.0 µM rotenone for 24 h. The cells were washed twice with 5 mL PBS washes. 1.0 mL of cold methanol (−80 °C) was immediately added to simultaneously lyse and quench the cells, which were then incubated at −80 °C for 15 min to facilitate cell lysis and metabolite extraction. The cells were detached from each dish using a cell scraper and cellular detachment was confirmed using an inverted microscope. The methanol and detached cell debris were transferred to 2.0 mL microcentrifuge tubes, which were centrifuged for 5 min at 15,294 g at 4 °C to separate the metabolite extract from the cell debris. The cell debris was then washed with 500 μL of 80 %/20 % methanol/water and then with 500 μL of 100 % ddH2O. Supernatants from each of the three extractions were finally combined into 2.0 mL microcentrifuge tubes.
Cell extract samples were then split into two portions: 1.8 mL for NMR analysis and 200 μL for MS analysis. The MS portions were diluted tenfold with a solution of H2O/methanol/FA (49.75:49.75:0.5) containing 20 µM reserpine as an internal mass reference. The NMR portions were placed in a RotoSpeed vacuum to remove the organic solvent, followed by freezing and lyophilization. Lyophilized NMR-bound metabolite extracts were then resuspended in 600 µL of 50 mM deuterated potassium phosphate buffer at pH 7.2 (uncorrected) containing 50 μM of 3-(trimethylsilyl)propionic acid-2,2,3,3-d4 (TMSP-d4) and transferred to 5 mm NMR tubes.
2.3 NMR data acquisition and preprocessing
The NMR data was collected and processed according to our previously described protocol (Zhang et al. 2013). A Bruker Avance DRX 500 MHz spectrometer equipped with a 5 mm triple-resonance cryogenic probe (1H, 13C, 15N) with a z-axis gradient, BACS-120 sample changer, and an automatic tuning and matching accessory was utilized for automated NMR data collection.
Following acquisition, the 1D 1H NMR spectra were processed in our MVAPACK software suite (http://bionmr.unl.edu/mvapack.php), which provides a uniform data handling environment that is highly tuned for NMR chemometrics (Worley and Powers 2014a). A 1.0 Hz exponential apodization function and a single round of zero-filling were applied prior to Fourier transformation. Spectra were then automatically phased and normalized using phase-scatter correction (PSC) (Worley and Powers 2014b). Finally, chemical shift regions containing spectral baseline noise or solvent signals were manually removed. Binning of the processed NMR spectra was performed using an adaptive intelligent binning algorithm that minimizes splitting signals between multiple bins (De Meyer et al. 2008).
2.4 Direct infusion ESI-Q-TOF–MS acquisition and preprocessing
2.4.1 Standard metabolite mixture and E. coli metabolome extracts
The standard metabolite mixtures were used to initially optimize DI-ESI–MS source conditions. DI-ESI–MS data were collected on a Synapt G2 HDMS quadrupole time-of-flight MS instrument (Waters, Milford, MA) equipped with an ESI source. All MS experiments were carried out at a flow rate of 10 μL/min for 1 min and mass spectra were acquired over a mass range of m/z 50 to 1200.
2.4.2 Human dopaminergic neuroblastoma cell extracts
Mass spectra of the SK-N-SH samples were acquired in positive ion mode over a mass range of m/z 50–1,200. Spectra were acquired for 0.5 min using the following optimized source conditions: 2.5 kV for ESI capillary voltage, 60 V for sampling cone voltage, 4.0 V for extraction voltage, 80 °C for source temperature, 250 °C for desolvation temperature, 500 L/h for desolvation gas, and 15 µL/min flow rate of injection.
The initial stages of mass spectral data processing were performed using MassLynx V4.1 (Waters Corp., Milford, MA). A background subtraction was performed on all spectra: reference spectra of either paraquat, MPP+, rotenone, or 6-OHDA in ddH2O/methanol/FA (49.75:49.75:0.5) at 10 ppm were used as backgrounds. Background subtraction of each spectrum was performed in a class-dependent manner (e.g. the MPP+ MS reference spectrum was used as background for MPP+ treated cells). As a result, mass spectral signals from the drugs themselves are guaranteed to not influence subsequent analyses. The background-subtracted mass spectra were then loaded into MVAPACK for binning and normalization. All mass spectra were linearly re-interpolated onto a common axis that spanned from m/z 50–1,200 in 0.003 m/z steps, resulting in 383,334 variables prior to processing. Based on the low probability of observing a metabolite in the mass range m/z 1,100–1,200 (Fig. S1 in Supplementary material), the region was removed prior to binning. Mass spectra were uniformly binned using a bin width of 0.5 m/z, resulting in a data matrix of 2,095 variables. Finally, the MS data matrix observations were normalized using probabilistic quotient (PQ) normalization (Dieterle et al. 2006).
2.5 Multivariate statistical analysis
Using functions available in the latest version of MVAPACK, the NMR and mass spectral data were joined into a single multiblock data structure and modeled using MB-PCA and MB-PLS. More specifically, the CPCA-W algorithm (Westerhuis et al. 1998) was used to generate the MB-PCA model. MB-PLS with super-score deflation (Westerhuis and Coenegracht 1997) was used to generate the MB-PLS model. Both blocks were scaled to unit variance prior to modeling, and equal contribution of each block to the models (fairness) was ensured by further scaling each block by the square root of its variable count (Smilde et al. 2003). For the purposes of comparison, PCA and PLS models of the independent NMR and MS data matrices were also constructed. All PLS models were trained on a binary discriminant response matrix (i.e., PLS-DA), in which untreated cells were assigned to one class and all drug-treated cells were assigned to a second class.
2.6 Cross validation of multivariate models
All PCA and MB-PCA models were cross-validated using a leave-one-out (LOOCV) procedure in MVAPACK during model fitting (Lei et al. 2014). PLS-DA and MB-PLS-DA models were cross-validated using a Monte Carlo leave-n-out (MCCV) procedure (Bove et al. 2005). The results of cross-validation were summarized by per-component Q 2 values, where the number of model components was chosen such that cumulative Q 2 was a strictly increasing function of component count. Response permutation tests of all supervised models were performed with 1,000 permutations each to assess the statistical significance of model R 2 Y and Q 2 values (Kamel and Hoppin 2004). CV-ANOVA significance tests (Eriksson et al. 2008) were also performed to supplement the results of the permutation tests. Results of all permutation tests, along with cross-validated scores plots of all supervised models, are provided in the supplementary information (Figs. S6–S11 in Supplementary material).
2.7 Metabolite identification by DI-ESI–MS and MS/MS
Metabolite identifications were achieved by obtaining accurate m/z and further verified using MS/MS experiments. A modified static mode nano-electrospray ionization (nESI) source was used for metabolite identification. Samples were loaded into home-pulled borosilicate emitters fabricated from PYREX 100 mm capillary melting point tubes (Corning, Tewksbury, MA, USA). Emitters were pulled with a vertical micropipette puller (David Kopf Instruments, Tujunga, CA, USA). Each emitter was examined under a microscope (American Optical Company, Buffalo, NY, USA) in order to maintain reproducible tip geometries. The capillary was filled with a metabolite extract and placed in a home-made sprayer mounted to the Synapt G2 nESI source XYZ stage such that the capillary potential was applied by a platinum wire in direct contact with the sample solution.
The optimum nESI source parameters were slightly different from those of the normal DI-ESI source. The nESI source parameters were as follows: capillary spray voltage 1.20 kV, sampling cone voltage 40 V, extraction voltage 5 V, and source temperature 80 °C. Spectra were collected for 2 min in positive ion mode over a mass range of m/z 50–1,200. MS/MS collision energies (CE) were optimized for each metabolite to yield maximum fragmentation. Mass calibration of the instrument was performed by external calibration with sodium acetate, which was infused under the same conditions as the samples. The mass signals of sodium acetate cluster ions (having the general formula [(C2H3O2Na)n + Na]+) occur every m/z 82.0031. Such cluster ions were used to externally calibrate the instrument from m/z 104.9928 (n = 1 cluster) to m/z 1171.0328 (n = 14 cluster). All metabolite spectra were smoothed, centroided, and internally mass corrected relative to the [M + H]+ ion for reserpine (m/z 609.2812) using MassLynx V4.1. Accurate m/z values were searched against the following online metabolite MS databases: Human Metabolome Database (HMDB, http://www.hmdb.ca/, 41,514 metabolites) (Wishart et al. 2007, 2009, 2013) and the general Metabolite and Tandem MS Database (METLIN, http://metlin.scripps.edu, 242,766 metabolites) (Smith et al. 2005) with a threshold window of 20 ppm. The wide window was used to guarantee a thorough search, but a more stringent mass tolerance (~1 ppm) was used when making the final assignment.
3 Results and discussion
3.1 Optimization of metabolomics sample preparation and MS source conditions
Acquisition of DI-ESI–MS data of the highest reliability, reproducibility and information content necessitated the identification of instrumental parameters that yielded maximal ion transmission efficiency. Using a standard metabolite mixture, we first optimized several critical ion source conditions, namely: SCV, ECV, desolvation temperature, desolvation gas flow rate, and cone gas flow rate. Each parameter was sequentially optimized by systematically varying its setting within a predefined range and searching for a maximum ion intensity based on the sum of all detectable spectral signal intensities. After an optimal setting was identified, the parameter value was held fixed while the next parameter was then varied. The process was repeated until an optimized setting was achieved for all five source parameters.
Initial optimization using the standard metabolite mixture indicated that changes to the SCV setting had the largest impact on ion transmission. Ion intensities increased from 5.18 × 103 to 2.69 × 105 for β-alanine, 1.83 × 104 to 1.39 × 106 for glutamine, 1.41 × 105 to 5.46 × 106 for l-histidine, 1.28 × 104 to 6.84 × 106 for caffeine, and 7.21 × 103 to 7.26 × 105 for l-asparagine as the SCV was reduced from 100 to 20 V. Ibuprofen was not observed in any of the mass spectra. Changes to all of the other source parameters were found to have a minimal impact on ion intensity: no other parameter increased the ion intensities by more than a factor of five. For example, the ion intensity only varied from 7.21 × 105 to 3.03 × 105 for caffeine when the desolvation gas flow was changed from 500 to 1,000 L/h. The optimal source parameters for the standard metabolite mixture were determined to be: SCV of 40 V, ECV of 6.0 V, desolvation temperature of 150 °C, desolvation gas flow of 500 L/h, and a cone gas flow of 0 L/h.
Further optimization of the SCV and ECV settings was pursued by applying DI-ESI–MS to a biological matrix of metabolites extracted from Mach1 E. coli. Three compounds from the cellular extract were randomly selected based on their equal distribution within the typical mass range (m/z 50–1,200) of known metabolites. These three compounds had molecular ion peaks corresponding to m/z 118.09, m/z 437.21, and m/z 704.53. The impact of ECV on ion intensity was tested over a range of 2.0–10 V, which identified an optimal ECV of 4.0 V. Importantly, the ion intensity of these selected molecular ion peaks were not significantly affected by varying ECV. ECV values optimized against the standard metabolite mixture and the E. coli cellular extract were found to be equivalent to within experimental error. The impact of SCV on ion intensity was also tested using E. coli cellular extracts. SCV was varied over a range of 20–100 V (Fig. S2A in Supplementary material), which identified an optimal SCV range of 40–60 V by examining a subset of ion peak intensities corresponding to m/z 118.09, m/z 437.21, and m/z 704.53 (Fig. S2B in Supplementary material). An SCV of 40 V, consistent with the value obtained with the standard metabolite mixture, was found to maximize the total information content because intense signals were observed for all metabolites.
Although ion intensity is also positively correlated with metabolite concentrations, simply injecting a highly concentrated metabolite sample may increase the likelihood of ion suppression (Annesley 2003; Skazov et al. 2006) due mostly to increased salt concentrations. The signals from low-mass and polar compounds are more likely to be suppressed by other metabolites as the total sample concentration increases. It was therefore necessary to determine an optimal sample size for DI-ESI–MS analysis of metabolomics samples in order to maximize both ion intensity and information content. E. coli cellular extracts that were optimized to maximize signal intensity in NMR-based metabolomics (Zhang et al. 2013) were used to determine the optimal sample size for DI-ESI–MS. A mass spectrum (Fig. S3A in Supplementary material) was collected for a series of sample dilutions (1:10, 1:25, 1:50, and 1:100) in ddH2O/methanol/FA (49.75:49.75:0.5). For each dilution factor, the total number of spectral peaks above the noise threshold was calculated (Fig. S4 in Supplementary material) and the relative intensities of molecular ion peaks at m/z 118.09, 437.21, and 705.53 were monitored. A bar graph summarizing these results is presented in Fig. S3B (Supplementary material). More spectral signals and higher signal intensities for the three monitored molecular ions were observed for the 10× sample relative to all the other dilution factors. Thus, a ten-fold dilution of a metabolomics sample previously optimized for 1D 1H NMR experiments was deemed suitable for DI-ESI–MS in our combined MS and NMR metabolic fingerprinting protocol. Put simply, a single sample may be prepared and split for NMR and MS analysis, where the preparation of the MS-bound sample only requires a ten-fold dilution into a compatible solvent, such as ddH2O/methanol/FA (49.75:49.75:0.5).
3.2 NMR and MS data pretreatment
Data preprocessing and pretreatment are critical components of any multivariate statistical analysis and have been extensively reviewed (Worley and Powers 2013). Our protocols for NMR data preprocessing have been previously reported (Halouska and Powers 2006; Zhang et al. 2011, 2013) and were utilized as a basis for NMR data handling.
A minimalistic set of pretreatment steps was pursued for both NMR and MS data matrices. To decrease the time required for computing PCA models, NMR spectra were AI-binned (De Meyer et al. 2008) and mass spectra were uniformly binned in preparation for PCA. The data were then normalized to correct for random errors in dilution factors or experimental parameters. Binned NMR and mass spectra were normalized using standard normal variate (SNV) and PQ normalization methods, respectively (Dieterle et al. 2006). Full-resolution NMR spectral data was used for PLS, which permitted the creation of backscaled pseudo-spectral loadings that greatly enhance model interpretability (Cloarec et al. 2005a, b). However, because the full-resolution mass spectral data matrix contained over 300,000 variables, binned mass spectra were used in PLS modeling to reduce computation time. While the NMR spectra were binned to mask chemical shift variations that reduce the effectiveness of PCA, binning of the mass spectra was performed solely to decrease the time required for model computation. For the mass spectral data, a uniform bin width of 0.5 m/z was used based on the mass distribution of all metabolites cataloged in the HMDB (Fig. S1 in Supplementary material). As noise is known to decrease the interpretability of scores (Halouska and Powers 2006), all spectral regions found by manual inspection to contain only baseline noise were removed prior to modeling. The final NMR data matrices for PCA and PLS contained 159 and 16,138 variables, respectively. Likewise, the MS data matrix contained 2,095 (PCA and PLS) variables.
Despite the marked reduction in MS variable count incurred from uniform binning, the resulting binned data matrix retained enough information to differentiate signals arising from distinct metabolites. Based on available data of accurate m/z of metabolites in the HMDB, 85 % of metabolites have an m/z difference with their “neighbor” metabolites (the metabolites with the closest m/z) greater than 0.5 m/z (Fig. S1 in Supplementary material). Also, the number of metabolites identified from a direct-infusion MS analysis of cell extracts generally ranges from 200 to 400 metabolites (Draper et al. 2014), which would be divided over 2,095 bins. This implies that on average the 0.5 m/z bin size would be expected to capture no more than one mass isotopic distribution or one metabolite per bin.
3.3 Classical PCA and PLS modeling
PCA of the binned NMR data matrix (N = 29, K = 159) resulted in 10 principal components having cumulative R 2 X (degree of fit) and Q 2 (predictive ability) metrics of 0.95 and 0.46, respectively. Overall, no patterns were readily discernable in the NMR PCA scores (Fig. 2a) due to high within-class variation in the data. However, scores for paraquat treatment were found to significantly separate from all other classes (p < 0.002) along PC1 (Worley et al. 2013). Scores from PCA of the binned MS data matrix (N = 29, K = 2,095) were found to exhibit markedly less within-class variation compared to the NMR data (Fig. 2b). Three significant components were identified from the binned MS data, yielding fairly low cumulative R 2 X and Q 2 metrics of 0.34 and 0.16. While paraquat treatment still separated from other drug treatments in MS PCA scores space, the greatest separations were observed between treated and untreated cells (p < 1.5 × 10−9). These differing patterns of separation in NMR and MS PCA scores suggested that multiblock analyses could provide further information, ideally separating both control and paraquat scores from all other classes.

Scores generated from (a) PCA of 1H NMR in vacuo, (b) PCA of DI-ESI–MS in vacuo, and (c) MB-PCA of 1H NMR and DI-ESI–MS. Separations between classes are greatly increased upon combination of the two datasets via MB-PCA. Symbols designate the following classes: Control (yellow circle), Rotenone (blue circle), 6-OHDA (red circle), MPP+ (green circle), and Paraquat (turquoise colour circle). Corresponding dendrograms are shown in (d–f). The statistical significance of each node in the dendrogram is indicated by a p value (Worley et al. 2013) (Color figure online)
PLS-DA of the full-resolution NMR (N = 29, K = 16,138) and MS (N = 29, K = 2,095) data matrices both resulted in two-component models. With the exception of the algorithmically forced separation between control and treatment classes, similar clustering patterns were observed when compared to the PCA scores. Leave-n-out cross-validation metrics from the NMR (R 2 Y = 0.92, Q 2 = 0.64) and MS (R 2 Y = 0.99, Q 2 = 0.93) PLS-DA models indicated reasonable levels of fit and predictive ability. Further validation by CV-ANOVA (Eriksson et al. 2008) indicated reliable models with p values of 1.5 × 10−6 and 2.9 × 10−15 for NMR and MS data, respectively. Response permutation tests for both PLS-DA models returned p values equal to zero, supporting the CV-ANOVA significance test results.
3.4 Multiblock PCA and PLS modeling
Identification of consensus directions in the NMR and MS data matrices that maximally captured data matrix variations (MB-PCA) or data-response correlations (MB-PLS) resulted in more informative models than those calculated against either NMR or MS in vacuo. Using MB-PCA, five significant components were identified (Q 2 = 0.23) that cumulatively explained comparable amounts of variation in the NMR (R 2 X = 0.85) and MS (R 2 X = 0.50) blocks relative to the individual PCA models. As expected, MB-PCA combined the information from both blocks to dramatically increase class separations in super-scores space (Fig. 2c). More specifically, both control and paraquat classes were separated from other drug treatments, predominantly along PC1. Furthermore, MPP+ treatment exhibited significant separation from 6-OHDA and Rotenone treatments, which was not expected from examination of the individual NMR or MS PCA scores.
MB-PLS of the data yielded similar improvements in model information content. Two significant components were identified (R 2 Y = 0.98, Q 2 = 0.89) that clearly separated untreated and paraquat treatment classes from all other classes in scores space. CV-ANOVA testing resulted in a p value of 1.7 × 10−12 and response permutation testing yielded a p value equal to zero, indicating a reliable MB-PLS-DA model.
3.5 Metabolite identification by combining NMR, accurate mass and MS/MS
Identification of key metabolites from a metabolomics sample is undoubtedly a nontrivial undertaking. The difficulties of metabolite assignment are further compounded by spectral overlap present in both 1D 1H NMR and DI-ESI–MS data. However, the combination of these two complementary forms of spectral information may aid in overcoming the ambiguities encountered during assignment. The ability of our approach to aid in metabolite identification was demonstrated using NMR and MS data obtained from the treatment of human dopaminergic neuroblastoma cells with known neurotoxic agents.
A first-pass identification of biologically important metabolites was performed by examination of the backscaled NMR block loadings from MB-PLS-DA (Fig. 3a). 1H NMR chemical shifts of loading ‘signals’ that contributed significantly to class separation were used to query the Human Metabolome Database (HMDB) (Wishart et al. 2013) for matching metabolites. To confirm the NMR results, accurate mass and MS/MS experiments were performed, guided by information obtained from the backscaled MS block loadings (Fig. 3b). Effectively, the MB-PLS-DA MS block loadings identified specific masses to pursue for more focused and detailed analyses. For accurate mass measurements, reserpine was used as an internal m/z reference, because it is located in a region containing a minimal number of known metabolites and it can be ionized in both positive and negative modes. Accurate masses were used to conduct elemental composition analyses with MassLynx 4.1 based on a restricted list of elements (C, H, O, N, P, S, Na and K), resulting in a set of possible molecular formulas and associated compounds. Metabolite assignments consistent with both accurate mass and NMR chemical shifts were retained for further study by collision-induced dissociation MS/MS. It is noteworthy that many compounds were identified as sodium adducts, which is expected for DI-ESI–MS (Lin et al. 2010), particularly when sample purification steps have been kept to a minimum.

Backscaled MB-PLS-DA first component loadings generated from (a) the 1H NMR block and (b) the DI-ESI–MS block that compare control with drug treatment. The peaks in the loadings are labeled with the same colored symbol (1H NMR, square; MS, circle) and were assigned to the following metabolites: lactate (red square), glutamate (lavender square), hexose (green square), citrate (blue square), heptose (blue circle), hexose (lavender circle), phosphoaspartate (green circle), and an ambiguous metabolite (red circle) (Color figure online)
As an illustration, examination of backscaled MS block loadings identified a significantly increased mass spectral signal at m/z 203.0534. This accurate mass is consistent with the molecular formula C6H12O6Na (theoretical exact m/z = 203.0532) to within 1 ppm. The corresponding potassium adduct of C6H12O6 was also observed at m/z 219.0266, which is within 2.5 ppm of theoretical exact m/z 219.0271. The molecular ion peak m/z 203.0534 was selected for further examination by MS/MS using collision-induced dissociation, which yielded product ions of m/z 123, 141, and 159, among others (Fig. 4). These three peaks were assigned to [C6H12O6–CO2 + Na]+, [C6H12O6–CO2–H2O + Na]+ and [C6H12O6–CO2–2H2O + Na]+ respectively. Together with the elemental composition suggested by accurate mass measurement, the neutral losses of CO2 and multiple H2O indicate a likelihood that the precursor ion was a polyhydroxy carboxylic acid. Possible structures are inset in Fig. 4. Using these procedures, our NMR analysis of human dopaminergic neuroblastoma cells treated with paraquat identified an increase in metabolites associated with the Pentose Phosphate Pathway (PPP) (Lei 2014).

Direct injection static nESI-MS/MS CID spectrum of m/z 203. Fragment ions at m/z 123, 141, and 159 are consistent with a polyhydroxy carboxylic acid, such as 2-deoxy-gluconate or fuconate (inset). A complete assignment of all peaks to the putative structure(s) was not possible, most likely due to selection of multiple isobaric and/or isomeric species prior to dissociation
4 Conclusions
We report an optimized protocol for combining 1D 1H NMR and DI-ESI–MS datasets for the purposes of high-throughput metabolic fingerprinting and profiling. By splitting metabolite extracts optimized for NMR acquisition and diluting the MS-bound aliquots ten-fold in H2O/methanol/FA (49.57:49.75:0.5), we obtained samples suitable for direct infusion electrospray ionization, thus avoiding the use of pre-source chromatographic separations. We also optimized several DI-ESI–MS ion source conditions in order to maximize the quality of the MS metabolomics data. The optimal source parameters were determined to be: SCV of 40 V, ECV of 4.0 V, desolvation temperature of 150 °C, desolvation gas flow of 500 L/h, and a cone gas flow of 0 L/h. The acquired mass spectra were preprocessed with background subtraction, followed by uniform binning with a 0.5 Da bin size and spectral noise region removal. Using multiblock bilinear factorization algorithms that capitalize on the availability of blocking information, we achieved greater levels of model interpretability with the NMR and MS data than available from single-block PCA and PLS methods. We also demonstrated the combined use of NMR and MS/MS data for the rapid and accurate identification of metabolites significantly perturbed in backscaled MB-PLS-DA loadings. In summary, we present a unique means of increasing metabolome coverage in a high throughput manner by leveraging the complementary information provided by MS and NMR without the encumbrances and liabilities of pre-source chromatographic separations. Our protocol for using combined NMR and MS metabolomics data was successfully demonstrated on a study examining the impact of neurotoxins known to induce dopaminergic cell death, an important model relevant to Parkinson’s disease.
References
Annesley, T. M. (2003). Ion suppression in mass spectrometry. Clinical Chemistry, 49, 1041–1044. doi:10.1373/49.7.1041.
Atherton, H. J., et al. (2006). A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR-α null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiological Genomics, 27, 178–186. doi:10.1152/physiolgenomics.00060.2006.
Barding, G. A., Beni, S., Fukao, T., Bailey-Serres, J., & Larive, C. K. (2013). Comparison of GC-MS and NMR for metabolite profiling of rice subjected to submergence stress. Journal of Proteome Research, 12, 898–909. doi:10.1021/pr300953k.
Beltran, A., et al. (2012). Assessment of compatibility between extraction methods for NMR- and LC/MS-based metabolomics. Analytical Chemistry, 84, 5838–5844. doi:10.1021/ac3005567.
Bove, J., Prou, D., Perier, C., & Przedborski, S. (2005). Toxin-induced models of Parkinson’s disease. NeuroRx, 2, 484–494.
Canelas, A. B., et al. (2009). Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Analytical Chemistry, 81, 7379–7389. doi:10.1021/ac900999t.
Chen, H., Pan, Z., Talaty, N., Raftery, D., & Cooks, R. G. (2006). Combining desorption electrospray ionization mass spectrometry and nuclear magnetic resonance for differential metabolomics without sample preparation. Rapid Communications in Mass Spectrometry, 20, 1577–1584. doi:10.1002/rcm.2474.
Cloarec, O., et al. (2005a). Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic H-1 NMR data sets. Analytical Chemistry, 77, 1282–1289. doi:10.1021/Ac048630x.
Cloarec, O., et al. (2005b). Evaluation of the orthogonal projection on latent structure model limitations caused by chemical shift variability and improved visualization of biomarker changes in H-1 NMR spectroscopic metabonomic studies. Analytical Chemistry, 77, 517–526. doi:10.1021/Ac048803i.
Crockford, D. J., et al. (2006). Statistical heterospectroscopy, an approach to the integrated analysis of NMR and UPLC-MS data sets: Application in metabonomic toxicology studies. Analytical Chemistry, 78, 363–371. doi:10.1021/ac051444m.
Dai, H., Xiao, C., Liu, H., Hao, F., & Tang, H. (2010). Combined NMR and LC-DAD-MS analysis reveals comprehensive metabonomic variations for three phenotypic cultivars of Salvia miltiorrhiza bunge. Journal of Proteome Research, 9, 1565–1578. doi:10.1021/pr901045c.
De Meyer, T., et al. (2008). NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm. Analytical Chemistry, 80, 3783–3790. doi:10.1021/Ac7025964.
Dettmer, K., Aronov, P. A., & Hammock, B. D. (2007). Mass spectrometry-based metabolomics. Mass Spectrometry Reviews, 26, 51–78. doi:10.1002/mas.20108.
Dieterle, F., Ross, A., Schlotterbeck, G., & Senn, H. (2006). Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in H-1 NMR metabonomics. Analytical Chemistry, 78, 4281–4290. doi:10.1021/Ac051632c.
Draper, J., Lloyd, A. J., Goodacre, R., & Beckmann, M. (2014). Flow infusion electrospray ionisation mass spectrometry for high throughput, non-targeted metabolite fingerprinting: A review. Metabolomics, 9, 4–29. doi:10.1007/s11306-012-0449-x.
Eriksson, L., Trygg, J., & Wold, S. (2008). CV-ANOVA for significance testing of PLS and OPLS (R) models. Journal of Chemometrics, 22, 594–600. doi:10.1002/Cem.1187.
Gu, H., Pan, Z., Xi, B., Asiago, V., Musselman, B., & Raftery, D. (2011). Principal component directed partial least squares analysis for combining nuclear magnetic resonance and mass spectrometry data in metabolomics: Application to the detection of breast cancer. Analytica Chimica Acta, 686, 57–63. doi:10.1016/j.aca.2010.11.040.
Halouska, S., & Powers, R. (2006). Negative impact of noise on the principal component analysis of NMR data. Journal of Magnetic Resonance, 178, 88–95. doi:10.1016/j.jmr.2005.08.016.
Jung, J.-Y., Jung, Y., Kim, J.-S., Ryu, D. H., & Hwang, G.-S. (2013). Assessment of peeling of Astragalus roots using 1H NMR- and UPLC-MS-based metabolite profiling. Journal of Agriculture and Food Chemistry, 61, 10398–10407. doi:10.1021/jf4026103.
Kamel, F., & Hoppin, J. A. (2004). Association of pesticide exposure with neurologic dysfunction and disease. Environmental Health Perspectives, 112, 950–958. doi:10.1289/ehp.7135.
Kanani, H., Chrysanthopoulos, P. K., & Klapa, M. I. (2008). Standardizing GC-MS metabolomics. Journal of Chromatography, B: Analytical Technologies in the Biomedical and Life Sciences, 871, 191–201. doi:10.1016/j.jchromb.2008.04.049.
Kell, D. B. (2004). Metabolomics and systems biology: Making sense of the soup. Current Opinion in Microbiology, 7, 296–307. doi:10.1016/j.mib.2004.04.012.
Kopka, J. (2006). Current challenges and developments in GC-MS based metabolite profiling technology. Journal of Biotechnology, 124, 312–322. doi:10.1016/j.jbiotec.2005.12.012.
Kuehnbaum, N. L., & Britz-McKibbin, P. (2013). New advances in separation science for metabolomics: Resolving chemical diversity in a post-genomic era. Chemical Reviews, 113, 2437–2468. doi:10.1021/cr300484s.
Lange, E., Tautenhahn, R., Neumann, S., & Gropl, C. (2008). Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics, 9, 375.
Lei, S., et al. (2014). Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chemical Biology. DOI:10.1021/cb400894a
Lenz, E. M., & Wilson, I. D. (2007). Analytical strategies in metabonomics. Journal of Proteome Research, 6, 443–458. doi:10.1021/pr0605217.
Lin, L., et al. (2010). Direct infusion mass spectrometry or liquid chromatography mass spectrometry for human metabonomics? A serum metabonomic study of kidney cancer. The Analyst, 135, 2970. doi:10.1039/c0an00265h.
Metz, T. O., et al. (2008). High-resolution separations and improved ion production and transmission in metabolomics. TrAC, Trends in Analytical Chemistry, 27, 205–214. doi:10.1016/j.trac.2007.11.003.
Mullen, A. R., et al. (2012). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature, 481, 385–388. doi:10.1038/nature10642.
Nicholson, J. K., Lindon, J. C., & Holmes, E. (1999). “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica, 29, 1181–1189.
Pan, Z., & Raftery, D. (2007). Comparing and combining NMR spectroscopy and mass spectrometry in metabolomics. Analytical and Bioanalytical Chemistry, 387, 525–527. doi:10.1007/s00216-006-0687-8.
Skazov, R. S., Nekrasov, Y. S., Kuklin, S. A., & Simenel, A. A. (2006). Influence of experimental conditions on electrospray ionization mass spectrometry of ferrocenylalkylazoles. European Journal of Mass Spectrometry, 12, 137–142. doi:10.1255/ejms.795.
Smilde, A. K., Westerhuis, J. A., & de Jong, S. (2003). A framework for sequential multiblock component methods. Journal of Chemometrics, 17, 323–337. doi:10.1002/cem.811.
Smith, C. A., et al. (2005). METLIN: a metabolite mass spectral database. Therapeutic Drug Monitoring, 27, 747–751.
Taylor, P. J. (2005). Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clinical Biochemistry, 38, 328–334. doi:10.1016/j.clinbiochem.2004.11.007.
t’Kindt, R., et al. (2010). Metabolomics to unveil and understand phenotypic diversity between pathogen populations. PLOS Neglected Tropical Diseases, 4, e904. DOI:10.1371/journal.pntd.0000904.
Westerhuis, J. A., & Coenegracht, P. M. J. (1997). Multivariate modelling of the pharmaceutical two-step process of wet granulation and tableting with multiblock partial least squares. Journal of Chemometrics, 11, 379–392. doi:10.1002/(SICI)1099-128X(199709/10)11:5<379::AID-CEM482>3.0.CO;2-8.
Westerhuis, J. A., Kourti, T., & Macgregor, J. F. (1998). Analysis of multiblock and hierarchical PCA and PLS models. Journal of Chemometrics, 12, 301–321. doi:10.1002/(SICI)1099-128X(199809/10)12:5<301::AID-CEM515>3.0.CO;2-S.
Wishart, D. S., et al. (2007). HMDB: The human metabolome database. Nucleic Acids Research, 35, D521–D526.
Wishart, D. S., et al. (2009). HMDB: A knowledgebase for the human metabolome. Nucleic Acids Research, 37, D603–D610. doi:10.1093/nar/gkn810.
Wishart, D. S., et al. (2013). HMDB 3.0—the human metabolome database in 2013. Nucleic Acids Research, 41, D801–D807. doi:10.1093/nar/gks1065.
Wold, S. (1987). PLS modeling with latent variables in two or more dimensions. In Proceedings of PLS Model Building: Theory and Applications. Symposium Frankfurt am Main, September 23–25, 1987.
Worley, B., Halouska, S., & Powers, R. (2013). Utilities for quantifying separation in PCA/PLS-DA scores plots. Analytical Biochemistry, 433, 102–104. doi:10.1016/j.ab.2012.10.011.
Worley, B., & Powers, R. (2013). Multivariate analysis in metabolomics. Current Metabolomics, 1, 92–107. doi:10.2174/2213235x11301010092.
Worley, B., & Powers, R. (2014a). MVAPACK: A complete data handling package for NMR metabolomics. ACS Chemical Biology, 9, 1138–1144. doi:10.1021/cb4008937.
Worley, B., & Powers, R. (2014b). Simultaneous phase and scatter correction for NMR datasets. Chemometrics and Intelligent Laboratory Systems, 131, 1–6. doi:10.1016/j.chemolab.2013.11.005.
Xu, Y., Correa, E., & Goodacre, R. (2013). Integrating multiple analytical platforms and chemometrics for comprehensive metabolic profiling: Application to meat spoilage detection. Analytical and Bioanalytical Chemistry, 405, 5063–5074. doi:10.1007/s00216-013-6884-3.
Xu, F., Zou, L., & Ong, C. N. (2009). Multiorigination of chromatographic peaks in derivatized GC/MS metabolomics: A confounder that influences metabolic pathway interpretation. Journal of Proteome Research, 8, 5657–5665. doi:10.1021/pr900738b.
Zhang, B., Halouska, S., Schiaffo, C. E., Sadykov, M. R., Somerville, G. A., & Powers, R. (2011). NMR analysis of a stress response metabolic signaling network. Journal of Proteome Research, 10, 3743–3754. doi:10.1021/pr200360w.
Zhang, B., et al. (2013). Revisiting protocols for the NMR analysis of bacterial metabolomes. Journal of Integrated OMICS, 2, 120–137.
Acknowledgments
We would like to thank Dr. Jiantao Guo for providing us with the Escherichia coli Mach1 cell line. This manuscript was supported in part by funds from the National Institute of Health (R01 AI087668, R21 AI087561, R01 CA163649, P20 RR-17675, P30 GM103335), the University of Nebraska, the Nebraska Tobacco Settlement Biomedical Research Development Fund, and the Nebraska Research Council. The research was performed in facilities renovated with support from the National Institutes of Health (RR015468-01).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Darrell D. Marshall, Shulei Lei and Bradley Worley have equally contributed to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Marshall, D.D., Lei, S., Worley, B. et al. Combining DI-ESI–MS and NMR datasets for metabolic profiling. Metabolomics 11, 391–402 (2015). https://doi.org/10.1007/s11306-014-0704-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11306-014-0704-4