
Abstract
Corynebacterium glutamicum, an important industrial producer, is a model microorganism. However, the limited gene editing methods and their defects limit the efficient genome editing of C. glutamicum. To improve the screening efficiency of second-cross-over strains of traditional SacB editing system, a universal pCS plasmid which harbors CRISPR-Cpf1 system targeting kan gene of SacB system was designed and established to kill the false positive single-cross-over strains remained abundantly after the second-cross-over events. The lethality of pCS plasmid to C. glutamicum carrying kan gene on its genome was as high as 98.6%. In the example of PodhA::PilvBNC replacement, pCS plasmid improved the screening efficiency of second-cross-over bacteria from 5% to over 95%. Then this pCS-assisted gene editing system was applied to improve the supply of precursors and reduce the generation of by-products in the production of 4-hydroxyisoleucine (4-HIL). The 4-HIL titer of one edited strain SC01-TD5IM reached 137.0 ± 33.9 mM, while the weakening of lysE by promoter engineering reduced Lys content by 19.0–47.7% and 4-HIL titer by 16.4–64.5%. These editing demonstrates again the efficiency of this novel CRISPR-Cpf1-assisted gene editing tool, suggesting it as a useful tool for improving the genome editing and metabolic engineering in C. glutamicum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum, a Gram-positive soil bacterium, has been widely used in industrial biotechnology to produce various organic acids and amino acids (Becker et al. 2018). With the development of molecular biology technology, the research on genetic engineering of C. glutamicum has been gradually deepened. Today, the most commonly used gene editing technology in C. glutamicum is still based on the homologous recombination system mediated by non-self-replicating suicide plasmid such as pK18mobsacB (Schäfer et al. 1994). In this system, gene editing is completed by two rounds of homologous recombination. First, the single-cross-over strain integrated with the whole plasmid is screened by antibiotic marker (usually kanamycin), and then the second-cross-over strain with the plasmid skeleton removed by growing in sucrose is screened by the conditionally lethal levansucrase (encoded by sacB) marker carried on the suicide plasmid. However, the high probability of false-positive clones with sacB-remaining and low probability of double-cross-over events limit the efficiency of editing (Kirchner and Tauch 2003; Nešvera and Pátek 2011). In order to improve the screening efficiency of the second-cross-over, rpsLmut (encoding small ribosomal protein S12P, resistance to streptomycin after mutations) and upp (encoding uracil phosphoribosyl transferase, which is lethal in the presence of 5-fluorouracil) have been developed as new screening markers instead of sacB (Ma et al. 2015; Wang et al. 2019), but the screening efficiency is still not up to the ideal state.
Recently, clustered regularly interspaced short palindromic repeats (CRISPR) technologies have been used for genome editing of C. glutamicum (Cho et al. 2017; Jiang et al. 2017; Liu et al. 2017; Peng et al. 2017). Under the guidance of the single guide RNA (sgRNA), CRISPR-associated proteins (Cas/Cpf1) precisely cleave complementary sequences and form double-strand breaks (DSB). Because there is no nonhomologous end-joining (NHEJ) mechanism in C. glutamicum (Resende et al. 2011), the DSB can only be repaired by homologue-directed DNA repair. Although CRISPR technology has been successfully applied for gene editing of C. glutamicum, it is still subject to many limitations in practice. First, the toxicity of high-level expression of Cas9/Cpf1 to C. glutamicum makes it difficult to generate transformants (Cho et al. 2017; Jiang et al. 2017; Liu et al. 2017; Peng et al. 2017). Second, the protospacer-adjacent motif (PAM) sequence limits the application of CRISPR system in small fragment editing such as promoter engineering and ribosomal binding site (RBS) engineering. Third, the editing efficiency is affected by the size of the knockout fragment, the total length of homologous arm, the relative length of homologous arm and inserted gene, and the design of sgRNA (Becker et al. 2018). Therefore, when editing with CRISPR system, it often takes a lot of unexpected time to do repetitive work.
4-Hydroxyisoleucine (4-HIL), an amino acid derivative, has the potential to treat type II diabetes because of its biological activities that promoting insulin secretion and reducing insulin resistance (Sauvaire et al. 1998; Yang et al. 2021). In our previous study, L-isoleucine dioxygenase (IDO) from Bacillus thuringiensis YBT-1520 was expressed in an L-isoleucine (Ile) producing strain C. glutamicum ssp. lactofermentum SN01 to achieve the de novo synthesis of 4-HIL (Shi et al. 2015). IDO can catalyze the oxidation of Ile and α-ketoglutarate (α-KG) to form 4-HIL and succinate, respectively by consuming O2. The titer of 4-HIL reached 139.82 ± 1.56 mM by knockout or overexpression of related genes to increase the supply of α-KG and oxygen (Shi et al. 2019, 2020). After that, the dynamic adjustment tools Lrp-PbrnFEN biosensor and PilvBNC attenuator were used to coordinate the supply of substrates, and L-lysine (Lys)-responsive riboswitch was used to regulate Lys synthesis. Finally, the titer of 4-HIL was increased to 177.3 ± 8.9 mM, and the content of by-product Lys was reduced to 6.1 ± 0.6 mM (Lai et al. 2022; Tan et al. 2020). Through the combination of static regulation and dynamic regulation to regulate the substrate supply in C. glutamicum YI, the 4-HIL of 34.2 g/L was obtained in 5 L bioreactor (Zhang et al. 2018). In addition, the synthetic pathway of Lys competes with that of Ile, so Lys has always been the main by-product in the production of 4-HIL. However, the deletion of the specific Lys exporter encoding gene lysE reduced the titers of both Lys and 4-HIL, while the deletion of ddh gene related to Lys synthesis failed to decrease the titer of Lys (Chen et al. 2022). In a research on Ile production, the introduction of point mutation at the promoter of dapA, a key gene for Lys synthesis, reduced its expression intensity, resulting in an increase in Ile titer and a decrease in Lys titer to 5.1 mM (Vogt et al. 2015). Therefore, slightly regulating the expression intensity of related genes by point mutation or promoter engineering may be more effective than directly deleting them.
In this study, in order to improve the screening efficiency of SacB editing system, CRISPR-Cpf1 system was designed and introduced to kill the false positive single-cross-over strains remained abundantly after the second-cross-over events. Based on this novel optimized editing system, the promoter of odhA was replaced by PilvBNC to increase the supply of α-KG. And ido was integrated into ldhA-pyk2 locus to reduce the content of pyruvate-derived L-alanine (Ala). In addition, the expression intensity of lysE was fine-tuned with PdapA−N of different intensity to reduce the content of by-product Lys. The main purpose of this study is to provide an efficient CRISPR-Cpf1-assisted gene editing tool and to apply this tool to obtain a strain overproducing 4-HIL and decreasing by-product Lys.
Materials and methods
Strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli JM109 and DH5α were used for constructing and propagating plasmids, while C. glutamicum ssp. lactofermentum SN01 was used as the initial strain for producing 4-HIL. SN01 was deposited in the China Center for Type Culture Collection (CCTCC) with accession number CCTCC M 2,014,410. E. coli strains were grown at 37 °C and 200 rpm in LB medium. C. glutamicum strains were cultivated at 30 °C and 200 rpm in LBB medium (5 g/L tryptone, 2.5 g/L yeast extract, 5 g/L NaCl, and 18.5 g/L brain heart infusion powder). If required, 30 mg/L kanamycin or 10 mg/L chloramphenicol was added to the media for selection.
Plasmid construction
The construction of pK18mobsacB editing plasmid was carried out in E. coli JM109. Supplementary Table 1 shows the primers used in this study. In order to construct a pK18mobsacB-PilvBNC-odhA plasmid for replacing odhA promoter with PilvBNC, the upstream and downstream homologous arms of odhA gene (odhA-U and odhA-D) and PilvBNC were amplified from C. glutamicum SN01 genomic DNA by primer pairs odhA-U-F/odhA-U-R, odhA-D-F/odhA-U-F and PilvBNC-F/PilvBNC-R, respectively. The upstream homologous arm, PilvBNC, and downstream homologous arm were connected by overlap-extension PCR. The pK18mobsacB was linearized by digestion at SalI site. ClonExpress MultiS One Step Cloning Kit (Vazyme, C113-01) was used to connect overlapping fragments and linearized pK18mobsacB to generate pK18mobsacB-PilvBNC-odhA plasmid. The other two groups of pK18mobsacB editing plasmids, four pK18mobsacB-ΔldhA-pyk2::X-ido and four pK18mobsacB-PdapA−N-lysE, were constructed in a similar way. For constructing the four pK18mobsacB-ΔldhA-pyk2::X-ido plasmids to integrate the ido gene or its expressing cassette at ldhA-pyk2 locus, the codon-optimized and Thr176Cys-mutated ido expression plasmids pIL-TNIM were constructed at first. The PbrnFEN-idoMUp and idoMDn fragments were amplified from pIL-NIU (Lai et al. 2022) by primer pairs PbrnFE-F/idoU-cys-R and idoU-cys-F/idoU-R, respectively, then overlapped, digested with KpnI and BamHI, and ligated into KpnI- and BamHI-digested pIL-NIU, generating pIL-TNIM. Next, one of the pIL-TNIM plasmids, i.e. pIL-TD5IM was used as template to amplify the PbrnFED5-idoM, dc-idoM, RBS-idoM, and idoM fragments by primer pairs IDO1-F/IDO-R, IDO2-F/IDO-R, IDO3-F/IDO-R, and IDO4-F/IDO-R, respectively. These fragments were then separately overlapped with upstream and downstream homologous arms to construct the four editing plasmids pK18mobsacB-ΔldhA-pyk2::PbrnFED5-idoM, pK18mobsacB-ΔldhA-pyk2::dc-idoM, pK18mobsacB-Δpyk2::RBS-idoM, and pK18mobsacB-ΔldhA-pyk2::idoM.
The construction of pCS plasmid was carried out in E. coli DH5α. Fragment 1 (containing Cpf1 expression cassette and terminator part of sgRNA expression cassette) and fragment 2 (including sgRNA expression cassette, E. coli replicon pSC101, and C. glutamicum temperature sensitive replicon pBL1ts) were amplified from plasmid pJYS3 (Jiang et al. 2017) with primer pairs Cpf1-F/sgRNA-R(N) and ori-F(N)/ori-R, respectively. A special sgRNA sequence targeting kan gene was introduced into primers gRNA-R(N) and ori-F(N). Fragment 3 [including chloramphenicol resistance gene (cat) expression cassette] was amplified by CmR-F/CmR-R using plasmid pDTW109 (Hu et al. 2013) as template. Fragment 2 and fragment 3 were linked together by overlap-extension PCR to get fragment 4. Fragment 1 and fragment 4 were linked by One Step Cloning Kit to generate a complete pCS(N) plasmid.
Gene editing process
Firstly, C. glutamicum was prepared into competent cells, and pK18mobsacB editing plasmid was transferred into competent cells by electroporation at 1.8 kV. Cells were incubated at 30 °C for about 2 h, then plated on LBB agar containing 30 mg/L kanamycin (LBBK) and incubated at 30 °C for about 36 h. Single-cross-over strains were confirmed by PCR amplification. Secondly, the single-cross-over strains were cultured in LBB liquid medium containing 10% sucrose (LBBS) for about 24 h, then prepared into competent cells, and transformed with pCS plasmid. The cells were incubated at 30 °C for about 1.5 h, plated on LBB agar containing 10 mg/L chloramphenicol (LBBC), and then incubated at 28 °C for about 36 h. The correct second-cross-over strains were verified by PCR. Finally, in order to cure the pCS plasmid in second-cross-over strain so as to conduct next round of genome editing, the strain was inoculated into LBB liquid medium and incubated at 37 °C for about 20 h. Cells were streaked onto LBB plates and cultured at 30 °C overnight. By scribing them both on LBB plates with or without chloramphenicol, the colonies whose pCS plasmids have been removed were confirmed and the edited strains were obtained. Finally, plasmid pIL-TD5IM was transformed into some edited strains to generate 4-HIL high-producing strains. Meanwhile, plasmids pIL-TNIM were also transformed into initial strain SN01 as controls.
Fatality rate determination
The kanamycin resistant marker was integrated into the aceA locus of C. glutamicum SN01 by homologous recombination to obtain ΔaceA::kan. The pCS(N) plasmid about 200 ng was transferred into SN01, diluted and plated on both LBB solid medium and LBBC solid medium. After about 36 h, the colonies on the solid medium were counted, and the transformation rate of pCS(N) was calculated by the ratio of colony number. Meanwhile, the pCS(N) plasmid about 200 ng was also transferred into ΔaceA::kan, diluted and plated on both LBB solid medium and LBBC solid medium. After about 36 h, the colonies on the solid medium were counted, and the lethality of pCS(N) to C. glutamicum containing kan gene in the chromosome could be calculated by the following formula.
4‑HIL fermentation
4-HIL producing strains were preconditioned in a 500-mL shake flask with 40 mL seed medium (25 g/L C6H12O6·H2O, 0.5 g/L (NH4)2SO4, 40 g/L corn steep liquor, 1 g/L KH2PO4, 0.5 g/L MgSO4, and 1.25 g/L urea) at 30 °C and 200 rpm for 16 h. For fermentation, strains were cultured in a 500-mL flask with 30 mL optimized fermentation medium (140 g/L C6H12O6·H2O, 30 g/L (NH4)2SO4, 10 g/L corn steep liquor, 1 g/L KH2PO4, 0.75 g/L MgSO4, 1.5 g/L FeSO4·7H2O, and 20 g/L CaCO3) at 30 °C and 200 rpm for 144 h. Every 24 h, 1 mL fermentation broth was taken to measure the cell density, pH, residual glucose, and amino acid concentration by the methods described previously (Shi et al. 2019).
Results
SacB editing system assisted by CRISPR-Cpf1
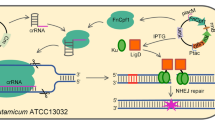
The workflow of CRISPR-Cpf1 assisted SacB editing system designed here was shown in Fig. 1A. The process of traditional SacB-mediated gene editing system applied in C. glutamicum is to integrate the whole plasmid into the genome through single-cross-over, and then to screen the second-cross-over strains through counter selection by sacB, which is the lethal gene in the presence of sucrose. However, the high spontaneous inactivation rate of sacB gene as a counter selection marker leads to a high false positive rate of gene editing results (Tan et al. 2012). Because there is no NHEJ in C. glutamicum (Resende et al. 2011), DSB formed by CRISPR system targeting genome sequence is fatal to C. glutamicum. Therefore, the screening efficiency of positive bacteria can be improved by introducing CRISPR system to kill the single-cross-over strains remained abundantly after the second-cross-over events and sucrose-resistant screening. Compared with Cas protein, Cpf1 protein has attracted much attention because of its low toxicity and low miss target rate (Jiang et al. 2017; Zhang et al. 2019). Therefore, CRISPR-Cpf1 was selected as the screening tool in this study. pJYS3 is the all-in-one CRISPR-Cpf1 plasmid for large gene deletions and insertions (Jiang et al. 2017). Here, the chloramphenicol resistant marker was used to replace the original kanamycin resistant marker of pJYS3 to avoid conflict with the resistant marker (kanamycin) in pK18mobsacB. In addition, we constructed three pCS series plasmids with Cpf1 expression box and sgRNA targeting the front, middle, and back parts of kan gene of the pK18mobsacB plasmid skeleton.

Schematic overview of SacB editing system assisted by CRISPR-Cpf1. A Process of CRISPR-Cpf1-assisted editing system and traditional SacB editing system. B Lethality of pCS(N) plasmids against strain with kan gene on chromosome. C Screening efficiency of traditional SacB editing system and CRISPR-Cpf1 assisted editing system for second-cross-over strain. pCS(N): temperature-sensitive plasmid carrying CRISPR-Cpf1 system targeting kan gene; SUC: only sucrose was used for screening in traditional system; SUC + CPL: after enrichment with sucrose, pCS(N) plasmid was transferred and transformants were screened on solid medium containing chloramphenicol
In order to detect the screening efficiency of pCS, the kanamycin resistant marker was integrated into the aceA site of C. glutamicum SN01. Different pCS(N) plasmids were transferred into SN01ΔaceA::kan to determine their lethality. Test results showed that the lethal rate of pCS(N) to bacteria carrying kan cassette was above 94.2%, with pCS(1) up to 98.6% (Fig. 1B), suggesting the feasibility of pCS for screening. To test the efficacy of the pCS(N) plasmid for screening out second-cross-over strains and to increase the supply of α-KG in 4-HIL production (Fig. 2), the pK18mobsacB-PilvBNC-odhA plasmid was constructed for replacing the promoter of odhA with Ile attenuator PilvBNC. Next, pK18mobsacB-PilvBNC-odhA was transferred into SN01 to obtain single-cross-over strains. The verified single-cross-over strains were incubated in LBBS liquid medium for about 24 h, then the second-cross-over strains were screened by two protocols. One is the traditional protocol that screened the second-cross-over strains directly on LBBS agar medium. The other is newly developed protocol that transformed with pCS(N) and then screened the second-cross-over strains on LBBC medium. Finally, the SC00 strain with the odhA promoter replaced by the PilvBNC was obtained through PCR verification. The results showed that for editing PodhA::PilvBNC in SN01, when traditional protocol was used for screening, the probability of the second-cross-over strains in all the verified colonies was only 5% (1/20), while when the pCS(1), pCS(2), and pCS(3)-assisted novel method was applied for screening, the probability of the second-cross-over strains in all the verified colonies was 100% (20/20), 95% (19/20), and 95% (19/20), respectively (Fig. 1C). This indicated that pCS plasmid could greatly improve the screening efficiency of the second-cross-over events. In addition, when the single-cross-over strains were cultured in LBB liquid medium without sucrose instead of in LBBS medium, then pCS(N) plasmid was transferred into this culture for assisting the screening of the naturally generated second-cross-over strains, the number of colonies on LBBC agar medium was less than 5, greatly lower than that cultured in LBBS medium (about 40 colonies). This indicated that preculturing the single-cross-over strains in medium containing sucrose could enrich the second-cross-over strains.

The de novo biosynthetic pathway and metabolic engineering strategies of 4-HIL in recombinant C. glutamicum ssp. lactofermentum SN01. Black crosses indicate gene deletion, green fonts indicate gene down-regulation, and red font indicates gene up-regulation
Integration of ido M at ldhA - pyk2 locus
Lactate dehydrogenase (encoded by ldhA) and pyruvate kinase 2 (encoded by pyk2) are involved in the metabolism of pyruvate (Fig. 2). The deletion of ldhA and pyk2 can reduce the formation of pyruvate-derived by-products and increase the carbon flow of 4-HIL synthesis (Shi et al. 2020; Zhang et al. 2018). The Thr176Cys mutation which introduces a disulfide bond in IDO can improve the thermal stability and enzyme activity of IDO (Qiao et al. 2019). In addition, in order to verify the production of 4-HIL with chromosomal integrated ido, this mutant ido (idoM) was integrated into the ldhA-pyk2 locus of SC00 in four different expression patterns (Fig. 3A). In the first pattern, the Ile-induced promoter PbrnFED5 was used to dynamically regulate the expression of idoM and the open reading frame (ORF) of ldhA-pyk2 was replaced by PbrnFED5-idoM expression cassette, resulting in SC01 strain. In the other three patterns, the endogenous promoter PldhA was used to control the expression of idoM. First, to form a bicistronic structure (Sun et al. 2020), the first 63 bp of ldhA gene was retained and linked with RBS-idoM (recorded as dc-idoM), resulting in SC02 strain. Second, the entire ORF of ldhA was retained and connected with RBS-idoM by a linker, resulting in SC03 strain. At last, PldhA was directly connected with idoM, resulting in SC04 strain. Before comparing these four strains, SC-TNIM carrying the PbrnFEN-idoM expression plasmid pIL-TNIM was tested for identify the best PbrnFEN-idoM expression cassette. Plasmids pIL-TNIM were constructed by inserting the PbrnFEN-idoM expression cassette into pJYW-4, an expression vector of C. glutamicum. The Ile-inducible promoters PbrnFEN include natural PbrnFE0 and modified PbrnFE1, PbrnFE5, PbrnFED5, PbrnFE7, PbrnFE9, and PbrnFE13 with varied strength (Lai et al. 2022; Tan et al. 2020).

Integration of idoM at ldhA-pyk2 locus. A Different patterns of idoM integration at ldhA-pyk2 locus. B 4-HIL and Ile titer of SC-TNIM. C Cell growth (solid lines) and glucose consumption (dotted lines) of SC01–SC04. D Amino acid titer of SC01–SC04 at 144 h
Among 7 SC-TNIM strains, SC-TD5IM carrying the PbrnFED5-controlled idoM exhibited the highest 4-HIL titer of 140.9 ± 11.5 mM (Fig. 3B), thereby PbrnFED5-idoM expression cassette was selected to replace the ldhA-pyk2 ORFs of SC00 in the first integration pattern. The 4-HIL fermentation of the idoM-integrated four strains in shake flask showed that after replacing the promoter of odhAwith PilvBNC and integrating ido at ldhA-pyk2 locus, the final OD of these strains was slightly lower than that of SC-TD5IM, but their glucose consumption was basically the same (Fig. 3C). Unfortunately, the single copy integration of idoM did not completely convert Ile into 4-HIL, in which the highest titer found in SC01 was only 14.8 ± 1.1 mM, which was 10.5% of that of the control strain SC-TD5IM, and 170.1 ± 40.6 mM of Ile was remained (Fig. 3D). While the 4-HIL titer of SC02–SC04 with idoM controlled by PldhA was only about 2.0 mM, and 143.2–158.0 mM Ile was remained. Compared with SC-TD5IM, the Ala accumulation of SC01–SC04 decreased, with that of SC02 decreased by 27.7%. Fortunately, the total titer of L-aspartate (Asp)-family amino acids [L-threonine (Thr), Ile, Lys, and 4-HIL] increased by 37.7%, 18.4%, 15.3%, and 11.9% in SC01, SC02, SC03, and SC04, respectively, indicating that the deletion of ldhA did increase the carbon flux of Asp-family amino acids synthesis. At the same time, the accumulation of Lys (34.8 ± 10.9 mM) in SC01 also increased by 9.0%, which is not conducive to the production of 4-HIL. Therefore, as Lys synthesis is a competitive pathway of 4-HIL synthesis, the weakening of genes related to Lys synthesis should be beneficial to the synthesis of 4-HIL.
Subtle regulation of lysE expression
The coding product of lysE is the exporter of Lys. Our recent research on 4-HIL synthesis showed that the deletion of lysE successfully reduced the accumulation of Lys by 66.7%, but it also reduced 4-HIL titer by 14% (Chen et al. 2022). Therefore, selecting a promoter with appropriate intensity to weaken lysE expression may be suitable for simultaneously reducing the accumulation of Lys and increasing the titer of 4-HIL (Fig. 2). PdapA exhibits different intensity after point mutation (Vasicová et al. 1999), and the application of these mutant PdapA−N promoters have been proved in a variety of gene regulation examples (Xu et al. 2019). So here, the promoter of lysE on chromosome was replaced by several PdapA−N promoters, i.e. PdapA−B27, PdapA−B6, PdapA−C13, and PdapA−C5 using CRISPR-Cpf1-assisted gene editing system to obtain strains SC05–SC08.
SC05–SC08 were fermented in shake flask. SC06 grew at a lower rate than SC01, while SC05 grew at a rate comparable to SC01, and SC07 and SC08 grew at a higher rate than SC01 (Fig. 4A). The glucose consumption rate of SC05 and SC06 were slightly slower than SC01 and other strains (Fig. 4A). Meanwhile, compared with SC01, the Lys accumulation of SC05–SC08 (13.1 ± 1.3 mM, 12.7 ± 0.6 mM, 23.1 ± 0.8 mM, and 28.7 ± 1.0 mM) decreased by 62.4%, 63.4%, 33.2%, and 17.4%, respectively (Fig. 4B), which indicated that the weakening of lysE expression was beneficial to reduce the accumulation of Lys. In addition, the 4-HIL titer of SC05 and SC06 (12.4 ± 1.0 mM and 7.9 ± 0.3 mM) decreased by 16.2% and 46.6%, while the 4-HIL titer of SC07 and SC08 (15.9 ± 0.9 mM and 15.2 ± 0.4 mM) increased slightly by 7.4% and 2.7%, respectively (Fig. 4B). Unfortunately, although both SC07 and SC08 have achieved the expectation of reducing Lys accumulation and increasing 4-HIL titer, a large amount of Ile (152.3 ± 3.4 mM and 169.7 ± 6.2 mM) was still remained in SC07 and SC08 and was not converted into 4-HIL. Therefore, plasmid pIL-TD5IM overexpressing PbrnFED5-controlled idoM was transformed into SC00, SC01, and SC05–SC08, generating the strains SC00-TD5IM, SC01-TD5IM and SC05-TD5IM –SC08-TD5IM.

Flask fermentation of lysE-attenuated strains. A Cell growth (solid lines) and glucose consumption (dotted lines). B 4-HIL, Ile, and Lys titer at 144 h
SC00-TD5IM, SC01-TD5IM, and SC05-TD5IM–SC08-TD5IM were fermented in shake flask. The growth rate of these strains was similar to that of SC-TD5IM (Fig. 5A). Except for SC05-TD5IM, which still did not consume glucose completely at the end of fermentation, other strains consumed all glucose at 96 h (Fig. 5A). After transformed with the expression plasmid carrying PbrnFED5-idoM, the remaining Ile in SC00, SC01, and SC05–SC08 was completely converted to 4-HIL (Fig. 5B). Unfortunately, compared with SC-TD5IM, the 4-HIL production of these genome-edited strains carrying pIL-TD5IM was decreased to different extents, with that of SC00-TD5IM (114.5 ± 12.2 mM) and SC01-TD5IM (137.0 ± 33.9 mM) decreased by 18.7% and 2.8%, respectively, while the 4-HIL titer of lysE-weakening strains decreased more significantly, with that of SC06-TD5IM (67.2 ± 21.8 mM) decreased by 47.7% and that of SC08-TD5IM (114.1 ± 29.9 mM) decreased by 19.0% (Fig. 5B). Meanwhile, the Lys accumulation of SC00-TD5IM (35.4 ± 4.4 mM) and SC01-TD5IM (39.3 ± 4.5 mM) increased by 11.1% and 23.4% respectively compared with SC-TD5IM, while that of lysE-weakening strains decreased by 16.4–64.5% compared with SC01-TD5IM. Among these strains, SC05-TD5IM accumulated the least Lys, which was only 14.0 ± 1.1 mM (Fig. 5B).

Flask fermentation of lysE-attenuated strains carrying pIL-TD5IM. A Cell growth (solid lines) and glucose consumption (dotted lines). B 4-HIL, Ile, and other amino acids titer at 144 h
Compared to SC-TD5IM, SC01-TD5IM showed a 2.7% increase in total Asp-family amino acids titer, a 2.8% decrease in 4-HIL titer, and a 23.4% increase in Lys accumulation. Lys is synthesized in C. glutamicum via a split pathway, whereas the succinylase branch works as the main pathway (Sonntag et al. 1993; Zhang et al. 2020), and the synthesis of Lys from Asp through this branch requires nine steps. Therefore, the increase of Lys titer in SC01-TD5IM would consume a large amount of carbon flux, thus reducing the carbon flux toward the 4-HIL synthesis. When the expression of lysE was weakened by promoter replacement, although the accumulation of Lys did decrease, the titer of 4-HIL did not increase, but instead, the accumulation of other by-products increased. The accumulation of L-glutamic acid increased from 1.7 ± 1.6 mM to 3.2–57.2 mM, the accumulation of Ala increased by 33.9–142.4%, and finally the total amount of Asp-family amino acids decreased by 17.0–54.1% (Fig. 5B).
Discussion
For the suicide plasmid mediated traditional genome editing methods used in C. glutamicum, the frequent occurrence of false positive colonies and the low probability of second-cross-over events limit their editing efficiency (Tan et al. 2012). CRISPR system is a new type of gene editing system, in which Cas9 or Cpf1 protein binds to the target sequence under the guidance of sgRNA to form DSB, and does not break the repaired sequence generated through homologous recombination, thereby achieving gene editing in C. glutamicum. The CRISPR-based system looks attractive because it ensures efficient, fast, and multiple genome editing, and theoretically produces only positive transformants. However, high expression of Cas9 may lead to the lack of transformants due to its lethality, and low expression of Cas9 may lead to the survival of false positive bacteria (Peng et al. 2017). In addition, the expression of Cas9 may cause a high mutation rate during replication, resulting in the nonsense mutation, insertion, knockout, and even complete deletion of cas9 genes from the plasmid, thereby make the editing failure. Furthermore, the gene editing plasmids with different sgRNA and homologous repair templates must be reconstructed for editing at different sites or with different purpose, and sometimes no suitable PAM sequence can be selected for editing small fragments. In order to avoid tedious reconstruction of different CRISPR editing plasmids and to obtain an efficient and universal editing system, we combined the traditional SacB-based genome editing system with CRISPR-Cpf1 counter selection system to establish a CRISPR-Cpf1-assisted SacB gene editing system. The new universal pCS plasmid expresses relatively low toxic cpf1 (Jiang et al. 2017) and targets kan gene of SacB system, thus can be used directly and repetitively for each editing event. Most importantly, the application of pCS greatly improved the screening efficiency of second-cross-over strains in SacB system (Fig. 1C, ≥ 95% vs. 5%), which was of great significance to improve the efficiency of gene editing in C. glutamicum.
In the process of using this CRISPR-Cpf1-assisted gene editing system, both SacB and CRISPR-Cpf1 function as counter selection markers for making only the second-cross-over strains survive, so the second-cross-over strains can also be selected by using pCS directly in theory. However, when only pCS plasmid was used for counter selection and cells were cultured in LBB but not LBBS medium, only 5 colonies were obtained on LBBC plate, much less than that obtained from selection via both pCS and SacB (about 40 colonies), so SacB also played a role in enriching second-cross-over bacteria in our new editing system. Selection via both CRISPR-Cpf1 and SacB ensures the survival of only second-cross-over strains. In addition, our CRISPR-Cpf1-assisted gene editing system has certain advantages compared with CRISPR-Cpf1 system. As pCS plasmid is a universal screening tool, so there is no need to design different sgRNA for different editing requirement and pCS can be used directly for each editing. The disadvantage of our CRISPR-Cpf1-assisted gene editing system is that the editing is based on two rounds of homologous recombination via pK18mobsacB plasmid and an extra counter selection via pCS(N) plasmid, so it will take more steps to edit, but the greatly increased screening efficiency of true-positive second-cross-over strains may instead save time on repetitive verification. Compared to this tool with high screening efficiency, tools with high overall editing efficiency would be more preferable for genome editing in C. glutamicum. To further improve the efficiency of recombination, recT gene can be introduced into our editing system in future.
When integrating idoM at ldhA-pyk2 locus, PldhA was used to control the expression of idoM. The fermentation results showed that the 4-HIL titer of SC02–SC04 with idoM under PldhA was much lower than that of SC01 with idoM under PbrnFED5 (Fig. 3D). The idoM transcription levels of SC01–SC04 at 16 and 60 h were analyzed (Fig. 6A). The results showed that at 16 h, only the idoM transcription level of SC03 was higher than that of SC01, while the idoM transcription level of SC02 and SC04 were lower than that of SC01. The difference between SC03 and SC02 or SC04 was that the ldhA was retained in SC03, so lactic acid could be still synthesized in SC03. It has been reported that lactic acid can activate PldhA (Toyoda and Inui 2021). However, at 60 h, the idoM transcription level of SC02–SC04 was significantly lower than that of SC01. The great decrease of idoM transcription level in SC03 may be due to the consumption of lactic acid at stationary phase (Shi et al. 2020) and the resulting decrease of PldhA intensity. To sum up, in the presence of lactic acid, PldhA did have a high strength, even higher than PbrnFED5, but along with the consumption of lactic acid, the strength of PldhA decreased greatly, thereby PldhA was not suitable for expressing idoM.

Transcriptional changes of related genes after editing. A Relative transcription level of idoM gene of SC01–SC04 at 16 and 60 h. Values are shown relative to the control strain SC01. B Relative transcription level of lysE gene of SC01 and SC05–SC08 at 16 h. Values are shown relative to the control strain SC01.
By transferring the expression plasmid carrying PbrnFED5-idoM into the genome edited strains, the excess Ile was converted to 4-HIL completely (Fig. 5B). This indicates that expression of exogenous genes by plasmids is indeed more effective than expression in chromosome, but expression on plasmids is unstable and existence of plasmids must be ensured by antibiotics. However, the use of related antibiotics increases production costs and product purification costs. The stable expression of exogenous genes on chromosomes is the inevitable trend of commercial production, but the single-copy integration of idoM can not completely convert Ile into 4-HIL (Figs. 3D and 4B), so homologous integration of idoM at multi-copy locus of C. glutamicum or multi-copy integration strategy using transposases (Li et al. 2021) shall be explored in the future.
The promoter replacement of lysE failed to achieve simultaneous decrease in Lys accumulation and increase in 4-HIL titer (Figs. 4B and 5B). The reason for this may be as follows. Although many PdapA−N promoters with different strengths have been explored, the modified weak promoters are all relatively too weak (Vasicová et al. 1999), and the lysE transcription levels of SC05–SC08 at 16 h were all less than 10% of that of SC01 (Fig. 6B), so the substitution of PlysE with these weak PdapA−N promoters resembles the effect of lysE knockout (Chen et al. 2022). Therefore, other weak promoters such as series of synthetic promoters (Rytter et al. 2014; Shen et al. 2017; Yim et al. 2013) or regulation of other genes related to the Lys synthetic pathway can also be considered. Recently, Lys content was decreased by about 70% via negative regulation of dapA by a Lys-OFF riboswitch (Lai et al. 2022), suggesting that dynamic down-regulation of Lys synthesis may be a more effective strategy for reducing Lys content. The difficulty for statically attenuating Lys synthesis is probably because that D,L-diaminopimelic acid, an intermediate metabolite of Lys biosynthetic pathway, is important for cell growth and peptidoglycan synthesis (Hochheim et al. 2017; Hutton et al. 2007; Wehrmann et al. 1998). In addition to dynamic regulation of dapA expression and Lys synthesis by Lys-OFF riboswitch, synergistically dynamic regulation of Ile synthesis by controlling ilvA with PilvBNC and conversion of Ile to 4-HIL by controlling idoU with various Lrp-PbrnFEN prevented Ile (as the substrate and inhibitor of IDO) from excessively accumulating and coordinated the 4-HIL synthetic pathway, thereby successfullly increased the 4-HIL titer of one of 6 strains to more than 170 mM (Lai et al. 2022). This suggests that dynamic regulation of precursor supply, product conversion, and by-product generation by multi-biosensors may be beneficial to improve strain performance for 4-HIL production. However, dynamic regulation by most biosensors was performed in plasmid (Lai et al. 2022), only the regulation of PdapA by Lys-OFF riboswitch was edited in chromosome. In particular, it took us half a year for editing PdapA::PdapA-Lys-OFF riboswitch by traditional SacB editing system. The novel CRISPR-Cpf1-assisted gene editing tool explored here may help us improve the editing efficiency in C. glutamicum.
Conclusion
In this study, we designed a pCS plasmid harboring CRISPR-Cpf1 and targeting kan gene. It greatly improved the screening efficiency of the second-cross-over strains in SacB system from 5% to over 95%. Then we applied this novel editing system to integrate idoM at ldhA-pyk2 locus in 4 manners, and obtained SC01 strain with 4-HIL titer of 14.8 ± 1.1 mM. We further applied this system to replace PlysE with 4 weak PdapA−N promoters of different intensities and reduced the accumulation of Lys to different degrees. Thereby, this system is a useful tool for efficient gene editing in C. glutamicum.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Becker J, Rohles CM, Wittmann C (2018) Metabolically engineered Corynebacterium glutamicum for bio-based production of chemicals, fuels, materials, and healthcare products. Metab Eng 50:122–141. https://doi.org/10.1016/j.ymben.2018.07.008
Chen R, Xiang Y, Yu X et al (2022) Comparative transcriptome analysis of global effect of ddh and lysE deletion on 4-hydroxyisoleucine production in Corynebacterium glutamicum. Syst Microbiol Biomanuf 2(3):542–554. https://doi.org/10.1007/s43393-022-00085-9
Cho JS, Choi KR, Prabowo CPS et al (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167. https://doi.org/10.1016/j.ymben.2017.06.010
Hochheim J, Kranz A, Krumbach K et al (2017) Mutations in MurE, the essential UDP-N-acetylmuramoylalanyl-D-glutamate 2,6-diaminopimelate ligase of Corynebacterium glutamicum: effect on L-lysine formation and analysis of systemic consequences. Biotechnol Lett 39(2):283–288. https://doi.org/10.1007/s10529-016-2243-8
Hu J, Tan Y, Li Y, Hu X, Xu D, Wang X (2013) Construction and application of an efficient multiple-gene-deletion system in Corynebacterium glutamicum. Plasmid 70(3):303–313. https://doi.org/10.1016/j.plasmid.2013.07.001
Hutton CA, Perugini MA, Gerrard JA (2007) Inhibition of lysine biosynthesis: an evolving antibiotic strategy. Mol Biosyst 3(7):458–465. https://doi.org/10.1039/b705624a
Jiang Y, Qian F, Yang J et al (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179. https://doi.org/10.1038/ncomms15179
Kirchner O, Tauch A (2003) Tools for genetic engineering in the amino acid-producing bacterium Corynebacterium glutamicum. J Biotechnol 104(1–3):287–299. https://doi.org/10.1016/s0168-1656(03)00148-2
Lai W, Shi F, Tan S, Liu H, Li Y, Xiang Y (2022) Dynamic control of 4-hydroxyisoleucine biosynthesis by multi-biosensor in Corynebacterium glutamicum. Appl Microbiol Biotechnol 106(13–16):5105–5121. https://doi.org/10.1007/s00253-022-12034-6
Li C, Swofford CA, Rückert C et al (2021) Heterologous production of α-Carotene in Corynebacterium glutamicum using a multi-copy chromosomal integration method. Bioresour Technol 341:125782. https://doi.org/10.1016/j.biortech.2021.125782
Liu J, Wang Y, Lu Y, Zheng P, Sun J, Ma Y (2017) Development of a CRISPR/Cas9 genome editing toolbox for Corynebacterium glutamicum. Microb Cell Fact 16(1):205. https://doi.org/10.1186/s12934-017-0815-5
Ma W, Wang X, Mao Y, Wang Z, Chen T, Zhao X (2015) Development of a markerless gene replacement system in Corynebacterium glutamicum using upp as a counter-selection marker. Biotechnol Lett 37(3):609–617. https://doi.org/10.1007/s10529-014-1718-8
Nešvera J, Pátek M (2011) Tools for genetic manipulations in Corynebacterium glutamicum and their applications. Appl Microbiol Biotechnol 90(5):1641–1654. https://doi.org/10.1007/s00253-011-3272-9
Peng F, Wang X, Sun Y et al (2017) Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb Cell Fact 16(1):201. https://doi.org/10.1186/s12934-017-0814-6
Qiao Z, Xu M, Shao M et al (2019) Engineered disulfide bonds improve thermostability and activity of L-isoleucine hydroxylase for efficient 4-HIL production in Bacillus subtilis 168. Eng Life Sci 20(1–2):7–16. https://doi.org/10.1002/elsc.201900090
Resende BC, Rebelato AB, D’Afonseca V et al (2011) DNA repair in Corynebacterium model. Gene 482(1–2):1–7. https://doi.org/10.1016/j.gene.2011.03.008
Rytter JV, Helmark S, Chen J, Lezyk MJ, Solem C, Jensen PR (2014) Synthetic promoter libraries for Corynebacterium glutamicum. Appl Microbiol Biotechnol 98:2617–2623. https://doi.org/10.1007/s00253-013-5481-x
Sauvaire Y, Petit P, Broca C et al (1998) 4-Hydroxyisoleucine: a novel amino acid potentiator of insulin secretion. Diabetes 47(2):206–210. https://doi.org/10.2337/diab.47.2.206
Schäfer A, Tauch A, Jäger W, Kalinowski J, Thierbach G, Pühler A (1994) Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145:69–73. https://doi.org/10.1016/0378-1119(94)90324-7
Shen J, Chen J, Jensen PR, Solem C (2017) A novel genetic tool for metabolic optimization of Corynebacterium glutamicum: efficient and repetitive chromosomal integration of synthetic promoter-driven expression libraries. Appl Microbiol Biotechnol 101:4737–4746. https://doi.org/10.1007/s00253-017-8222-8
Shi F, Niu T, Fang H (2015) 4-Hydroxyisoleucine production of recombinant Corynebacterium glutamicum ssp. lactofermentum under optimal corn steep liquor limitation. Appl Microbiol Biotechnol 99(9):3851–3863. https://doi.org/10.1007/s00253-015-6481-9
Shi F, Zhang S, Li Y, Lu Z (2019) Enhancement of substrate supply and ido expression to improve 4-hydroxyisoleucine production in recombinant Corynebacterium glutamicum ssp. lactofermentum. Appl Microbiol Biotechnol 103(10):4113–4124. https://doi.org/10.1007/s00253-019-09791-2
Shi F, Fan Z, Zhang S, Wang Y, Tan S, Li Y (2020) Optimization of ribosomal binding site sequences for gene expression and 4-hydroxyisoleucine biosynthesis in recombinant Corynebacterium glutamicum. Enzyme Microb Technol 140:109622. https://doi.org/10.1016/j.enzmictec.2020.109622
Sonntag K, Eggeling L, De Graaf AA, Sahm H (1993) Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum. Quantification by 13 C- and 1H-NMR spectroscopy. Eur J Biochem 213(3):1325–1331. https://doi.org/10.1111/j.1432-1033.1993.tb17884.x
Sun M, Gao X, Zhao Z et al (2020) Enhanced production of recombinant proteins in Corynebacterium glutamicum by constructing a bicistronic gene expression system. Microb Cell Fact 19(1):113. https://doi.org/10.1186/s12934-020-01370-9
Tan Y, Xu D, Li Y, Wang X (2012) Construction of a novel sacb-based system for marker-free gene deletion in Corynebacterium glutamicum. Plasmid 67(1):44–52. https://doi.org/10.1016/j.plasmid.2011.11.001
Tan S, Shi F, Liu H et al (2020) Dynamic control of 4-hydroxyisoleucine biosynthesis by modified L-isoleucine biosensor in recombinant Corynebacterium glutamicum. ACS Synth Biol 9(9):2378–2389. https://doi.org/10.1021/acssynbio.0c00127
Toyoda K, Inui M (2021) The ldhA gene encoding fermentative L-lactate dehydrogenase in Corynebacterium glutamicum is positively regulated by the global regulator GlxR. Microorganisms 9(3):550. https://doi.org/10.3390/microorganisms9030550
Vasicová P, Pátek M, Nesvera J, Sahm H, Eikmanns B (1999) Analysis of the Corynebacterium glutamicum dapA promoter. J Bacteriol 181(19):6188–6191. https://doi.org/10.1128/JB.181.19.6188-6191.1999
Vogt M, Krumbach K, Bang WG et al (2015) The contest for precursors: channelling L-isoleucine synthesis in Corynebacterium glutamicum without byproduct formation. Appl Microbiol Biotechnol 99(2):791–800. https://doi.org/10.1007/s00253-014-6109-5
Wang T, Li Y, Li J et al (2019) An update of the suicide plasmid-mediated genome editing system in Corynebacterium glutamicum. Microb Biotechnol 12(5):907–919. https://doi.org/10.1111/1751-7915.13444
Wehrmann A, Phillipp B, Sahm H, Eggeling L (1998) Different modes of diaminopimelate synthesis and their role in cell wall integrity: a study with Corynebacterium glutamicum. J Bacteriol 180(12):3159–3165. https://doi.org/10.1128/JB.180.12.3159-3165.1998
Xu N, Wei L, Liu J (2019) Recent advances in the applications of promoter engineering for the optimization of metabolite biosynthesis. World J Microbiol Biotechnol 35(2):33. https://doi.org/10.1007/s11274-019-2606-0
Yang J, Ran Y, Yang Y et al (2021) 4-Hydroxyisoleucine alleviates macrophage-related chronic inflammation and metabolic syndrome in mice fed a high-fat diet. Front Pharmacol 11:606514. https://doi.org/10.3389/fphar.2020.606514
Yim SS, An SJ, Kang M, Lee JH, Jeong KJ (2013) Isolation of fully synthetic promoters for high-level gene expression in Corynebacterium glutamicum. Biotechnol Bioeng 110(11):2959–2969. https://doi.org/10.1002/bit.24954
Zhang C, Li Y, Ma J et al (2018) High production of 4-hydroxyisoleucine in Corynebacterium glutamicum by multistep metabolic engineering. Metab Eng 49:287–298. https://doi.org/10.1016/j.ymben.2018.09.008
Zhang J, Yang F, Yang Y, Jiang Y, Huo YX (2019) Optimizing a CRISPR-Cpf1-based genome engineering system for Corynebacterium glutamicum. Microb Cell Fact 18(1):60. https://doi.org/10.1186/s12934-019-1109-x
Zhang Y, Sun X, Wang Q et al (2020) Multicopy chromosomal integration using CRISPR-associated transposases. ACS Synth Biol 9(8):1998–2008. https://doi.org/10.1021/acssynbio.0c00073
Funding
This work was supported by the program of State Key Laboratory of Food Science and Technology (SKLF-ZZA-201904).
Author information
Authors and Affiliations
Contributions
FS conceived and designed research. RC and WL conducted experiments. RC and FS analyzed data. RC, FS, YX and GJ wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Compliance with ethics requirements
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, R., Shi, F., Xiang, Y. et al. Establishment of CRISPR-Cpf1-assisted gene editing tool and engineering of 4-hydroxyisoleucine biosynthesis in Corynebacterium glutamicum. World J Microbiol Biotechnol 39, 266 (2023). https://doi.org/10.1007/s11274-023-03705-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-023-03705-1