
Abstract
Objectives
Corynebacterium glutamicum (C. glutamicum) has been harnessed for multi-million-ton scale production of glutamate and lysine. To further increase its amino acid production for fermentation industry, there is an acute need to develop next-generation genome manipulation tool for its metabolic engineering. All reported methods for genome editing triggered with CRISPR-Cas are based on the homologous recombination. While, it requires the generation of DNA repair template, which is a bottle-neck for its extensive application.
Results
In this study, we developed a method for gene knockout in C. glutamicum via CRISPR-Cpf1-coupled non-homologous end-joining (CC-NHEJ). Specifically, CRISPR-Cpf1 introduced double-strand breaks in the genome of C. glutamicum, which was further repaired by ectopically expressed two NHEJ key proteins (Mycobacterium tuberculosis Ku and ligase D). We provide the proof of concept, for CC-NHEJ, by the successful knockout of the crtYf/e gene in C. glutamicum with the efficiency of 22.00 ± 5.56%, or something like that.
Conclusion
The present study reported a novel genome manipulation method for C. glutamicum.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Corynebacterium glutamicum is well known as the amino acid-producing workhorse of fermentation industry, being used for multi-million-ton scale production of glutamate and lysine for more than 60 years (Sanchez et al. 2017; Zahoor et al. 2012). To further increase its production efficiency, genome editing based metabolic engineering may be harnessed (Krumbach et al. 2019). In the past decades, several genome-editing techniques have been developed, employing meganucleases, zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN) and clustered regularly interspaced short palindromic repeats (CRISPR) system. To date, successes of bacterial genome editing using one type of CRISPR-Cas, CRISPR-Cpf1(also named with CRISPR-Cas12a) have been reported, including C. glutamicum(Cho et al. 2017; Jiang et al. 2017; Kim et al. 2020; Lee and Lee 2021; Peng et al. 2017; Zhang et al. 2020, 2019), Escherichia coli(Yan et al. 2017) and Streptomyces(Li et al. 2018). In these studies, CRISPR-Cas triggered double-strand breaks (DSBs) disrupt the continuity of chromosomes (Sfeir and Symington 2015). There are two major pathways for repair of DSBs, homologous recombination (HR) and error-prone non-homologous end joining (NHEJ). The HR provides a precision DNA repair with the presence of a donor DNA template to maintain the integrity of genome (Cromie et al. 2001). DSBs also could be repaired by NHEJ in eukaryotic cells (Lieber et al. 2006). While, most prokaryotic cells, including C. glutamicum (Castaneda-Garcia et al. 2017; Ishino et al. 2018), lack the NHEJ pathway (Shuman and Glickman 2007). Therefore, to perform gene inactivation or replacement in C. glutamicum, HR-based genetic engineering strategies have been extensively applied. However, efficient HR requires an exogenous donor DNA fragment as the repair template (Jager et al. 1992; Suzuki et al. 2005). To improve the editing efficiency, antibiotic resistance markers have been harnessed. While, subsequent removal of the selection marker requires additional experiments (Jager et al. 1992), which is not practical for large-scale editing at the genome level. Therefore, a simple and effective genome editing method is highly desired.
In eukaryotes, the core components of NHEJ are the Ku70/Ku80 heterodimer, which binds to DNA ends with high affinity and protects them from degradation, and DNA Ligase IV (Dnl4/Lig4), which catalyzes end ligation (Chiruvella et al. 2013). Functional prokaryotic NHEJ genes/proteins have been identified from three different types of bacteria: bacillus, mycobacteria, and pseudomonas (Bowater and Doherty 2006; Della et al. 2004; Gong et al. 2005; Weller et al. 2002; Zhu and Shuman 2005). Therefore, theoretically, genome editing in NHEJ-defect organisms would benefit via introducing NHEJ-related proteins from bacteria. Several reports showed the proof of concept, including the boosted efficiency of the CRISPR-Cas9 system in Streptomyces coelicolor by co-expressing a DNA ligase D (LigD) from Salmonella paratyphi-C (Tong et al. 2015), inactivation of E. coli genes in a HR-independent manner without the use of selective markers but with bacterial NHEJ (Su et al. 2016; Zheng et al. 2017).
In this study, we hypothesized that genome manipulation of C. glutamicum may be achieved with the introduction of an ectopically expressed bacterial NHEJ. We show that DSBs triggered with CRISPR-Cpf1 can be successfully repaired in the presence of Mycobacterium tuberculosis (Mtb) expressing NHEJ system. This report provides a platform for efficient manipulation of C. glutamicum genome.
Materials and methods
Strains and bacterial cultivation
Bacterial strains and plasmids used in this study are listed in Table 1. The E. coli DH10B was used as the host strain for molecular cloning. Strains for cloning were grown in Luria–Bertani (LB) medium (1% (w/v) tryptone, 0.5% (w/v) yeast extract, and 1% (w/v) NaCl). The C. glutamicum type strain ATCC 13032 was used as wild type. C. glutamicum strains were grown at 30 °C in BHIS medium (3.7% (w/v) brain heart infusion, 9.1% (w/v) sorbitol). For the selection of plasmids and strains, kanamycin (50 mg/L for E. coli, 25 mg/L for C. glutamicum), ampicillin (100 mg/L for E. coli), and chloramphenicol (50 mg/L for E. coli, 10 mg/L for C. glutamicum) were used. Agar was added at 1.5 g/L for plates.
Plasmid generation
Plasmids used in this study are listed in Table 1, and all primers are summarized in Supplementary Table 1 and Table 2. To ectopically express Mtb Ku and ligase D (LigD) in C. glutamicum, the corresponding encoding genes under the control of IPTG-inducible promoter Ptac were synthesized and cloned into pJYS3△crtYf plasmid (Jiang et al. 2017). The crRNA expression cassette and E. coli replicon pSC101 was replaced by an E. coli replicon p15A to construct pFY1-NHEJ. To generate plasmid pFY2, the crRNA-expression unit, which contained two Aar I restriction sites for insertion of the target sequence and driven by the synthetic constitutive promoter J23119, was synthesized and replaced the Ku-LigD expression cassette of pFY1-NHEJ plasmid. A pair of oligonucleotide DNAs that contained the crRNA sequence targeting gene crtYf/e was annealed and ligated into pFY2; then, the repair template was amplified by PCR from the genome of C. glutamicum and inserted into pFY2-crRNA to construct pFY2-cRNA-HA_△crtYf/e. The original plasmid, pEP2 (Messerotti et al. 1990), was modified by the replacement of a Kanamycin resistant marker to a chloramphenicol resistant marker to obtain pFY3. The pFY4 plasmid harbouring the crRNA-expression unit was constructed using Gibson Assembly of the PCR products amplified from pFY3 and pFY2. For the pFY4-crRNA_△crtYf/e, the crRNA sequence targeting gene crtYf/e was annealed and ligated into pFY4. DNA sequencing confirmed the desired specific sequence in the constructs.
The lethal-reporter system
DSBs disruption of the continuity of chromosomes by functional CRISPR-Cpf1-crRNA complex is lethal to C. glutamicum that lacks NHEJ (Castaneda-Garcia et al. 2017; Ishino et al. 2018; Zhang et al. 2019). The lethality rate of CRISPR-Cpf1-mediated genome editing can verify the efficiency of crRNA. The survival ratio for each crRNA is calculated by counting colonies formed. This ratio was further normalized by determining the colony number after transformation with a negative control crRNA plasmid.
Genome editing protocol
Electrocompetent C. glutamicum was prepared as previously described (Chen et al. 2014). In the CRISPR-Cpf1-coupled non-homologous end-joining experiment, competent cells of C. glutamicum harboring pFY1-NHEJ were prepared. To induce the expression of Ku and LigD, C. glutamicum was treated with IPTG (final concentration of 1 mM). Before electroporation, plasmid-free C. glutamicum cells or those harboring the pFY1-NHEJ plasmid were thawed on ice, mixed with 500 ng of the pFY2-crRNA-HA_△crtYf/e or pFY4-crRNA_△crtYf/e, and then transferred into 4 °C pre-cooled electroporation cuvettes (Bio-Rad, Hercules, USA). Electroporation was performed at 25 μF, 1.25 kV/cm, and 200Ω. Cells were immediately transferred into 1 mL of pre-warmed (46 °C) BHIS medium and heat-shocked for 6 min at 46 °C without shaking. The cells were grown to recover for 2–4 h at 30 °C with shaking at 200 rpm. Cells were then spread on BHIS containing 25 μg/mL kanamycin (BHIS-kar), or 25 μg/mL kanamycin and 10 μg/mL chloramphenicol (BHIS-kar-cmr), and incubated for 2 days.
DNA isolation
Genomic DNA was isolated from C. glutamicum using MiniBEST Bacteria Genomic DNA Extraction Kit Ver.3.0 (Takara, Beijing, China), and quantified with a NS9300 Ulrtramicro Spectrometer (Allfine Medlab, Guangdong, China). The fragments harboring the targeted sites were amplified with specific primers (Table S3), and the amplicons were purified with a FastPure Gel DNA Extraction Mini Kit (Vazyme, Hangzhou, China).
Editing efficiency calculation
To evaluate the efficiency of gene editing, we selected the crtYe/f gene in C. glutamicum as a reporter gene, because its inactivation results in a color change from yellow to red, which could be directly observed by eye (Krubasik et al. 2001). The editing efficiency was calculated by dividing the number of edited single colonies on the selection plate by the total number of colonies on the control plate.
Results
Design of the CRISPR-Cpf1 coupled non-homologous end-join (CC-NHEJ)
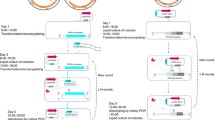
So far, there was no report for CRISPR-Cas mediated genome editing in the absence of a donor DNA (exogenous DNA repair template) in C. glutamicum. To address it, here we sought to establish a novel method to manipulate its genome (Fig. 1). Because it is reported that Cpf1 could trigger DSBs in C. glutamicum (Zhang et al. 2019), here we generated a plasmid (pFY1-NHEJ), which has the gene expression cassette for Cpf1, also it contained IPTG-inducible gene expression of Mtb NHEJ (Table1). The Mtb NHEJ was originally from Mycobacterium tuberculosis, which includes only two key proteins, Ku and ligase D (LigD) (Della et al. 2004). The Mtb NHEJ has been shown to be functional in some bacteria, including Mycobacterium smegmatis and E. coli (Sun et al. 2018a; Zheng et al. 2017). Another plasmid (pFY2) expressing crRNAs for Cpf1 targeting genome was also assembled. As to the genome editing procedure, the plasmid pFY1-NHEJ was electroporated into the host. The Mtb NHEJ and Cpf1 (here is one type of Cpf1, FnCpf1) were ectopically expressed in C. glutamicum. Then the crRNA expression plasmid was electrotransformed into the C. glutamicum harboring pFY1-NHEJ, which may result in the DSBs at desired genomic loci. The NHEJ pathway would repair the DSBs, and under the selection condition, the survived cells would be knockout triggered with the CRISPR-Cpf1 at the target genomic loci (Fig. 1). The candidate edited colonies would be further confirmed.

Schematic strategy of CC-NHEJ for one-step gene inactivation in C. glutamicum. FnCpf1 and Mtb NHEJ proteins (Mtb Ku and Mtb LigD) were pre-expressed in host cells, and then crRNA was transformed into the host cells. The crRNA-Cpf1 complex cuts the double-strand DNA proximal to a PAM site, generating DSBs. The Ku homodimer bound to DNA ends and recruited LigD, probably via a direct physical interaction on the DNA between Ku and LigD, resulting in ligation of the two ends of DNA. Imprecise repair of DSBs leads to the frameshift mutation. Only the DSBs-repaired colonies would be survived
Knockout of crtYf/e gene on the chromosome using CC-NHEJ
It is reported that C. glutamicum contains glycosylated C50 carotenoid decaprenoxanthin would be in yellow (Heider et al. 2012). Carotenoids are yellow to red-colored pigments originating from the terpenoid biosynthetic pathway. The mutant △crtY lacking the final reaction in the synthesis of decaprenoxanthin, i.e., the introduction of two ε-ionone groups into the acyclic flavuxanthin catalyzed by gene products of crtYf/e, accumulated flavuxanthin and exhibited a red color (Heider et al. 2012; Netzer et al. 2010), in contrast to cells with wild-type crtYf/e of yellow-colored. Kim et al. combined oligonucleotide-directed mutagenesis with negative screening by CRISPR-Cpf1 system to target crtEb, then calculating editing efficiency through color changes after lycopene accumulation (Kim et al. 2020). Zhang et al. also targeted the gene crtYf on carotenoid biosynthesis cluster to achieve efficient gene editing (Lee and Lee 2021; Zhang et al. 2020), therefore, crtYf is a valid proof-of-concept target. To measure the efficiency of knockout in C. glutamicum, we took advantage of crtYf/e gene as a reporter because it is easy to distinguish the wild-type and crtYf/e knockout mutant colonies. Specifically, in the presence of Cpf1-crRNA complex and NHEJ, if Cpf1-crRNA and NHEJ are both functional, wild-type crtYf/e would be disrupted and the cells(colonies) would be in red due to the accumulated flavuxanthin, otherwise, crtYf/e gene would still be intact and colonies would remain in yellow (Fig. 2a). Thus, the editing efficiency of △crtYf/e could be easily assessed by quantifying the fraction of red colonies.

CC-NHEJ mediated crtYf/e knockout. a The scheme of crtYf/e knockout as a visual reporter to assess editing efficiency. b Schematic representation of pFY1-NHEJ and pFY4-crRNA-△crtYe/f plasmid used for experiment. c The C. glutamicum competent cells harboring pFY1-NHEJ plasmids were transformed with pFY4-crRNA-△crtYe/f plasmid (500 ng). The images were obtained at 48 h post transformation (△crtYe/f colored in red; WT colored in yellow). d CC-NHEJ mediated indels formation at target sites in crtYf/e. Red nucleotides indicated PAM sequence, pink nucleotides indicated crRNA sequence, black lines indicated indels. See also Supplementary Fig. 2
To identify activated crRNA for genome editing of Cpf1 endonuclease, we first designed three crRNAs targeting crtYf/e gene (Supplementary Fig. 1a and Supplementary Table 1). Then we used a lethal-reporter system (Zhang et al. 2019) to screen the crRNA with relative higher activity, which was a fast and convenient way to evaluate the activity of different crRNA (see also materials and methods). We inserted three crRNAs (crRNA1, crRNA2 and crRNA3) into Cpf1 backbone plasmid to construct three pCpf1_△crtYe/f plasmids, respectively. The plasmid pCpf1 without crRNA insertion was used as its negative control. After transformation and editing, the survival ratio for each crRNA was calculated by the number of colonies on the plate divided by the number of colonies on the plate developed from transformants containing pCpf1 (Supplementary Fig. 1b). After testing, we found that crRNA3 leads to the lowest survival ratio, which indicated that its targeting efficiency is relative higher, compared with that of crRNA1 or crRNA2. Thus, crRNA3 was selected for the further experiment.
To investigate the effects of the CC-NHEJ, crtYf/e gene was chosen as the reporter gene. As shown in Fig. 2b, an expression vector containing the crRNA3 was constructed and named pFY4-crRNA-△crtYe/f, carrying the chloramphenicol (cmr) resistance marker. The pFY1-NHEJ plasmid with the IPTG-inducible Mtb NHEJ, Cpf1 protein-encoding gene, and the kanamycin (kar) resistant marker was transformed into C. glutamicum, generating the C. glutamicum pFY1-NHEJ competent cells. With the selection, we found that colonies of the CC-NHEJ group turned red (Fig. 2c). Some individual red colonies were randomly picked, cultured and the genomic DNA was extracted. PCR primers (Supplementary Table 3) were designed to amplify a product size of ~ 800-bp harboring the target site. PCR products were inserted into cloning vector and then transformed into E. coli competent cells. Individual colonies were sequenced and ten representative sequencing results are shown in Fig. 2d. Deletions of different lengths within the target regions were observed (Fig. 2d and Supplementary Fig. 2). Taken together, these data indicate that the introduction of ectopically expression of Mtb NHEJ may enable the C. glutamicum to perform NHEJ repair. The CC-NHEJ may be harnessed for inactivation of genes in the genome of C. glutamicum.
Comparison of genome editing achieved with HR and CC-NHEJ
To parallel compare the editing efficiency of the CC-NHEJ and HR, we performed a HR-based genome editing study, which contains three elements: a constitutive Cpf1 expression cassette, a crRNA expression cassette and a donor DNA template for repairing the DSBs triggered with the Cpf1-crRNA (Fig. 3a). Survived colonies appeared on selected plates after 24 to 36 h incubation at 30 °C, and the colonies were counted (Fig. 3b). Eight red single colonies were randomly picked and individually incubated in BHIS medium (kar) overnight and their genomic DNA was extracted. The crtYe/f gene knockout was verified for each colony by PCR, using primers that bind upstream of the 5′ homologous arm and downstream of the 3′ homologous arm (Fig. 3c and Supplementary Table 3). The PCR results showed that the eight red single colonies were successfully edited; approximately 36.0% of the colonies were correctly edited, according to their color (Fig. 3c). Then, we compared the efficiency via HR and NHEJ. The crtYe/f gene knockout efficiencies were 33.60 ± 7.76% and 22.00 ± 5.56% for HR and CC-NHEJ, respectively (Fig. 3d). These findings indicate that the HR-based genetic engineering approach would lead to relative higher efficiency than that of CC-NHEJ. While, CC-NHEJ greatly simplifies the editing procedure because it does not require a DNA repair template.

Comparison of HR and CC-NHEJ. a Schematics of the donor DNA and targeting strategy for CRISPR/Cpf1-mediated homology recombination and crtYf/e would be deleted after editing. Black lines indicated sections of homology between the genomic locus and the donor DNA. Positions of PCR primers used for detecting of crtYf/e gene knockout were shown. b The C. glutamicum competent cells were transformed with pFY2-crRNA-HA-△crtYe/f plasmid (500 ng), and images were obtained at 48 h post transformation (△crtYe/f colored in red; WT colored in yellow). c PCR validation of crtYf/e gene deletion using homology recombination. The F13/R13 primers bind outside of the homologous arms. The amplified fragments are 3025 bp (wild-type) and 2320 bp (△crtYe/f strain), respectively. d Comparison of editing efficiency via HR and CC-NHEJ for crtYf/e knockout
Discussion
Genetic engineering strategies based on the CRISPR-Cas system have been successfully performed in C. glutamicum, promoting its application for fermentation industry (Cho et al. 2017; Jiang et al. 2017). Jiang et al. introduced CRISPR-Cpf1 into C. glutamate for the first time, and the recombinant efficiency of single-stranded DNA reached 100%, the efficiency of gene integration and knockout was 5–15% (Jiang et al. 2017). Zhang et al. combined the CRISPR-Cpf1 system with SacB to improve editing efficiency, the efficiency of 7.5 kb large fragment knockout reached 100%, and the efficiency of 705 bp gene knockout and insertion of 3 kb gene at the same time reached 61% (Zhang et al. 2020). Kim et al. also achieved 99.7% gene editing using CRISPR-Cpf1 and mismatched crRNA (Kim et al. 2020). Cas9 protein is toxic to C. glutamate, which was solved by codon optimization of Cas9 protein (Cho et al. 2017), using inducible promoter (Peng et al. 2017) or integrating it into the genome (Wang et al. 2018) for low-level expression. Cho et al. introduced CRISPR-Cas9 combined with recT into C. glutamate for the first time (Cho et al. 2017). Peng et al. used the inducible expression system to express the CRISPR-Cas9 system, with gene deletion and insertion efficiency of 60–100% (Peng et al. 2017). Wang et al. combined CRISPR-Cas9 with RecET and integrated it into the genome, achieving up to 20 kb gene knockout and 7.5 kb gene insertion (Wang et al. 2018). Existing gene editing tools have effectively implemented C. glutamate gene editing, and this study, as a complementary research work, expanded the C. glutamate gene editing tools.
We introduced NHEJ from Mycobacterium tuberculosis, and successfully knockout crtYe/f gene. Compared with HR-based genome editing, the CC-NHEJ method does not require the construction of a DNA repair template. Thus, only plasmids to target gene are essential for the genome editing. We acknowledge that, compared with HR-based genome editing, the gene-editing efficiency of the CC-NHEJ is slightly lower. We also tried single plasmid (all-in-one) for gene editing, which consists of Cpf1 and crRNA, and Mtb NHEJ, while, for some reason, we could not get the positive colonies. Further optimization may be required, including the optimization of NHEJ proteins from additional species, promoters to drive NHEJ gene(s) expressions, CRISPR-Cas expression cassettes, small-molecule enhancer, and optimized codon for ectopically expressed genes. Also, additional studies for testing more genes would be required for optimization of the platform.
The delivery of ectopically expressed genes is plasmids in the present study. The plasmids may be incorporated into the genome of C. glutamicum, especially with the presence of DSBs triggered with Cpf1, which may be confounding experimental results. Thus, another approaches, i.e., RNP formed with recombinant proteins and RNAs may be considered because RNP form in C. glutamicum may eradicate the DNA contamination (Kim et al. 2014). In addition, the base-editing (Gaudelli et al. 2017; Komor et al. 2016) and primer editing (Anzalone et al. 2019) may be additional approach for the manipulation of C. glutamicum genome.
The metabolic engineering of industrial strains often requires editing multiple genes simultaneous. Cpf1 has been harnessed as a tool for genome manipulation to simultaneously target multiple genes with only a single customized crRNA array in human cells (Sun et al. 2018b) and this strategy may be adopted for the manipulation of C. glutamicum. If these ectopically expressed NHEJ and Cpf1 genes could be inserted into the specific genomic locus of C. glutamicum, only a single customized crRNA array to target multiple genes would trigger genome editing, which would greatly boost functional gene dissection and directional molecular metabolic engineering of C. glutamicum.
In conclusion, we illustrated that, without donor DNA, C. glutamicum genome editing via CC-NHEJ could be successfully achieved in the present study. Our study would pave a way for the genome mining and metabolic engineering of C. glutamicum.
References
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A et al (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–157
Bowater R, Doherty AJ (2006) Making ends meet: repairing breaks in bacterial DNA by non-homologous end-joining. Plos Genet 2:93–99
Castaneda-Garcia A, Prieto AI, Rodriguez-Beltran J, Alonso N, Cantillon D, Costas C, Perez-Lago L, Zegeye ED, Herranz M, Plocinski P et al (2017) A non-canonical mismatch repair pathway in prokaryotes. Nat Commun 8:14246
Chen T, Zhu N, Xia H (2014) Aerobic production of succinate from arabinose by metabolically engineered Corynebacterium glutamicum. Bioresour Technol 151:411–414
Chiruvella KK, Liang ZB, Wilson TE (2013) Repair of double-strand breaks by end joining. Csh Perspect Biol 5:a012757
Cho JS, Choi KR, Prabowo CPS, Shin JH, Yang D, Jang J, Lee SY (2017) CRISPR/Cas9-coupled recombineering for metabolic engineering of Corynebacterium glutamicum. Metab Eng 42:157–167
Cromie GA, Connelly JC, Leach DR (2001) Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell 8:1163–1174
Della M, Palmbos PL, Tseng HM, Tonkin LM, Daley JM, Topper LM, Pitcher RS, Tomkinson AE, Wilson TE, Doherty AJ (2004) Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science (New York, NY) 306:683–685
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551:464–471
Gong CL, Bongiorno P, Martins A, Stephanou NC, Zhu H, Shuman S, Glickman MS (2005) Mechanism of nonhomologous end-joining in mycobacteria: a low-fidelity repair system driven by Ku, ligase D and ligase C. Nat Struct Mol Biol 12:304–312
Heider SA, Peters-Wendisch P, Wendisch VF (2012) Carotenoid biosynthesis and overproduction in Corynebacterium glutamicum. BMC Microbiol 12:198
Ishino S, Skouloubris S, Kudo H, I’Hermitte-Stead C, Es-Sadik A, Lambry JC, Ishino Y, Myllykallio H (2018) Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication. Nucleic Acids Res 46:6206–6217
Jager W, Schafer A, Puhler A, Labes G, Wohlleben W (1992) Expression of the Bacillus subtilis sacB gene leads to sucrose sensitivity in the gram-positive bacterium Corynebacterium glutamicum but not in Streptomyces lividans. J Bacteriol 174:5462–5465
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y et al (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179
Kim S, Kim D, Cho SW, Kim J, Kim JS (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019
Kim HJ, Oh SY, Lee SJ (2020) Single-Base Genome Editing in Corynebacterium glutamicum with the Help of Negative Selection by Target-Mismatched CRISPR/Cpf1. J Microbiol Biotechnol 30:1583–1591
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424
Krubasik P, Takaichi S, Maoka T, Kobayashi M, Masamoto K, Sandmann G (2001) Detailed biosynthetic pathway to decaprenoxanthin diglucoside in Corynebacterium glutamicum and identification of novel intermediates. Arch Microbiol 176:217–223
Krumbach K, Sonntag CK, Eggeling L, Marienhagen J (2019) CRISPR/Cas12a mediated genome editing to introduce amino acid substitutions into the mechanosensitive channel MscCG of Corynebacterium glutamicum. ACS Synth Biol 8:2726–2734
Lee HJ, Lee SJ (2021) Advances in accurate microbial genome-editing CRISPR technologies. J Microbiol Biotechnol 31:903–911
Li L, Wei K, Zheng G, Liu X, Chen S, Jiang W, Lu Y (2018) CRISPR-Cpf1-assisted multiplex genome editing and transcriptional repression in Streptomyces. Appl Environ Microbiol. https://doi.org/10.1128/AEM.00827-18
Lieber MR, Yu K, Raghavan SC (2006) Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair (Amst) 5:1234–1245
Messerotti LJ, Radford AJ, Hodgson AL (1990) Nucleotide sequence of the replication region from the Mycobacterium-Escherichia coli shuttle vector pEP2. Gene 96:147–148
Netzer R, Stafsnes MH, Andreassen T, Goksoyr A, Bruheim P, Brautaset T (2010) Biosynthetic pathway for gamma-cyclic sarcinaxanthin in Micrococcus luteus: heterologous expression and evidence for diverse and multiple catalytic functions of C(50) carotenoid cyclases. J Bacteriol 192:5688–5699
Peng F, Wang X, Sun Y, Dong G, Yang Y, Liu X, Bai Z (2017) Efficient gene editing in Corynebacterium glutamicum using the CRISPR/Cas9 system. Microb Cell Fact 16:201
Sanchez S, Rodríguez-Sanoja R, Ramos A, Demain AL (2017) Our microbes not only produce antibiotics, they also overproduce amino acids. J Antibiot (Tokyo). https://doi.org/10.1038/ja.2017.1142
Sfeir A, Symington LS (2015) Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem Sci 40:701–714
Shuman S, Glickman MS (2007) Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol 5:852–861
Su T, Liu F, Gu P, Jin H, Chang Y, Wang Q, Liang Q, Qi Q (2016) A CRISPR-Cas9 assisted non-homologous end-joining strategy for one-step engineering of bacterial genome. Sci Rep 6:37895
Sun BB, Yang JJ, Yang S, Ye RD, Chen DJ, Jiang Y (2018a) A CRISPR-Cpf1-assisted non-homologous end joining genome editing system of Mycobacterium smegmatis. Biotechnol J 13:1700588
Sun H, Li F, Liu J, Yang F, Zeng Z, Lv X, Tu M, Liu Y, Ge X, Liu C et al (2018b) A single multiplex crRNA array for FnCpf1-mediated human genome editing. Mol Ther 26:2070–2076
Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H (2005) New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl Environ Microb 71:8472–8480
Tong Y, Charusanti P, Zhang L, Weber T, Lee SY (2015) CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029
Wang B, Hu Q, Zhang Y, Shi R, Chai X, Liu Z, Shang X, Zhang Y, Wen T (2018) A RecET-assisted CRISPR-Cas9 genome editing in Corynebacterium glutamicum. Microb Cell Fact. https://doi.org/10.1186/s12934-018-0910-2
Weller GR, Kysela B, Roy R, Tonkin LM, Scanlan E, Della M, Devine SK, Day JP, Wilkinson A, d’Adda di Fagagna F et al (2002) Identification of a DNA nonhomologous end-joining complex in bacteria. Science (New York, NY) 297:1686–1689
Yan MY, Yan HQ, Ren GX, Zhao JP, Guo XP, Sun YC (2017) CRISPR-Cas12a-assisted recombineering in bacteria. Appl Environ Microb. https://doi.org/10.1128/AEM.00947-17
Zahoor A, Lindner SN, Wendisch VF (2012) Metabolic engineering of Corynebacterium glutamicum aimed at alternative carbon sources and new products. Comput Struct Biotechnol J 3:e201210004–e201210004
Zhang J, Yang F, Yang Y, Jiang Y, Huo YX (2019) Optimizing a CRISPR-Cpf1-based genome engineering system for Corynebacterium glutamicum. Microb Cell Fact 18:60
Zhang J, Qian F, Dong F, Wang Q, Yang J, Jiang Y, Yang S (2020) De Novo Engineering of Corynebacterium glutamicum for l-proline production. ACS Synth Biol 9:1897–1906
Zheng X, Li SY, Zhao GP, Wang J (2017) An efficient system for deletion of large DNA fragments in Escherichia coli via introduction of both Cas9 and the non-homologous end joining system from Mycobacterium smegmatis. Biochem Bioph Res Co 485:768–774
Zhu H, Shuman S (2005) A primer-dependent polymerase function of pseudomonas aeruginosa ATP-dependent DNA ligase (LigD). J Biol Chem 280:418–427
Funding
The authors would like to acknowledge financial support of the National Natural Science Foundation of China (No. 31871247), Central Public-interest Scientific Institution Basal Research Fund (No. Y2019PT10, Y2020XK18) and Chinese Academy of Agricultural Sciences grants (No. CAAS-Y2019YJ07-03).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, FY., Wei, N., Zhang, ZH. et al. Genome editing of Corynebacterium glutamicum mediated with Cpf1 plus Ku/LigD. Biotechnol Lett 43, 2273–2281 (2021). https://doi.org/10.1007/s10529-021-03195-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-021-03195-x