
Abstract
This study aimed to characterize a powerful antifungal component from bacteria. Bacillus subtilis strain XB-1, which showed maximal inhibition of Monilinia fructicola, was isolated and identified, and an antifungal protein was obtained from it. Ammonium sulfate precipitation, ion exchange chromatography, and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were used to purify and identify the proteins secreted by B. subtilis XB-1. Analyses revealed that purified fraction V had the strongest antifungal effect, with the largest pathogen inhibition zone diameter of 4.15 cm after 4 days (P < 0.05). This fraction showed a single band with a molecular weight of approximately 43 kDa in SDS-PAGE. Results from SDS-PAGE and liquid chromatography electrospray ionization tandem mass spectrometry analyses demonstrated that fraction V was likely a member of the chitosanase family. These results suggest that B. subtilis XB-1 and its antifungal protein may be useful in potential biocontrol applications.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brown rot is one of the most destructive diseases of stone and pome fruits. The incidence of this disease can be up to 80% both pre- and post-harvest in Spain and United States (Gell et al. 2008). Monilinia spp., particularly Monilinia fructicola, are the major pathogenic fungi causing brown rot (Poniatowska et al. 2013; Eguen et al. 2015). Currently, management of this disease relies heavily on the use of synthetic chemical fungicides (Yang et al. 2012). However, the negative impacts of chemical fungicides on the environment and public health, as well as the increasing development of pathogen resistance, have created demand for new strategies for fruit disease control (Ma et al. 2003; Casals et al. 2012; Dukare et al. 2018). Therefore, safer and more ecologically friendly alternative fungicides to control postharvest diseases and decay are being developed globally.
Among possible approaches, biocontrol via antagonistic microbes is regarded as a promising and attractive option (Mizumoto and Sawa 2007; Dukare et al. 2018). In recent years, bacterial and fungal antagonists including Bacillus, Pseudomonas, Pichia, and lactic acid bacteria have been identified as biocontrol agents for postharvest pathogens (Haas and Defago 2005; Lastochkina et al. 2019). Generally, these antagonists occupy the same niche as the pathogens, and also produce a wide range of antibiotic substances (Liu et al. 2012). For example, Bacillus spp. are reported to inhibit pathogens by producing antibiotics, enzymes, and antifungal volatile compounds (Wang et al. 2014; Kilani-Feki et al. 2016; Liu et al. 2018). However, the complexity of microbes and their metabolites creates difficulty in determining the most abundant and effective antifungal compounds.
The objectives of this study were to screen effective antifungal strains from among rhizosphere microbes to identify potential biocontrol agents and determine their main antifungal proteins against M. fructicola. The proteins secreted by the most effective strain were precipitated with ammonium sulfate, purified through fast protein liquid chromatography and gel filtration chromatography, and identified using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and liquid chromatography electrospray ionization tandem mass spectrometry (LC–ESI–MS/MS).
Materials and methods
Microorganisms and culture conditions
Soil samples collected from maize roots on the experimental farm of Beijing University of Agriculture (Beijing, China) were diluted tenfold and incubated at 37 °C on an orbital shaker at 160 rpm for 10 min. Serial dilutions of 10- to 1000-fold in sterile water were made, and 0.1 mL of each dilution was placed onto a lysogeny broth (LB) agar medium plate and incubated at 37 °C until most bacterial colonies became visible (Testa et al. 2003). A total of 24 colonies with distinctive morphologies were picked and streaked on agar nutrient media to obtain pure cultures.
The fungal strains (Botrytis cinerea, Colletotrichum gloeosporioides, Verticillium dahliae Kleb., Monilinia laxa, Ralstonia solanacearum, Polyporus hirsutus, Fusarium moniliforme, and M. fructicola) were isolated from various hosts and identified by the Department of Plant Science and Technology, Beijing University of Agriculture (Beijing, China). They were incubated on potato dextrose agar (PDA) plates for 4 days at 27 °C in an incubator.
Determination of antagonistic activity of the isolated bacteria
Each of the test bacterial strains was grown in LB liquid medium at 37 °C for 24 h, and M. fructicola was grown on PDA plates at 28 °C for 4 days prior to the bioassay tests. The antifungal effect was determined by placing a 5-mm diameter fungal colony in the center of a PDA plate, with 1 μL of the test bacterial suspension from LB culture 20 mm away on each side. Colony growth and the type of interaction were examined daily under a stereomicroscope (Innocenti et al. 2003). Inhibition was determined from the inhibition zone between the bacterial and fungal strains (Hernandez-Rodriguez et al. 2008). From these analyses, the isolate designated XB-1 exhibited the strongest antifungal activity. This isolate was then tested against a range of fungi to determine the spectrum of its antifungal activity.
Amplification of the 16S rRNA gene of isolate XB-1 via polymerase chain reaction (PCR)
To identify and further characterize strain XB-1, we combined standard biochemical methods with 16S rRNA gene sequence analysis. Chromosomal DNA was prepared for PCR using a bacterial genomic DNA preparation kit (Tiangen, Beijing, China). PCR was performed using the general bacterial primers 27f (5′-AGAGTTTGATCCTGGCTCAG-3′) (Invitrogen, Carlsbad, CA, USA) and 1492r (5′-GGTTACCTTGTTACGACTT-3′) (Invitrogen). Primers bound to conserved regions of this gene, whereas the divergent regions were sequenced for identification. PCR was performed using the MyCycler S1000 (Bio-Rad, Hercules, CA, USA). Amplification was carried out in a 25-μL reaction tube using a PCR master mix kit (Invitrogen) according to the manufacturer’s instructions, with the following reaction conditions: an initial denaturation step at 95 °C for 5 min, followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 60 s, followed by a final extension step at 72 °C for 5 min. The PCR products were purified with an Axygen kit (Invitrogen). The purified products were sequenced using the same primers on an Applied Biosystems 3730XL genetic analyzer (Foster City, CA, USA) according to the manufacturer’s instructions. The sequences obtained were subjected to a BLAST search against the GenBank database.
Purification and identification of the antifungal protein
In order to obtain more secondary metabolites, the strains that showed strong antagonistic activity against M. fructicola were cultured at 37 °C for 48 h at 160 rpm in LB medium, followed by centrifugation at 12,000 rpm and 4 °C for 15 min. The bacterial suspensions prepared for efficacy assays were divided into eight tubes with different saturations of ammonium sulfate (30%, 40%, 50%, 60%, 70%, 80%, 90%, and 100%), and incubated at 4 °C for 48 h. The precipitate was collected through centrifugation at 12,000 rpm and 4 °C for 15 min, dissolved in distilled water, and dialyzed using a 25-kDa filter (Millipore, Burlington, MA, USA) at 4 °C for 8 h to remove (NH4)2SO4. The washing procedure was repeated three times to obtain the crude protein, the activity of which was then evaluated. Crude protein (10 mL) was dissolved in Tris–HCl buffer (pH 8.0) and applied to fast protein liquid chromatography (AKTA Avant 25, GE Healthcare, Chicago, IL, USA) using a diethyl-aminoethyl (DEAE) anion exchange column (GE Healthcare) equilibrated with 20 mmol/L Tris–HCl buffer (pH 8.0). The buffer was eluted with a linear gradient of NaCl (0–1.0 M) at a flow rate of 1 mL/min. Each fraction was collected in a volume of 10 mL and monitored at 280 nm, and pooled fractions were freeze-dried and subsequently investigated for antifungal activity. The active fraction exhibiting maximum antifungal activity was further purified through gel filtration chromatography. That fraction was dissolved in distilled water and loaded onto a Sephadex G-25 gel filtration column (2.5 cm × 75 cm) that had been previously equilibrated with distilled water. The column was then eluted with distilled water at a flow rate of 1 mL/min. Each 5-mL fraction was monitored at 280 nm; pooled fractions were freeze-dried and tested for antifungal activity. Pooled fractions were concentrated using a rotary evaporator, and their antifungal activities were investigated. Lyophilized proteins were resolved through SDS-PAGE and stained with Coomassie blue. The molecular weight of the antifungal protein was determined by comparing its electrophoretic mobility against those of marker proteins purchased from Solarbio (Beijing, China). The sequence of the purified protein was determined through ESI–MS/MS (Kumar et al. 2012).
Accurate molecular mass and peptide sequencing of the purified protein was carried out via ESI–MS and MS/MS in positive ion mode. The purified protein was dissolved in 75% aqueous acetonitrile solution of HPLC grade, and then loaded into the FIA type 3200 QTRAP mass spectrometer (Applied Biosystems). The sample flow rate was 20 μL/min. The drying (35 psi) and ESI nebulizing (45 psi) gas was high purity nitrogen. Spectra were recorded over the mass/charge (m/z) range of 200–1000. The peptide sequence obtained was subjected to a search against the Mascot database.
Antifungal activity of the precipitated and purified culture filtrates
The precipitated and purified bacterial culture filtrates from the antagonistic bacteria were concentrated via rotary evaporation and filtered through sterile 0.45-μm Millipore filters. The inhibitory effects of these crude extracts toward M. fructicola were assessed using a method modified from that of Hastings and Kirby (1966). Briefly, 10 mL of extract was first mixed with 90 mL of molten PDA culture (45 °C) and then spread on 90-mm plates. Mycelial disks (5 mm in diameter) from 4-days-old fungal cultures were placed in the center of each plate. The control experiment was conducted using standard PDA medium without bacterial extracts. All plates were incubated at 28 °C for 4 days. Two perpendicular measurements of the diameter of the fungal colony were taken using a stereo zoom microscope. The inhibitory ability was calculated by measuring the diameter in two perpendicular directions on the fungal culture. The experiment was repeated three times, with five replicates per treatment.
Statistical analysis
All assays for antifungal activity were conducted in triplicate. The data are reported as mean ± standard deviation. Statistical analyses were performed using SPSS 10.0 software (SPSS, Chicago, IL, USA). Significant differences were determined based on 95% confidence intervals (P < 0.05).
Results
Isolation and determination of the antagonistic strains
A total of 24 colonies with morphological differences were picked and streaked on agar nutrient media to obtain pure cultures, seven of which showed antifungal activity. Their antifungal effects against M. fructicola are presented in Fig. 1a. The inhibitory zone between isolate XB-1 and M. fructicola was 10.9 mm after 4 days of incubation (Fig. 1b), the largest among the seven isolated strains, indicating the strongest antifungal ability (P < 0.05). The spectrum of its antifungal ability was also examined. The strain also showed strong inhibitory effects against B. cinerea, M. laxa, and F. moniliforme (Table 1). As a result, strain XB-1 was selected for further study.

Antifungal effects of bacteria on M. fructicola after 4 days of incubation. a The inhibitory zones of seven different bacteria on M. fructicola. b The antifungal effect of strain XB-1. Means followed by different letters in a indicate significant differences among strains at P < 0.05
PCR amplification and identification of the 16S rRNA gene of XB-1
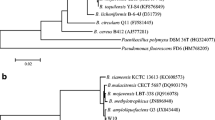
The phylogenetic relationships of strain XB-1 with other bacteria can be inferred through alignment analysis with homologous nucleotide sequences of known bacteria. After amplification and sequencing, the sequences obtained were subjected to a BLAST search against the GenBank database. The 16S rRNA gene sequence of XB-1 was searched in GenBank, and was highly homologous to that of Bacillus subtilis. Further, among all B. subtilis sequences, the highest homology was with B. subtilis (99.93%, Accession number: NR 102783.2). A phylogenetic tree was constructed using MEGA7 software, selecting bacteria with high homology (Fig. 2). The conclusion was that the strain XB-1 belongs to B. subtilis, and thus it was named B. subtilis XB-1. To further explore its active metabolites, we extracted the antifungal substances of B. subtilis XB-1 and performed the following analyses.

Dendrogram based on the 16S rRNA gene sequence of strain XB-1 constructed using the neighbor-joining method. The GenBank accession number of each type strain’s 16S rRNA gene sequence is shown after the type strain
Isolation of antifungal metabolites from B. subtilis XB-1
Crude protein from B. subtilis XB-1 culture was precipitated in vitro using (NH4)2SO4 solutions of various saturation levels. Figure 3a shows the diameters of the pathogen in the bioassay to analyze the inhibitory abilities of the precipitates. The greatest inhibitory effect, with a diameter of 3.91 cm after 4 days, was exhibited by the precipitate in 70% saturated solution (P < 0.05). Details of the effects identified through this assay are depicted in Fig. 3b.

Inhibitory effects of crude and purified proteins on M. fructicola. a Crude proteins precipitated at various saturation levels of (NH4)2SO4 (2–4 days). b Crude proteins precipitated at various saturation levels of (NH4)2SO4 (4 days). c Proteins purified with a DEAE anion exchange column (2–4 days). d Proteins purified with a Sephadex G-25 gel filtration column (2–4 days). Means followed by different letters in C and D indicate significant differences among proteins over the same incubation time at P < 0.05
Following dialysis and lyophilization, the precipitate in 70% saturation (NH4)2SO4 was isolated using an anion exchange column at pH 8.0 with a linear gradient concentration of NaCl (0–1 M). Further purification of the protein was performed using ion exchange chromatography and the product was monitored with an antifungal assay. The results of the antifungal assay, shown in Fig. 3c, indicated that fraction F4 had the highest activity, with a pathogen diameter of 4.5 cm, which was the smallest among all fractions at all measurement times (P < 0.05), despite the diameter of the pathogen increasing over time. Fraction F4 was further purified using a gel filtration column. As shown in Fig. 4a, seven peaks were eluted from this fraction. Among these peaks, fraction V showed the highest level of activity (P < 0.05), with diameters of 2.24 cm after 2 days, 2.91 cm after 3 days, and 4.15 cm after 4 days (Fig. 3d).

Proteins purified with a Sephadex G-25 gel filtration column. a Absorbance spectrum of Sephadex G-25 gel filtration chromatography. b SDS-PAGE analysis of fraction V from Sephadex G-25 gel filtration chromatography
To verify the purity and size of the active component, SDS-PAGE was used. The SDS-PAGE gel showed a single band corresponding to 43 kDa, suggesting high purity of the protein (Fig. 4b). The active fractions obtained after chromatography were subjected to ESI–MS/MS for peptide sequencing. The resulting protein sequence was searched against the Mascot database using BLAST. The best overall match was with chitosanase, and therefore we named it chitosanase-like protein. We predicted that this protein was either a type of chitosanase or a member of the chitosanase family, and that this protein was responsible for the antifungal activity exhibited by the isolate.
Discussion
In this study, several useful strains were identified in the genus Bacillus through repeatedly screening isolates from soil. The strong and broad antifungal activity of B. subtilis XB-1 showed that strain XB-1 is a promising bacterium for biocontrol purposes in agriculture. Similar potential of B. subtilis has been reported by other researchers (Ahmad et al. 2017; Tan et al. 2019). To identify the most effective antifungal component, a number of antifungal proteins were isolated from strain XB-1, which were selected as candidates due to their wide spectra of antifungal activities (Wang et al. 2016). Several proteins, including fractions V, VI and VII (Fig. 3d), inhibited the growth of M. fructicola, but fraction V was the most effective. Based on its peptide fingerprint and Mascot search results, the isolated antifungal protein was identified as a chitosanase-like protein. Whereas the molecular weight of most chitosanases ranges from 20 to 75 kDa (Gao et al. 2008; Thadathil and Velappan 2014), the novel protein from B. subtilis XB-1 (43 kDa) differed from chitosanases reported previously. Moreover, the molecular weights of other functional proteins isolated from B. subtilis, including bacisubin (41.9 kDa) (Liu et al. 2007), B.s G87 (50.8 kDa) (Tan et al. 2013), flagellin-like protein (32 kDa) (Ren et al. 2013), and vegetative catalase protein (55.4 kDa) (Srikhong et al. 2018), were similar to or lower than that of the novel protein. Therefore, fraction V may represent a novel chitosanase that supports the main antifungal activity of B. subtilis XB-1.
According to the results of previous studies on the antifungal components of Bacillus, effective proteins can be classified into three categories: (1) antimicrobial peptides, including cyclic lipopeptides and linear peptides (Cao et al. 2009); (2) biosurfactants; and (3) enzymes, such as hydrolases, redox enzymes, and some antimicrobial proteins (Ren et al. 2013). In the present study, we found that the novel protein belongs to the chitosanase family, which is a group of enzymes that can catalyze the cytoderm via hydrolysis, consistent with known mechanisms. This protein family has been observed performing endohydrolysis of β-1,4-linkages between D-glucosamine residues in a partly acetylated chitosan (Su et al. 2006). Chitosan is an essential component of the cell wall in most fungi, including M. fructicola. As a protein in the chitosanase family, the purified component likely has a common conserved sequence and catalytic site with reported chitosanases, supporting a powerful antifungal effect based on chitosan hydrolysis. Above all, B. subtilis XB-1 and the newly purified protein have strong potential for applications in ecological agriculture.
References
Ahmad Z, Wu J, Chen L, Dong W (2017). Isolated Bacillus subtilis strain 330-2 and its antagonistic genes identified by the removing PCR. Sci Rep 7:1777. https://doi.org/1777.10.1038/s41598-017-01940-9
Cao XH, Liao ZY, Wang CL, Yang WY, Lu MF (2009) Evaluation of a lipopeptide biosurfactant from Bacillus natto TK-1 as a potential source of anti-adhesive, antimicrobial and antitumor activities. Braz J Microbiol 40:373–379. https://doi.org/10.1590/S1517-838220090002000030
Casals C, Elmer PAG, Vinas I, Teixido N, Sisquella M, Usall J (2012) The combination of curing with either chitosan or Bacillus subtilis CPA-8 to control brown rot infections caused by Monilinia fructicola. Postharvest Biol Technol 64:126–132. https://doi.org/10.1016/j.postharvbio.2011.06.004
Dukare AS, Paul S, Nambi VE, Gupta RK, Singh R, Sharma K, Vishwakarma RK (2018) Exploitation of microbial antagonists for the control of postharvest diseases of fruits: a review. Crit Rev Food Sci Nutr 59(9):1498–1513. https://doi.org/10.1080/10408398.2017.1417235
Eguen B, Melgarejo P, De Cal A (2015) Sensitivity of Monilinia fructicola from Spanish peach orchards to thiophanate-methyl, iprodione, and cyproconazole: fitness analysis and competitiveness. Eur J Plant Pathol 141:789–801. https://doi.org/10.1007/s10658-014-0579-2
Gao XA, Ju WT, Jung WJ, Park RD (2008) Purification and characterization of chitosanase from Bacillus cereus D-11. Carbohydr Polym 72:513–520. https://doi.org/10.1016/j.carbpol.2007.09.025
Gell I, De Cal A, Torres R, Usall J, Melgarejo P (2008) Relationship between the incidence of latent infections caused by Monilinia spp. and the incidence of brown rot of peach fruit: factors affecting latent infection. Eur J Plant Pathol 121:487–498. https://doi.org/10.1007/s10658-008-9268-3
Haas D, Defago G (2005) Biological control of soil-borne pathogens by fluorescent pseudomonads. Nat Rev Microbiol 3:307–319. https://doi.org/10.1038/nrmicro1129
Hastings J, Kirby K (1966) The nucleic acids of Drosophila melanogaster. Biochem J 100:532–539
Hernandez-Rodriguez A, Heydrich-Perez M, Acebo-Guerrero Y, Velazquez-del Valle MG, Hernandez-Lauzardo AN (2008) Antagonistic activity of Cuban native rhizobacteria against Fusarium verticillioides (Sacc.) Nirenb. in maize (Zea mays L.). Appl Soil Ecol 39:180–186. https://doi.org/10.1016/j.apsoil.2007.12.008
Innocenti G, Roberti R, Montanari M, Zakrisson E (2003) Efficacy of microorganisms antagonistic to Rhizoctonia cerealis and their cell wall degrading enzymatic activities. Mycol Res 107:421–427. https://doi.org/10.1017/S0953756203007640
Kilani-Feki O, Ben Khedher S, Dammak M, Kamoun A, Jabnoun-Khiareddine H, Daami-Remadi M, Tounsi S (2016) Improvement of antifungal metabolites production by Bacillus subtilis V26 for biocontrol of tomato postharvest disease. Biol Control 95:73–82. https://doi.org/10.1016/j.biocontrol.2016.01.005
Kumar NSS, Nazeer RA, Jaiganesh R (2012) Purification and identification of antioxidant peptides from the skin protein hydrolysate of two marine fishes, horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber). Amino Acids 42:1641–1649. https://doi.org/10.1007/s00726-011-0858-6
Lastochkina O, Seifikalhor M, Aliniaeifard S, Baymiev A, Pusenkova L, Garipova S, Kulabuhova D, Maksimov I (2019) Bacillus spp.: efficient biotic strategy to control postharvest diseases of fruits and vegetables. Plants 8:97. https://doi.org/10.3390/plants8040097
Liu YF, Chen ZY, Ng TB, Zhang J, Zhou MG, Song FP, Lu F, Liu YZ (2007) Bacisubin, an antifungal protein with ribonuclease and hemagglutinating activities from Bacillus subtilis strain B-916. Peptides 28:553–559. https://doi.org/10.1016/j.peptides.2006.10.009
Liu J, Sui Y, Wisniewski M, Droby S, Tian SP, Norelli J, Hershkovitz V (2012) Effect of heat treatment on inhibition of Monilinia fructicola and induction of disease resistance in peach fruit. Postharvest Biol Technol 65:61–68. https://doi.org/10.1016/j.postharvbio.2011.11.002
Liu C, Yin X, Wang Q, Peng Y, Ma Y, Liu P, Shi J (2018) Antagonistic activities of volatiles produced by two Bacillus strains against Monilinia fructicola in peach fruit. J Sci Food Agric 98:5756–5763. https://doi.org/10.1002/jsfa.9125
Ma ZH, Yoshimura MA, Michailides TJ (2003) Identification and characterization of benzimidazole resistance in Monilinia fructicola from stone fruit orchards in California. Appl Environ Microbiol 69:7145–7152. https://doi.org/10.1128/Aem.69.12.7145-7152.2003
Mizumoto K, Sawa H (2007) Two βs or not two βs: regulation of asymmetric division by β-catenin. Trends Cell Biol 17:465–473. https://doi.org/10.1016/j.tcb.2007.08.004
Poniatowska A, Michalecka M, Bielenin A (2013) Characteristic of Monilinia spp. fungi causing brown rot of pome and stone fruits in Poland. Eur J Plant Pathol 135:855–865. https://doi.org/10.1007/s10658-012-0130-2
Ren JJ, Shi GL, Wang XQ, Liu JG, Wang YN (2013) Identification and characterization of a novel Bacillus subtilis strain with potent antifungal activity of a flagellin-like protein. World J Microb Biot 29:2343–2352. https://doi.org/10.1007/s11274-013-1401-6
Srikhong P, Lertmongkonthum K, Sowanpreecha R, Rerngsamran P (2018) Bacillus sp. strain M10 as a potential biocontrol agent protecting chili pepper and tomato fruits from anthracnose disease caused by Colletotrichum capsici. Biocontrol 63:833–842. https://doi.org/10.1007/s10526-018-9902-8
Su CX, Wang DM, Yao LM, Yu ZL (2006) Purification, characterization, and gene cloning of a chitosanase from Bacillus species strain S65. J Agric Food Chem 54:4208–4214. https://doi.org/10.1021/jf0600556
Tan ZQ, Lin BY, Zhang RY (2013) A novel antifungal protein of Bacillus subtilis B25. Springerplus 2:1–6. https://doi.org/10.1186/2193-1801-2-543
Tan TM, Zhu JX, Shen AR, Li JL, Yu YT, Zhang MJ, Zhao MR, Li ZM, Chen J, Gao CS, Cheng Y, Guo LT, Yan L, Sun XP, Zeng LB, Yan Z (2019) Isolation and identification of a Bacillus subtilis HZ-72 exhibiting biocontrol activity against flax seedling blight. Eur J Plant Pathol 153:825–836. https://doi.org/10.1007/s10658-018-1595-4
Testa MM, de Valladares RR, de Cardenas ILB (2003) Antagonistic interactions among Fusobacterium nucleatum and Prevotella intermedia with oral lactobacilli. Res Microbiol 154:669–675. https://doi.org/10.1016/j.resmic.2003.09.007
Thadathil N, Velappan SP (2014) Recent developments in chitosanase research and its biotechnological applications: a review. Food Chem 150:392–399. https://doi.org/10.1016/j.foodchem.2013.10.083
Wang Z, Wang Y, Zheng L, Yang X, Liu H, Guo J (2014) Isolation and characterization of an antifungal protein from Bacillus licheniformis HS10. Biochem Biophys Res Commun 454:48–52. https://doi.org/10.1016/j.bbrc.2014.10.031
Wang NN, Yan X, Gao XN, Niu HJ, Kang ZS, Huang LL (2016) Purification and characterization of a potential antifungal protein from Bacillus subtilis E1R-J against Valsa mali. World J Microb Biot 32:63. https://doi.org/10.1007/s11274-016-2024-5
Yang LY, Zhang JL, Bassett CL, Meng XH (2012) Difference between chitosan and oligochitosan in growth of Monilinia fructicola and control of brown rot in peach fruit. Lwt-Food Sci Technol 46:254–259. https://doi.org/10.1016/j.lwt.2011.09.023
Acknowledgements
This work was financially supported by the Science Foundation of Changzhou University (Grant No. ZMF18020316), Hebei Cixin Environmental Technology Co., Ltd, China (Grant No. 2018K0948), and Shaanxi Xintiandi Grass Industry Co., Ltd, China (Grant No. 2018K0947).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ren, J., He, W., Li, C. et al. Purification and identification of a novel antifungal protein from Bacillus subtilis XB-1. World J Microbiol Biotechnol 35, 150 (2019). https://doi.org/10.1007/s11274-019-2726-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2726-6