
Abstract
The growth of Phytophthora capsica, Rhizoctonia solani, Fusarium graminearum, Fusarium oxysporum and Botrytis cinerea were all inhibited by the fermentation supernatant of Bacillus licheniformis TG116, a biocontrol strain isolated from Typhonium giganteum Engl. previously with broad-spectrum resistance to plant pathogens. The fermentation supernatant of the TG116 has a great stability on temperature and UV, and shows the biological activity of protease and cellulase. The antifungal protease produced by B. licheniformis TG116 was purified to homogeneity by ammonium sulfate precipitation, DEAE Sepharose Fast Flow column chromatography and Sephadex G-50 column chromatography. The inhibition of protease by the three surfactants increased with increasing concentration inhibition. Among these surfactants, EDTA showed the strongest inhibition, with only 25% protein activity at a concentration of 1.1 mmol·L−1. Gene amplification verified the presence of a gene fragment of serine protease in the strain TG116. The antimicrobial substance isolated from the fermentation broth of TG116 is a serine protease component.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diseases caused by pathogenic fungi lead to serious waste of resources and economic losses in the pre-harvest, post-harvest, transportation and preservation of fruit and vegetables, and in animal husbandry (Leiter et al. 2017). In agriculture, growers use broad-spectrum chemical fungicides to control plant diseases caused by pathogenic fungi, however, these fungicides not only make pollution to environment but also may remain in plant products, which have been of the biggest concern to human health. Therefore, the use of biological control agents may be a safe alternative to the control of plant diseases (Prapasri et al. 2018). Biological control is an internationally growing popular and widespread approach to alleviating the negative impact of plant pathogens on agricultural practice and food security (Ciancio et al. 2016).
Endophytes protect plants from phytopathogens by invading and colonizing plants, allowing plants to grow normally and healthily (Huang et al. 2016). Endophyte mostly invades plant roots from the earth and spreads to leaves, flowers, and fruit via the vascular system (Hardoim et al. 2008; S et al. 2011). These facultative or obligatory endophytes colonize part or all of the plant's life cycle and take advantages of this strategic encounter (Hardoim et al. 2015). They not only help plants grow, but they also protect them from pathogenic germs. Plant growth rates are boosted by endophytes, which enable phytohormone synthesis, nitrogen fixation, phosphate solubilization, and ammonium ion generation (Agarwal et al. 2020). They protect the host by interacting directly with infections and creating many antifungal compounds, or by competing for nutrition in colonized tissues as biological control agents (Ryan et al. 2010). Endophytes also exert indirect control by inducing inducible systemic resistance (ISR) in the plant host, which is the result of multiple endophyte metabolites that initiate plant systemic resistance against pathogenic organisms (Verhagen et al. 2011).
The important characteristic of endophytes is the secretion of lytic enzymes that degrade many biopolymers in the environment. Those cellulase, protease and chitinase, allow endophytic bacteria to enter plant tissues and form stable colonies, providing a clear competitive advantage for bacteria with this ability (Compant et al. 2005).
The inhibition and killing effect of biocontrol microorganisms on pathogenic fungi are related to the structure of the fungal cell walls (Calonje et al. 2000). As the cell wall composition of pathogenic fungi is complex, the degradation of pathogenic fungi cell wall is an important aspect. However, most fungal cell walls contain chitin, protein, β-1.3-Glucan and lipids (Bowman and Free 2010). Due to the complex composition of the fungal cell wall, its degradation may require the interaction of multiple enzymes. Antifungal proteins have a more pronounced effect on pathogenic fungi, and when the cell wall of the pathogenic fungus is degraded by hydrolases, it exhibits bacteriostatic or bactericidal effects.
Studies have shown that most Bacillus can produce secondary metabolites with broad spectrum antibiotic activity and a very diverse structure. Therefore, Bacillus have broad application prospects in agricultural applications and medical applications (Moyne et al. 2004).
B. licheniformis TG116, an endophytic bacterium isolated from Typhonium giganteum Engl. has been reported in previous studies on its spectral inhibition against plant pathogens (Ling et al. 2014). The nucleotide sequence accession number for the 16S rRNA nucleotide sequences of the TG116 is MN696247. In this study, the enzyme production of the aseptic filtrate of B. licheniformis TG116 were characterized. An important role that extracellular serine protease played in the process of biological control, and its enzymatic properties were also investigated in this study.
Materials and methods
Strains and medium
B. licheniformis TG116, cultured as described previously study (Ling et al. 2014), which was used as a protease producer. The fermentation medium for TG116 was composed of peptone (0.5%), yeast powder (0.5%), and glucose (2%), with a pH value of 7.5. TG116 was cultured in a 1 L fermenter and incubated at 37 °C for 72 h. Pathogens P. capsici, R. solani, F. graminearum, F. oxysporum, B. cinerea and C. capsici were maintained on PDA medium.
Chemical reagents
The DEAE Sepharose Fast Flow, used in this research, was produced by Shanghai Rachel Biotechnology Co., Ltd. (Shanghai, China). Molecular primers were synthesized by the Beijing Genomics Institute (Beijing, China). Other reagents were purchased from Shanghai Watson Biotechnology Co. Ltd. (Shanghai, China).
Enzyme activity determination in fermentation broth
The activity of each enzyme such as cellulase, protease and chitinase was determined qualitatively by the ability of the bacterium to form clear zone on solid media. TG116 was cultured in LB liquid medium for 24 h, and sterilized by filtration to obtain a sterile fermentation broth (Youcef-Ali et al. 2014).
Cellulase activity
The activity of cellulase was assayed according to the method of Kumar et al. by measuring the formation of different hydrolyzed ring sizes (Kumar et al. 2014). The crude enzyme solution was inoculated into solid LB medium containing CMC-Na and incubated at 30 °C for 48 h. After removal, they were stained with Congo red (1% w/v) for 30 min and then decolorized with 0.9% NaCl for 40 min to observe the appearance of hydrolytic circles.
Protease activity analysis of the TG116 antifungal protein
The activity of protease was detected by the Oxford Cup method. Based on previous study, the Oxford cups were put on the solid medium containing 1% skimmed milk powder, 200 µL crude enzyme solution were added to the Oxford cups. All the plates were incubated at 30 °C for 48 h.
Chitinase activity
The activity of chitinase was detected by the Oxford Cup method. Briefly, 200 µL crude enzyme solution in triplicate was added into the Oxford cups in solid medium containing 2% colloidal chitin. All the plates were incubated at 30 °C for 48 h.
Purification of protease
Culture solutions were centrifuged (17,400 g, 20 min) and ammonium sulfate was added to the supernatant to a final concentration of 40% to precipitate other components. After centrifugation, ammonium sulfate was increased to 70% to obtain a crude protease, which was then dialyzed in a dialyzer with a molecular weight cut-off of 1000 Da. After the dialyzer was freeze-dried, the crude extract was redissolved in PBS, and the supernatant was filtered with a 0.22 μm filter and stored at − 20 °C for use. All purification steps were carried out at 4 °C.
The enzyme solution was loaded onto a DEAE Sepharose Fast Flow column (16 mm × 400 mm) equilibrated with 10 mmol·L−1 Tris–HCl buffer (pH 7.3), and the protease was eluted from the column with NaCl concentration of 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5 and 1 mol·L−1 gradient solution. Each fraction was collected and the absorbance value was measured at 280 nm, and the elution curve was made with the NaCl elution volume as the horizontal coordinate and the absorbance value as the vertical coordinate. The second peak sample was further separated by Sephadex G-50 molecular sieve chromatography. After elution with sterile water, the antifungal activity of each eluted peak was measured.
The positive fraction was evaporated and dissolved in 200 μL methanol, which was then divided into 20 μL aliquots for injection. A high-performance liquid chromatography (HPLC) Agilent 1100 System (Agilent Technologies) was used with a SunFireTM C18 reverse phase column (250 × 4.6 mm, 5 μm; Waters) and a methanol/water gradient at a flow rate of 0.2 mL·min−1 to perform the study (Zhao et al. 2012).
Antimicrobial activity assay
The diffusion plate method (according to Shi et al. 2015) with slightly modified was used to determine the antimicrobial activity of the samples. Briefly, the plant pathogens P. capsici, R. solani, F. graminearum, F. oxysporum, B. cinerea and C. capsici were placed in the center of the Petri dish prepared agar medium and the sterile Oxford cup was then quickly placed in a petri dish 3 cm from the center of the pathogen, then 200 μL of sample was added after solidification of the medium. After incubation for 24 h at a temperature suitable for growth of the strain, the diameter of the zone of inhibition was measured in each case. Three parallel assays were performed for each sample.
Characterization of the purified antimicrobial protease
Molecular mass analysis
The purity and molecular mass of the antimicrobial protease were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE, 12% (w/v) separating gel and 4% stacking gel) as described by Pang et al. (Pang et al. 2021) with some modification. The samples were boiled in the buffer for 5 min. Electrophoresis was performed at a constant current (20 mA) at 4 °C. After then, the gels were stained Coomassie Brilliant Blue R-250 for 30 min and decolorized with decolorizing solution for three times. The molecular mass of the purified protease was evaluated by comparing its electrophoretic mobility to marker proteins ran on the same gel.
Effect of crude protease solution on pathogenic fungi
The fungal hyphae from the periphery of the zone of inhibition produced by the C. capsici were observed under light microscope (400 ×) for morphological changes. Hyphae cultured under normal conditions as a control.
Enzyme assay
Detection of TG116 protein enzyme activity by revised Jin assay (Jin et al. 2011), one unit of enzyme activity is expressed as hydrolysis of casein per minute to yield 1 μg of tyrosine. A growing sample of B. licheniformis broth (100 mL) was taken from a 500 L fermenter every 4 h and centrifuged at 1679 g for 10 min. The supernatant is used as a source of extracellular protease.
Effect of surfactant active agents on protease activity
To study the effect of surfactant on protease activity, EDTA, Tris and SDS solutions were prepared at concentrations of 0.1, 0.3, 0.5, 0.7, 0.9 and 1.0 mmol·L−1. SDS, Tris and EDTA were mixed with the enzyme solution in the ratio of 0.1:2, and the reaction was carried out at 40 °C for 10 min. The protease enzyme activity was measured at 40 °C by the Folin-Phenol colorimetric method.
Mass spectrometry identification
The band corresponding to the antifungal protein was excised from an SDS-PAGE gel and sent to Huada Gene Company (Beijing, China) for the determination of its constituent peptides by matrix assisted laser desorption/ionization-time of flight-mass spectrometry (MALDI–TOF-MS).
Cloning and amino acid sequence of the TG116 antifungal protein
The whole genome of B. licheniformis TG116 was used as a template to amplify the Pase using primer pairs PaseF (5'-GGTATTACGGGCGTCCAG-3') and PaseR (5'-AAGAAGTGCGGCATCAGG-3'). The PCR reaction conditions were: pre-denaturation at 94 °C for 2 min, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s, and extension at 72 °C for 30 s, followed by a final extension step at 72 °C for 2 min before holding at 4 °C.
Agarose gel electrophoresis was used to detect the PCR products before they were sent for sequencing by Huada Gene Company (Beijing, China), to sequence and submit the obtained gene sequence to NCBI. Alignments of protein sequences were performed with the BLAST program from the NCBI (Thompson et al. 1994).
The nucleotide sequence of the gene encoding the extracellular serine protease gene has been submitted to the GenBank database.
Analysis of protein sequences
The protein encoded Pase were analyzed using the BLAST 2.0 program from the NCBI database. The presence and delimitation of protein domains were accomplished using the conserved domain database (CDD) (Marchler-Bauer et al. 2012).
Statistical analysis
Stability evaluation of the purified protein was performed in triplicate, and the mean diameters of inhibitory zones and standard deviations were determined. Data from different treatments were analyzed by ANOVA. P < 0.05 was considered statistically significant.
Results
Determination of activity and stability of sterile fermentation broth of TG116
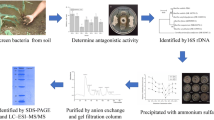
A qualitative study was conducted on the ability of TG116 to produce cell wall degrading enzymes. As the results shown in Fig. 1AB, the strain produced protease and cellulase but no chitinase activity as shown in Fig. 1C. Figure 1D shows crude protein of TG116 has good thermal stability, and its antifungal activity remains basically unchanged with the increase of temperature; when the temperature reaches 100 °C, the residual activity of crude protein still maintains more than 95% of its original activity. With the increase of UV irradiation time, the bacteriostatic activity began to decrease slowly. After 6 h of irradiation, the antifungal activity began to decrease rapidly. After 10 h of irradiation, the residual protease activity still retained 60% of the original bacteriostatic activity, so the TG116 crude protein had a certain UV tolerance in Fig. 1E.

A Protease activity of fermentation broth. B Cellulase activity of fermentation broth. C Chitinase activity of fermentation broth. D, E Activity and stability of sterile fermentation Broth of TG116, Vertical bars represent standard deviation (n = 3)
Inhibitory spectrum of the crude protein
The crude protein showed broad-spectrum antimicrobial activity against some fungi as shown in Fig. 2, the protein has been de-genetically expressed against several pathogens such as P. Capsici, C. Capsici, R. Solani, F. Graminearum, F. Oxysporum and B. Cinerea. As shown in Fig. 3B compared with control Fig. 3A, the hyphae of C. capsici were distorted to bend, blend and break.

Bacteriostatic effect of plant pathogen by the crude protein (100 mL) of TG116. Note: A: Phytophthora capsici, B: Colletotrichum capsici, C: Rhizoctonia solani, D: Fusarium graminearum, E: Fusarium oxysporum, F: Botrytis cinerea

Effects of the antifungal protein from endophytic B. licheniformis TG116 on hyphal growth of Colletotrichum capsici observed by the optical microscope (400 ×). Note: (A) Hypha from untreated Colletotrichum capsici colony; (B) abnormal hyphal growth of Colletotrichum capsici treated with the antifungal protein
Purification and molecular weight determination of TG116 protease
The filtered culture supernatant of TG116 was separated and precipitated with saturated ammonium sulfate, and the result (Fig. 4) showed that the optimal saturation of the ammonium sulfate-precipitating antifungal protein was 70%. Then the protein precipitated by ammonium sulfate was concentrated and added to the DEAE-sepharose fast flow column. There were 12 peaks in the collection solution due to the different content of NaCl, and the five components (H, I, J, K, L) had antifungal effect (Fig. 5), and the antifungal effect is shown in Fig. 6. Elution peak (I) was analyzed by SDS-PAGE. The fraction (I) was further purified by Sephadex G-50 molecular sieve chromatography to give a pure compound. (I) as shown in Fig. 7 lane C, the fraction included low impurity levels. After thorough dialysis, concentrate (I) and apply sterile water to the Sephadex G-50 molecular sieve column and elute the active peak (M) with sterile water (Fig. 8), the elution profiles were monitored by spectrophotometry at 280 nm, and its antifungal effect of is shown in Fig. 9. As shown in Fig. 10, the target band size was calculated to be approximately 86.17 kDa according to the different mobility of the standard proteins. The crude extract was concentrated and applied to a DEAE Sepharose Fast Flow (25 mm × 200 mm) column equilibrated with 0.02 mol·L−1 Tris–HCl buffer (pH 8.5). After washing the column, active peak C was eluted using a stepwise gradient of 0.15–1 mol·L−1 NaCl in 0.02 mol·L−1 Tris–HCl (pH 8.5) buffer at a flow rate of 1.0 mL·min−1. The elution profiles were monitored by spectrophotometry at 280 nm. Of the five active fractions, high performance liquid chromatography (I) was used to analyze the fraction, and acetonitrile of different concentrations was used as eluent. Three different components were added to show similar results to previous Sephadex G-50 molecular sieve chromatography as shown in Fig. 11.

Determination of optimal salting conditions for TG116 antifungal protein

Ion exchange chromatography of crude extract from Bacillus licheniformis TG116. Note: A: Not adsorbed; B: 0.05 mol·L−1; C: 0.1 mol·L−1; D: 0.15 mol·L−1; E: 0.2 mol·L−1; F: 0.25 mol·L−1; G: 0.3 mol·L−1; H: 0.35 mol·L−1; I: 0.4 mol·L−1; J: 0.45 mol·L−1; K: 0.5 mol·L−1; L: 1 mol·L.−1

The bacteriostatic effect of components H, I, J, K and L separated by DEAE Sepharose Fast Flow, aseptic water as a control

Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis of TG116 protease. Note: Lane Marker molecular weight markers; Lanes A Sterile fermentation broth total protein; Lanes BM fraction eluted from Sephadex G-50; Lane C I fraction eluted from DEAE Sepharose Fast Flow; respectively. The separated bands were visualized after Coomassie brilliant blue R-250 staining

Sephadex G-50 molecular sieve chromatography of M. Note: The column was eluted with distilled water, at a rate of 2 mL·min.−1

The bacteriostatic effect diagram of component M was obtained by Sephadex G-50 molecular sieve chromatography

Standard protein migration rate standard curve

HPLC chromatogram of I fraction
Mass spectrometry analysis and identification of the TG116 antifungal protease
The results of mass spectrometry are shown in the Table 1. The top 10 proteins with the highest matching rate are oxalate decarboxylase, peptidase T, 2-cysteine peroxidase, aminopeptidase, extracellular serine protease, dihydrolipoyl dehydrogenase, putative cytosolic aminopeptidase, the putative aminopeptidase YSDC, the di-zinc aminopeptidase and the translation elongation factor TU. According to the migration rate of the standard protein, the relative molecular weight of M component was 86.17 kDa, which was the closest to the molecular weight of extracellular serine protease in the mass spectrometry analysis results, and the group was determined to be divided into extracellular serine protease.
The resistance to surfactant active agents
Figure 12 shows the effect of different surfactants on the activity of the protease. The results show that the tested surfactants have an inhibitory effect on protease activity, and the inhibitory effect became stronger with the increase of concentration. Among these surfactants, EDTA has the strongest inhibitory effect, and when the concentration reaches 1.1 mmol·L−1, the protein activity is only 25%.

Effects of surfactants on protease activity, Vertical bars represent standard deviation (n = 3)
Pase gene PCR results
According to the PCR amplification of the previous primer pairs, the length of the target cloned fragment was 1600 bp. The product was sent to Beijing Huada Gene Sequencing Company for analysis, and 1617 bp gene fragment was obtained. The results showed that the entire TG116 gene contained the gene sequence of the protease, named as Pase, and submitted to Genback (MK659579). Compared with the gene sequence encoding extracellular serine protease (KZD86682.1), the similarity was 98.20%. The use of ORF to predict the protein primary structure of nucleic acid information indicated that the sequence is 1506 bp in length and encodes 501 amino acid residues with a complete protein coding region.
Through blast homology alignment, the protein sequence was found to belong to the protease gene superfamily, which was similar to the expected result. The protein encoded by the Pase gene has three conserved sequences (Fig. 13): from 8 to 94 bp, associated with a peptidase inhibitor i9 (peptidase inhibitor i9) with an e-value of 1.61e-10, acting as a molecular chaperone and promoting folding of mature peptidase; from 131–497 bp, a peptidase s8 family member (peptidase s8 family) with an e-value of 1.28e-82, as a lytic protein, can exist both inside and outside the cell and can be used at extreme temperatures and pH; from 308 to 450 bp, is a serine protease on cells surface (PA_C5a_like) with an e-value of 5.86e-45, the domain participates in substrate binding, promotes conformational changes, and affects the stability of the site to the substrate.

Conserved domain prediction of Pase protein
Discussion
In this paper, we describe the use of Bacillus licheniformis TG116 isolated from Typhonium giganteum Engl. as well as the secondary metabolites secreted as antifungal products against plant pathogens. This strain showed strong antifungal and antagonistic activities, and the cell-free culture supernatants significantly inhibited yeast growth (inhibition zone ≥ 19 mm). As described by Barakate (Barakate et al. 2002), this strong activity expressed by the large repression zone on agar plates indicated that the isolate produced water-soluble antimicrobial metabolites. It is well known that several Bacillus spp. can produce biologically active molecules, including antifungal substances (Caldeira et al. 2011; Qi-Qin et al. 2012; Zhao et al. 2010) and the present study was in a good coordination with the previous findings.
The serine protease produced by TG116 has antifungal effects on plant fungal pathogens by inhibiting spore germination and mycelial dispersal. As with Chitarra, antifungal compounds produced by Bacillus subtilis YM 10–20 inhibited spore germination of Penicillium roqueforti (Chitarra et al. 2010). Deepak found that antifungal proteins from Urginea indica bulbs completely inhibited spore germination and mycelial growth of Fusarium oxysporum (Deepak et al. 2003). Analysis of the functional domains of the TG116 antifungal protein identified many domains, but the main domain is the peptidase S8 domain, consisting of amino acids 131–497. The analysis of the functional structural domains of the W10 antifungal protein in Ji's study was dominated by the peptidase S8 structural domain formed by amino acids 152–437 (Ji et al. 2020). And Cheng showed that this structural domain is a characteristic structural domain in the serine protease family of B. subtilis (Cheng 2010). The functional domain analysis of TG116 antifungal protein found that the inhibitor I9 region was between amino acids 8–94.The inhibitor I9 region or commonly referred to as the proto-domain, was found to have specific inhibitory activity towards the adjacent peptidase S8 structural domain until it was autocatalytically cleaved during maturation of the enzyme (Baker et al. 1992; Huang et al. 1997). This structural domain has also been found to act as a molecular chaperone, assisting the mature serine peptidase to fold correctly into its active conformation (Li et al. 1995; Shinde et al. 1997; Yabuta et al. 2001). PA structural domain between amino acids 308–450. This structural domain binds to substrates and promotes conformational changes that affect the stability of the site to the substrate (Siezen and Leunissen, 1997). The PA domain consists of 120 to 160 amino acids inserted between the His and Ser active site residues (Luo and Hofmann 2001). In addition, for the I9 domain (I9), its main function is to maintain the inactive state of the zymogen and prevent the substrate from entering the active site (Bryan 2002). The interaction between the PA and I9 domains leads to N-terminal cleavage, which will open the way for the substrate to enter the catalytic site and promote protease activity (Bergeron et al. 2000).
Microbial proteases secreted by bacteria breakdown proteins into their constituent monomers, accounting for approximately 40% of the total global enzyme production (Gupta et al. 2002). Most microbial proteases from the genus Bacillus are the most widely developed industrial enzymes, mainly used in uncertain agents (Gupta et al. 1999). All of these enzymes exhibited stability in the presence of various detergent components and were active under different conditions. Since the commercialization of the first alkaline protease Carlsberg from B. licheniformis in the 1960s used as an additive in detergent, proteases that are more alkaline have been purified and characterized, and significant biological activity and stability, extensive substrate specificity, short fermentation times and downstream flow application techniques have been demonstrated (Haddar et al. 2009).
Endophytic bacteria and host plants coexist to manage various environmental stresses. The rich biodiversity and biosynthetic diversity of endophytes make them a treasure trove of undeveloped natural products (Gaiero et al. 2013). Microbial production of a variety of chemical scaffolds with extensive plant interactions or protective properties may be desirable to provide a preference for endogenous life. The identification of these promising isolates can be of great use in a variety of fields, including clinical, industrial, and agricultural fields (Jasim et al. 2016).
Conclusion
In this study, B. licheniformis TG116 was verified that it has strong antifungal activity, especially against C. capsici. B. licheniformis TG116 had the potential to produce cell-wall-degrading enzymes but did not show any chitinase activities. Therefore, these bioactive ingredients may provide an alternative resource for the drug control of plant pathogens. The results of this study provided evidence of the potential presence of the protein encoded by the Pase gene in B. licheniformis TG116. This protein played an important role in protecting plants from harmful pathogens. Thus, the antifungal properties of proteins may be beneficial against plant pathogen diseases, demonstrating their ability as effective biocontrol agents for the effective management of these plant pathogen diseases. Further research is needed to determine the ability of this Bacillus strain to control diseases caused by other plant pathogens.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Agarwal H, Dowarah B, Baruah PM, Bordoloi KS, Agarwala NJMR (2020) Endophytes from Gnetum gnemon L can protect seedlings against infection of the phytopathogenic bacterium R. solanacearum as well as promote plant growth in tomato. Microbiol Res 238:126503
Baker D, Silen JL, Agard DA (1992) Protease pro region required for folding is a potent inhibitor of the mature enzyme. Proteins: Structure, Function, and Bioinformatics 12(4):339–344
Barakate M, Ouhdouch Y, Oufdou K, Beaulieu C (2002) Characterization of rhizospheric soil streptomycetes from Moroccan habitats and their antimicrobial activities. World J Microb Biot 18(1):49–54
Bergeron F, Leduc R, Day R (2000) Subtilase-like pro-protein convertases: from molecular specificity to therapeutic applications. J Mol Endocrinol 24:1–22
Bowman SM, Free SJ (2010) The structure and synthesis of the fungal cell wall. Bioessays News Rev Mol Cell Dev Biol 28(8):799–808
Bryan PN (2002) Prodomains and protein folding catalysis. Chem Rev 102(12):4805–4816
Caldeira AT, Arteiro JMS, Coelho AV, Roseiro JC (2011) Combined use of LC–ESI–MS and antifungal tests for rapid identification of bioactive lipopeptides produced by Bacillus amyloliquefaciens CCMI 1051. Process Biochem 46(9):1738–1746
Calonje M, Novaes-Ledieu M, Bernardo D, Ahrazem O, Mendoza CG (2000) Chemical components and their locations in the Verticillium fungicola cell wall. Can J Microbiol 46(2):101
Cheng GY (2010) Characterization of a spore-associated protease and anextracellular serine protease from Thermoactinomyces sp. Wuhan Univ, Hubei, CDF ((in Chinese))
Chitarra GS, Breeuwer P, Nout MJR, Aelst ACV, Abee T (2010) An antifungal compound produced by Bacillus subtilis YM 10–20 inhibits germination of Penicillium roqueforti conidiospores. J Appl Microbiol 94(2):159–166
Ciancio A, Pieterse C, Mercado-Blanco J (2016) Editorial: harnessing useful rhizosphere microorganisms for pathogen and pest biocontrol. Front Microbiol 7:162
Compant S, Mitter B, Juan Gualberto C-M, Helmut G, Angela S (2011) Endophytes of grapevine flowers, berries, and seeds: identification of cultivable bacteria, comparison with other plant parts, and visualization of niches of colonization. Microb Ecol 62(1):188–197
Compant S, Reiter B, Sessitsch A, Nowak J, Clement C, Ait Barka E (2005) Endophytic Colonization of Vitis vinifera L. by Plant Growth-Promoting Bacterium Burkholderia sp. Strain PsJN. Appl Environ Microb 71(4):1685–1693
Gaiero JR, McCall CA, Thompson KA, Day NJ, Best AS, Dunfield KE (2013) Inside the root microbiome: bacterial root endophytes and plant growth promotion. Am J Bot 100(9):1738–1750
Gupta R, Gupta K, Saxena R, Khan S (1999) Bleach-stable, alkaline protease from Bacillus sp. Biotechnol Lett 21(2):135–138
Gupta R, Beg Q, Lorenz P (2002) Bacterial alkaline proteases: molecular approaches and industrial applications. Appl Microbiol Biot 59(1):15–32
Haddar A, Agrebi R, Bougatef A, Hmidet N, Sellami-Kamoun A, Nasri M (2009) Two detergent stable alkaline serine-proteases from Bacillus mojavensis A21: purification, characterization and potential application as a laundry detergent additive. Bioresource Technol 100(13):3366–3373
Hardoim PR, Overbeek LSv, Elsas JDv, (2008) Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol 16(10):463–471
Hardoim PR, van Overbeek LS, Berg G et al (2015) The hidden world within plants: ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol Mol Biol Rev 79(3):293–320
Huang HW, Chen WC, Wu CY et al (1997) Kinetic studies of the inhibitory effects of propeptides subtilisin BPN’ and Carlsberg to bacterial serine proteases. Protein Eng Des Sel 10(10):1227–1233
Huang Y, Kuang Z, Wang W, Cao L (2016) Exploring potential bacterial and fungal biocontrol agents transmitted from seeds to sprouts of wheat. Biol Control 98:27–33
Jasim B, Sreelakshmi S, Mathew J, Radhakrishnan E (2016) Identification of endophytic Bacillus mojavensis with highly specialized broad spectrum antibacterial activity. Biotech 6(2):187
Ji Z-L, Peng S, Chen L-L, Liu Y, Yan C, Zhu F (2020) Identification and characterization of a serine protease from Bacillus licheniformis W10: A potential antifungal agent. Int J Biol Macromol 145:594–603
Jin Y-G, Li H-L, Ma M-H, Wang J, Kim H-N, Oh D-H (2011) Purification and characterization of an extracellular protease from Bacillus pumilus CN8. Food Hyg Safe Sci 26(1):76–81
Kumar L, Kumar D, Nagar S et al (2014) Modulation of xylanase production from alkaliphilic Bacillus pumilus VLK-1 through process optimization and temperature shift operation. Biotech 4(4):345–356
Leiter é, Gáll T, Csernoch L, Pócsi I, (2017) Biofungicide utilizations of antifungal proteins of filamentous ascomycetes: current and foreseeable future developments. Biocontrol 62(2):125–138
Li QQ, Ye YF, Fu G, Yuan GQ, Miao JH, Lin W (2012) Identification of Antifungal Substance (Iturin A_2) Produced by Bacillus subtilis B47 and Its Effect on Southern Corn Leaf Blight. J Integr Agr 11(1):90–99
Li Y, Hu Z, Jordan F, Inouye M (1995) Functional Analysis of the Propeptide of Subtilisin E as an Intramolecular Chaperone for Protein Folding: refolding and inhibitory abilities of propeptide mutants. J Biol Chem 270(42):25127-25132 Luo X, Hofmann K (2001) The protease-associated domain: a homology domain associated with multiple classes of proteases. Trends Biochem Sci 26(3):147-148
Ling L J, Lei L, Feng Lei, He Nan, Ding L. (2014) Isolation and identification of endophytic bacterium TG116 from Typhonium giganteum and its antimicrobial characteristics (in Chinese). Jof Northwest Normal University 50(50)
Marchler-Bauer A, Zheng C, Chitsaz F et al (2012) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41(D1):D348–D352
Moyne AL, Cleveland TE, Tuzun S (2004) Molecular characterization and analysis of the operon encoding the antifungal lipopeptide bacillomycin D. Fems Microbiol Lett 234(1):43–49
Pang Y, Yang J, Chen X, et al. (2021) An Antifungal Chitosanase from Bacillus subtilis SH21. Molecules 26(7):1863
Prapasri S, Kongyuth L, Rapeewan S, Panan R (2018) Bacillus sp. strain M10 as a potential biocontrol agent protecting chili pepper and tomato fruits from anthracnose disease caused by Colletotrichum capsici. Biocontrol 63(11):833–842
Shi B, Zheng H, Huang J, Luo X, Luo X (2015) Purification and partial characterization of a thermostable antimicrobial protein fromBacillus subtilisFB123. World J of Microbiol & Biotechnol 31(8):1285–1290
Shinde UP, Liu JJ, Inouye M (1997) Protein memory through altered folding mediated by intramolecular chaperones. Nature 389(6650):520–522
Siezen RJ, Leunissen JA (1997) Subtilases: the superfamily of subtilisin-like serine proteases. Protein Sci 6:501–523
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680
Verhagen B, Trotelazia P, Jeandet P (2011) Improved resistance against botrytis cinerea by grapevine-associated bacteria that induce a prime oxidative burst and phytoalexin production. Phytopathology 101(7):768–777
Yabuta Y, Takagi H, Inouye M, Shinde U (2001) Folding pathway mediated by an intramolecular chaperone: propeptide release modulates activation precision of pro-subtilisin *. J Biol Chem 276(48):44427–44434
Youcef-Ali M, Chaouche NK, Dehimat L et al (2014) Antifungal activity and bioactive compounds produced by Bacillus mojavensis and Bacillus subtilis. Afr J Microbiol Res 8(6):476–484
Zhao Z, Wang Q, Wang K, Brian K, Liu C, Gu Y (2010) Study of the antifungal activity of Bacillus vallismortis ZZ185 in vitro and identification of its antifungal components. Bioresour Technol 101(1):292–297
Zhao J, Guo L, Zeng H et al (2012) Purification and characterization of a novel antimicrobial peptide from Brevibacillus laterosporus strain A60. Peptides 33(2):206–211
Funding
This work was supported by Lanzhou Science and Technology Plan Project (2018–1-104); Special Fund Project for Guiding Science and Technology Innovation and Development in Gansu Province (2019ZX-05); Industrial support Plan Project of Colleges and Universities in Gansu Province (2020C-21).
Author information
Authors and Affiliations
Contributions
LL: Put forward experimental ideas and provide funds; WC, KJ: Complete the experiment and write the first draft of the paper; ZJ, HL, CY, MP: The processing of Experimental data and the making of related Chart; LL: revise the manuscript. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ling, L., Cheng, W., Jiang, K. et al. The antifungal activity of a serine protease and the enzyme production of characteristics of Bacillus licheniformis TG116. Arch Microbiol 204, 601 (2022). https://doi.org/10.1007/s00203-022-03216-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00203-022-03216-x