
Abstract
In the current study, two peptides with antioxidant properties were purified from skin protein hydrolysates of horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber) by consecutive chromatographic fractionations including ion exchange chromatography and gel filtration chromatography. By electron spray ionization double mass spectrometry (ESI-MS/MS), the sequence of the peptide from the skin protein hydrolysate of horse mackerel was identified to be Asn-His-Arg-Tyr-Asp-Arg (856 Da) and that of croaker to be Gly-Asn-Arg-Gly-Phe-Ala-Cys-Arg-His-Ala (1101.5 Da). The antioxidant activity of these peptides was tested by electron spin resonance (ESR) spectrometry using 1-diphenyl-2-picryl hydrazyl (DPPH·) and hydroxyl (OH·) radical scavenging assays. Both peptides exhibited higher activity against polyunsaturated fatty acid (PUFA) peroxidation than the natural antioxidant α-tocopherol. These results suggest that the two peptides isolated from the skin protein hydrolysates of horse mackerel and croaker are potent antioxidants and may be effectively used as food additives and as pharmaceutical agents.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Free radicals are defined as molecules having an unpaired electron in their outermost orbits. They are generally unstable and reactive in nature. Reactive oxygen species (ROS) include superoxide anion radical (O2), hydroxyl radical (OH·), hydrogen peroxide (H2O2), singlet oxygen (1O2), and hypochlorous acid (HOCl) (Dahl and Richardson 1978). ROS are produced inside the cell under physiological and pathological conditions (Evans and Halliwell 2001) in response to external stimuli and to chemicals. Cytotoxicity of free radicals is deleterious to mammalian cells, but is also effective in killing pathogens by activating macrophages and other phagocytes of the immune system (McCord 2000). Uncontrolled generation of free radicals causes havoc in biological system by damaging membrane lipids, proteins and DNA (Wiseman and Halliwell 1996). These effects are believed to be involved in aging and in many health disorders such as diabetes mellitus, cancer, neurodegenerative and inflammatory diseases (Pryor and Ann 1982; Butterfield et al. 2002). The key antioxidants that decrease the impact of ROS by their specific mechanisms are enzymes, such as superoxide dismutase (SOD), glutathione peroxidase (GPx), glutathione reductase (GR) and catalase (CAT), as well as readily oxidized substrates, such as glutathione (GSH) and ascorbic acid (AA).
A number of natural and synthetic antioxidants directly scavenge free radicals (Dahl and Richardson 1978). However, the use of synthetic antioxidants, such as butylated hydroxyl anisole (BHA) and butylated hydroxyl toluene (BHT), has been suspected to threaten health by causing liver damage and carcinogenesis (Ito et al. 1986). Hence the development of natural, safer antioxidants, which have synergistic effects of amino acids, peptides and proteins, has attracted considerable attention. Protein hydrolysates are the major source of bioactive peptides, which are short-chain peptides with certain biological properties such as enzyme inhibition, antioxidant ability, or immunomodulatory, anti-hypertensive and anti-thrombotic effects etc. (Jung et al. 2005; Suetsuna 1998; Chen et al. 1995). These peptides are inactive within the sequence of the parent protein, however, after enzymatic hydrolysis, their bioactivity will be released (Gill et al. 1996). These peptides usually contain 3–20 amino acid residues, and their activity is dependent on their amino acid composition and sequence (Pihlanto 2001). Antioxidant activities have been found in peptides purified from hydrolyzed proteins of squid muscle (Rajapakse et al. 2005), milk (Pihlanto 2006), porcine collagen (Lertittikul et al. 2007), bull frog skin (Qian et al. 2008), round scad mince (Thiansilakul et al. 2007), water buffalo horn (Liu et al. 2010), black scabbard fish (Batista et al. 2010), pink perch and flying fish muscle (Shabeena and Nazeer 2010).
Among various marine bio-resources available in Tamilnadu coastal line, the antioxidant activity of fish-derived peptides has not yet been adequately studied. Therefore, the aim of this study was to identify potential antioxidant peptides derived from the skin of two marine fishes—horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber)—and to characterize their amino acid sequences. To this purpose, the skins of both fishes were enzymatically hydrolyzed in an in vitro digestion procedure mimicking conditions in the gastrointestinal tract, and the activity of the resulting peptides against different free radicals was tested.
Materials and methods
Proteases for enzymatic hydrolysis (pepsin, trypsin and α-chymotrypsin), α-linoleic acid, ammonium thiocyanate, α-tocopherol, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), FeSO4, and H2O2 were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Other commercially available chemicals and reagents were of analytical grade.
Sample collection
Horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber) were collected from Royapuram sea coast (13°6′26″N 80°17′43″E), Tamilnadu, India. Skin was dissected from the fish, wiped with blotting paper and weighed. The organs were minced separately using a grinder and stored in plastic bags at −20°C until used.
Proximate analysis
Proximate (moisture, ash, lipid and protein) composition was determined on wet weight basis. Moisture content was determined by placing approximately 2 g of sample into a pre-weighted aluminum dish (AOAC 1991). Samples were then dried in an oven at 105°C until a constant mass was obtained. Ash content was estimated by charring the pre-dried sample in a crucible at 600°C until a white ash was formed (AOAC 1991). The total crude protein (N × 6.25) in raw material was determined using the Kjeldahl method (AOAC 1991). Lipid content was determined gravimetrically using the Bligh and Dyer (1959) method.
In vitro digestion
The digestion process was carried out using the method described by Kapsokefalou and Miller (1991) with necessary modifications. A 100 ml aliquot of 4% (w/v) fish skin suspension in protein isolating solution (0.1 M phosphate buffer, pH 6.4) was brought with HCl to pH 2.5 to represent the stomach digestion milieu. Pepsin was added at an enzyme-to-substrate ratio of 1/100 (w/w), and the mixture incubated on a shaker at 37°C for 2 h. The pH was then set to 8 with NaOH to obtain the conditions of small intestine digestion. Both trypsin and α-chymotrypsin were supplemented at an enzyme-to-substrate ratio of 1/100 (w/w), and the solution was further incubated at 37°C for 2.5 h. Samples were taken at the start and at the end of the treatment, and their pH was quickly adjusted to 6.5. After centrifugation at 10,000×g for 15 min at 4°C, the supernatants were collected and cooled at −80°C. The frozen samples were lyophilized to obtain dry powder.
Reducing power assay
The ability of the hydrolysates to reduce iron(III) was determined according to the method of Yen and Chen (1995). An aliquot of 1 ml of each hydrolysate at the concentration of 1 mg/ml was mixed with 2.5 ml of 0.2 M phosphate buffer (pH 6.6) and 2.5 ml of 1% potassium ferricyanide. The mixture was incubated at 50°C for 30 min, followed by addition of 2.5 ml of 10% (w/v) trichloroacetic acid. The mixture was then centrifuged at 1,650×g for 10 min. Finally 2.5 ml of the supernatant were mixed with 2.5 ml of distilled water and 0.5 ml of 0.1% (w/v) ferric chloride. After 10 min of reaction the absorbance of the resulting solution was measured at 700 nm. Reducing power was proportional to absorbance of the reaction mixture.
Chelating activity on Fe2+
The chelation of Fe2+ was measured by the method of Decker and Welch (1990) with a slight modification. An aliquot of 4.7 ml of sample at the concentration of 1 mg/ml was mixed with 0.1 ml of 2 mM FeCl2 and 0.2 ml of 5 mM ferrozine. The reaction mixture was allowed to stand for 20 min at room temperature. The absorbance was then read at 562 nm. The chelating activity was calculated as follows;
The blank was prepared in the same manner except that distilled water was used instead of the sample. EDTA was used as positive control.
Measurement of the antioxidative activity in the linoleic acid model system
The antioxidative activity was measured in the α-linoleic acid model system according to the methods of Osawa and Namiki (1985). Briefly, a sample (corresponding to 1 mg of lyophilized peptides) was dissolved in 10 ml of 50 mM phosphate buffer (pH 7.0), and added to a solution containing 0.13 ml of linoleic acid and 10 ml of 99.5% ethanol. The total volume was then adjusted to 25 ml with distilled water, and the mixture was incubated in a storage bottle with a screw cap at 40 ± 2°C in the dark. The degree of oxidation was evaluated by measuring the ferric thiocyanate values according to the method of Mitsuta et al. (1996): an aliquot of 100 μl of the above solution was mixed with 4.7 ml of 75% ethanol, 0.1 ml of 30% ammonium thiocyanate, and 0.1 ml of 2 × 10−2 M ferrous chloride solution in 3.5% HCl. After 3 min, the absorbance was read at 500 nm.
Assays using electron spin resonance (ESR) spectroscopy
DPPH radical scavenging activity (RSA)
The DPPH RSA of protein hydrolysates was measured using the method described by Nanjo et al. (1996). An aliquot of 60 μl of peptide solution at the concentration of 1 mg/ml (or ethanol itself as control) was added to 60 μl of DPPH (60 μM) in ethanol solution. After mixing vigorously for 10 s, the solution was transferred into a 100 μl quartz capillary tube, and the spin adduct was measured exactly 2 min later in an EMX ESR spectrometer equipped with rectangular cavity (Bruker, Germany). The experimental conditions employed were as follows: magnetic field, 336.5 ± 5 mT; power, 5 mW; modulation frequency, 9.41 GHz; amplitude, 1 × 1000; sweep time, 30 s. DPPH radical scavenging ability was calculated with the following equation, in which H and H 0 are relative peak height of radical signals with and without sample, respectively.
Hydroxyl radical scavenging activity
Hydroxyl radicals were generated by iron-catalyzed Fenton Haber–Weiss reaction and the generated hydroxyl radicals were rapidly reacted with the nitrone spin trap DMPO (Rosen and Rauckman 1984). The resultant DMPO–OH adducts was detectable with an ESR spectrometer. The peptide solution (0.2 ml) was mixed with DMPO (0.3 M, 20 μl), FeSO4 (10 mM, 20 μl) and H2O2 (10 mM, 20 μl) in a phosphate buffer solution (pH 7.4), and then transferred into a 100 μl quartz capillary tube. After 2.5 min, the ESR spectrum was recorded using an EMX ESR spectrometer equipped with rectangular cavity (Bruker, Germany). The experimental conditions employed were as follows: magnetic field, 336.5 ± 5 mT; power, 1 mW; modulation frequency, 9.41 GHz; amplitude, 1 × 200; sweep time, 4 min. Hydroxyl radical scavenging ability was calculated with the following equation, in which H and H 0 are relative peak height of radical signals with and without sample, respectively.
Purification of the antioxidant peptide
Ion exchange chromatography (IEC)
The lyophilized protein hydrolysates were dissolved in 20 mM sodium acetate buffer (pH 4.0) at a concentration of 20 mg/ml, and loaded for fast protein liquid chromatography (FPLC) on a XK 26 DEAE anion exchange column equilibrated with 20 mM sodium acetate buffer (pH 4.0). Elution was with a linear gradient of NaCl (0–1.5 M) in the same buffer at a flow rate of 1 ml/min. Each fraction, collected at a volume of 6 ml, was monitored at 280 nm. Pooled fractions were then concentrated using a rotary evaporator, and antioxidant activities were investigated. The fractions having strong antioxidant properties were lyophilized, and subjected to further separation.
Gel filtration chromatography (GFC)
The fractions exhibiting the highest antioxidant activity were further purified after dissolution in distilled water by loading onto a Sephadex G-25 gel filtration column (2.5 × 75 cm) previously equilibrated with distilled water. The column was then eluted with distilled water at a flow rate of 1 ml/min. Each fraction collected at a volume of 3 ml was monitored at 280 nm. Pooled fractions were then concentrated using a rotary evaporator, and antioxidant activities were investigated. The fractions having strong antioxidant properties were lyophilized, and subjected to ESI-MS/MS to determine the sequence of the purified peptides.
Identification of peptides by ESI-MS/MS
The fraction with the highest antioxidant activity after GFC was dissolved in 75% acetonitrile/25% water of HPLC grade, and loaded into a FIA type 3200 QTRAP mass spectrometer (Applied Biosystem). The sample was passed at a flow rate of 20 μl/min via the electro spray interface, which was operated in the positive electrospray ionization (ESI+ ve) mode. The gas used for drying (35 psi) and ESI nebulization (45 psi) was high-purity nitrogen. Spectra were recorded over the mass/charge (m/z) range 50–1200. About three spectra were averaged in the MS and multiple MS (MS/MS) analyses. Peptide sequencing was performed by manual calculation.
Statistical analyses
All the assays for antioxidant activities of hydrolysates were conducted on three replicates. Data were expressed as mean ± standard deviation. The statistical analysis was performed using SPSS 10.0 software (SPSS Inc. Chicago, IL, USA). Significant differences were determined with 95% confidence interval (P < 0.05).
Results and discussion
Proximate composition
Proximate composition of horse mackerel and croaker skin is shown in Table 1. Moisture, ash, crude protein, lipid and carbohydrate contents were tabulated as per their wet weight basis. The protein content was 13 and 14.3%, respectively; usually, marine fish contains between 8 and 21% of crude protein (Chandrasekhar and Deosthale 1993) and the results observed for our samples fall in this interval.
Reducing power assay
The protein hydrolysates of horse mackerel and croaker skin were prepared by in vitro digestion mimicking gastrointestinal conditions, and were then lyophilized for further studies. Crude hydrolysates were tested for their reducing ability and metal chelating activity. The reducing power assay is often used to evaluate the ability of an antioxidant to donate electrons (Yildirim et al. 2000), a mechanism through which it can stabilize the free radicals. Both crude hydrolysates showed good antioxidant activity in a dose-dependent manner at a concentration of 1 mg/ml; the activity was comparable to that of standard compounds such as α-tocopherol and BHT, as depicted in Fig. 1. Several papers have also reported that the reducing power of peptides increases with sample amount (Zhu et al. 2006; Klompong et al. 2007; Bougatef et al. 2010).

Reducing power and metal chelating ability of horse mackerel (HM) and croaker (CR) skin protein hydrolysate; Mean ± standard deviation; n = 3
Chelating activity on Fe2+
Crude skin protein hydrolysates of horse mackerel and croaker were tested for their metal chelating ability at a concentration of 1 mg/ml (Fig. 1). Transition metals, such as Fe, Cu, and Co, react very quickly with peroxides by acting as one-electron donors to form alkoxyl radicals (Gordon 2001). Therefore, chelation of transition metal ions would retard the oxidation process (Sherwin 1990). In comparison with other ions, ferrous ion is a key active species responsible for ROS formation in cells, leading to increased levels of lipid peroxidation (Huang et al. 2002). In the Decker and Welch (1990) test, the formation of a violet complex by ferrozine and Fe2+ is interrupted in the presence of a chelating agent. Both crude hydrolysates showed good chelating activity (52.3 and 57.2%, respectively) and the results were comparable with those obtained with EDTA (P < 0.05). Our results were thus similar to those reported for hemp and alfalfa leaf protein hydrolysates (Tang et al. 2009; Xie et al. 2008). Peptides are well known metal chelators and this is one of the antioxidant mechanisms for many active peptides (Peng et al. 2009); the activity is affected by size and amino acid sequence.
Purification of the antioxidant peptide
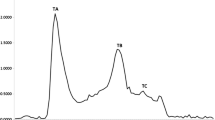
The skin protein hydrolysates were fractionated on an anion exchange column at pH 4.0 with a linear gradient of NaCl concentration resulting, with both samples, in the elution of four peaks (1–4) as depicted in Fig. 2. All the fractions were tested against DPPH and hydroxyl radicals; as shown in Table 2, fraction 2 of horse mackerel and fraction 4 of croaker skin hydrolysates showed good activity. The active fractions were further fractionated on a gel filtration column. GFC had also been used for the purification of peptides from marine rotifer (Byun et al. 2009), water buffalo horn (Liu et al. 2010), and sardinelle (Bougatef et al. 2010). As shown in Fig. 3, three peaks were eluted in both cases (A, B and C). The fractions were tested for their radical quenching efficiency and those showing antioxidant activity were concentrated for further characterization.

Separation of antioxidant peptides from horse mackerel and croaker skin protein hydrolysate by XK 26 DEAE anion exchange chromatography

Separation of antioxidant peptides from active fractions of horse mackerel and croaker skin protein hydrolysate after XK 26 DEAE anion exchange chromatography by Sephadex G-25 gel filtration chromatography
ESR spectroscopy
ESR spectroscopy with a proper radical trapper is a sensitive method to detect RSA of proteins and peptides. It is a sensitive, direct and accurate spectroscopic tool to monitor reactive species generated at room temperature. The high sensitivity of ESR allows the detection of even low concentration of radicals with peptide samples (Peng et al. 2009). In this study, to investigate the discriminating nature of the antioxidant activity, a small ROS (OH·) and a relatively large radical (DPPH·) were used; the protein hydrolysate prepared from the skin of horse mackerel and croaker were effective in quenching both of these radicals (Table 2). Therefore, both samples were subjected to RSA tests against DPPH· and OH· species; in these tests croaker protein hydrolysate showed a higher activity (65.3 and 41.2%) than horse mackerel (56.4 and 36.3%). Testing was repeated after purification for all the fractions collected from ion exchange and GFC. After every step the activity increased, as reported for their systems by Peng et al. (2009), Liu et al. (2010), Batista et al. (2010), and Qian et al. (2008). For the peptides isolated from fish skin hydrolysates showed the increase in scavenging activity against both the radicals amounted to ~14–16%. Radical quenching is a primary mechanism to inhibit oxidative processes. Therefore, the results obtained from the ESR spectroscopy associate a strong antioxidant activity to the peptides purified from horse mackerel and croaker skin protein hydrolysates.
Identification of peptides by ESI-MS/MS
The two active peptides were loaded onto ESI-MS/MS to characterize the sequence of their amino acids. The peptide purified from horse mackerel skin protein hydrolysate was identified as the hexapeptide Asn-His-Arg-Tyr-Asp-Arg (856 Da) and that purified from croaker skin protein hydrolysate was identified as the decapeptide Gly-Asn-Arg-Gly-Phe-Ala-Cys-Arg-His-Ala (1101.5 Da), respectively (Figs. 4, 5). Antioxidant peptides identified from fish sources have been reported in the literature to have molecular weights between 500 and 1500 Da (Jun et al. 2004; Ranathung et al. 2006; Wu et al. 2003 ). Bioactive peptides usually contain 2–20 amino acid residues per molecule (Pihlanto 2001), and the lower the molecular weight, the higher their chance to cross the intestinal barrier and exert biological effects (Roberts et al. 1999). The active peptide isolated from the skin of horse mackerel contains both essential and non-essential amino acids. The presence of aromatic amino acids like tyrosine allows direct electron transfer to ROS (Qian et al. 2008). In addition this peptide contains aspartic acid, which seems to play a vital role as observed in several antioxidative peptides (Rajapakse et al. 2005). Conversely, the good ROS quenching activity observed for the antioxidant peptide isolated from croaker skin may be due to the presence of cysteine, phenylalanine, glycine and alanine, which are known for their potential antioxidant activity. This peptide is also rich in alanine (approximately 20% of the purified peptide sequence), a hydrophobic amino acid residue; it is presumed that the presence of hydrophobic amino acids in individual peptides may contribute to their lipid peroxidation inhibitory activity by increasing peptide solubility in lipid hence facilitating the in situ interaction with radical species (Chen et al. 1996). Furthermore, both the isolated peptides contain histidine, which enhances the scavenging activity due to the proton-donation ability of the histidine imidazole group (Chen et al. 1996).

Characterization of the antioxidant peptide from horse mackerel skin protein hydrolysate: a mass spectrum of the purified fraction C; b MS/MS spectrum of ion m/z 856. By manual calculation, the sequence of this peptide is displayed with the fragmentations observed in the spectrum. For clarity, only a, b, and y″ ions are labeled

Characterization of the antioxidant peptide from croaker skin protein hydrolysate: a mass spectrum of the purified fraction A; b MS/MS spectrum of ion m/z 1101.5. By manual calculation, the sequence of this peptide is displayed with the fragmentations observed in the spectrum. For clarity, only a, b, and y″ ions are labeled
Measurement of the antioxidative activity in the linoleic acid model system
Peroxidation of lipids is a complex process that involves formation and propagation of lipid radicals and lipid hydroperoxides in the presence of oxygen (Ames 1983). The activity of the two purified peptides from horse mackerel and croaker skin protein hydrolysates against the peroxidation of α-linoleic acid was investigated and compared to that of α-tocopherol, a natural antioxidative agent. As shown in Fig. 6, the control (without antioxidant) had—as expected—the highest absorbance value in the Osawa and Namiki (1985) test, indicating the highest degree of oxidation among the samples after a 7-day incubation, whereas the peptide from croaker skin protein hydrolysate had the lowest absorbance followed by α-tocopherol and by the peptide from horse mackerel skin protein hydrolysate. Cheng et al. (2003) reported that phenolic compounds afford their protective actions in lipid peroxidation by scavenging the lipid-derived radicals (R·, RO· or ROO·) in a heterogeneous lipid phase. According to this model, the presence of hydrophobic properties is very important for a compound to inhibit lipid peroxidation. From their sequence, both purified peptides contain several hydrophobic residues; thanks to this feature, both could easily interact with lipid molecules and scavenge lipid-derived radicals through proton donation. As reported by Halliwell and Gutteridge (1989) the beneficial effects of antioxidants are mediated by the prevention of oxidative damage with the interruption of the radical chain reaction involved in lipid peroxidation. Our results show that the peptides isolated from horse mackerel and croaker skin protein hydrolysates are very successful in this process, which makes them of great interest as antioxidant compounds.

Antioxidative activities of purified peptide in α-linoleic acid emulsion system measured by the ferric thiocyanate method
Conclusion
By-products of marine fish processing are used in many industries and their commercial applications are expanding every year. However, their applicability as bioactive compounds and their nutraceutical value are not extensively studied. Based on our results in this study, it appears that the low molecular weight peptides released from horse mackerel and croaker skin by in vitro digestion have good antioxidant properties. The purified antioxidant peptides also are potent free radical scavengers and effectively inhibit lipid peroxidation. With these properties they may be expected to protect against oxidative damage in living systems in relation to aging, carcinogenesis, and neurodegenerative diseases. Therefore, our data suggest that horse mackerel and croaker skin present a potential as nutraceutical and bioactive material. Further detailed studies on peptide fractions will be needed to evaluate their in vivo antioxidant activities.
References
Ames BN (1983) Dietary carcinogens and anticarcinogens: oxygen radicals and degenerative disease. Science 221:1256–1264
AOAC (1991) Official methods of analysis, 16th edn. Association of Official Analytical Chemists, Washington, DC
Batista I, Ramos C, Coutinho J, Bandarra NM, Nunes ML (2010) Characterization of protein hydrolysates and lipids obtained from black scabbard fish (Aphanopus carbo) byproducts and antioxidative activity of the hydrolysates produced. Process Biochem 45:18–24
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917
Bougatef A, Nedjar-Arroume N, Manni L, Ravallec R, Barkia A, Guillochon D, Nasri M (2010) Purification and identification of novel antioxidant peptides from enzymatic hydrolysates of sardinelle (Sardinella aurita) by-products proteins. Food Chem 118:559–565
Butterfield DA, Castenga A, Pocernich CB, Drake J, Scapagnini G, Calabrese V (2002) Nutritional approaches to combat oxidative stress in Alzheimer’s disease. J Nutr Biochem 13:444–461
Byun HG, Lee JK, Park HG, Jeon JK, Kim SK (2009) Antioxidant peptides isolated from the marine rotifer, Brachionus rotundiformis. Process Biochem 44:842–846
Chandrasekhar K, Deosthale YG (1993) Proximate composition, amino acid, mineral and trace element content of the edible muscle of 20 Indian fish species. J Food Comp Anal 6:195–200
Chen HM, Muramoto K, Yamaguchi F (1995) Structural analysis of antioxidative peptides from soybean β-conglycinin. J Agric Food Chem 43:574–578
Chen HM, Muramoto K, Yamauchi F, Nokihara K (1996) Antioxidant activity of designed peptides based on the antioxidative peptide isolated from digests of a soy bean protein. J Agric Food Chem 44:2619–2623
Cheng Z, Ren J, Li Y, Chang W, Chen Z (2003) Establishment of a quantitative structure activity relationship model for evaluating and predicting the protective potentials of phenolic antioxidants on lipid peroxidation. J Pharm Sci 92:475–484
Dahl MK, Richardson T (1978) Photogeneration of superoxide anion in serum of bovine milk and in model systems containing riboflavin and amino acid. J Dairy Sci 61:400–407
Decker EA, Welch B (1990) Role of ferritin as a lipid oxidation catalyst in muscle food. J Agric Food Chem 38:674–677
Evans P, Halliwell B (2001) Micronutrients: oxidant/antioxidant status. Br J Nutr 85:67–74
Gill I, Lopez-Fadino R, Jorba X, Vulfson EN (1996) Biological active peptides and enzymatic approach to their production. Enzyme Microb Technol 18:162–183
Gordon M (2001) Antioxidants and food stability. In: Pokorny J, Yanishlieva N, Gordon M (eds) Antioxidant in food. CRC Press, New York, USA, pp 7–21
Halliwell B, Gutteridge JMC (1989) Free radicals in biology and medicine, 2nd edn. Clarendon Press, Oxford, pp 1–81
Huang X, Dai J, Fournier J, Ali AM, Zhang Q, Frenkel K (2002) Ferrous ion autoxidation and its chelation in iron-loaded human liver Hep G2 cells. Free Radic Biol Med 32:84–92
Ito N, Hirose M, Fukushima S, Tsuda H, Shirai T, Tatematsu M (1986) Studies on antioxidants: their carcinogenic and modifying effects on chemical carcinogenesis. Food Chem Toxicol 24:1099–1102
Jun SY, Park PJ, Jung WK, Kim SK (2004) Purification and characterization of an antioxidative peptide from enzymatic hydrolysate of yellow fin sole (Limanda aspera) frame protein. Eur Food Res Technol 219:20–26
Jung WK, Rajapakse N, Kim SK (2005) Antioxidative activity of low molecular peptide derived from the sauce of fermented blue mussel, Mytilus edulis. Eur Food Res Technol 220:535–539
Kapsokefalou M, Miller DD (1991) Effects of meat and selected food components on the valence of nonheme iron during in vitro digestion. J Food Sci 56(2):352–355
Klompong V, Benjakul S, Kantachote D, Shahidi F (2007) Antioxidative activity and functional properties of protein hydrolysate of yellow stripe trevally (Selaroides leptolepis) as influenced by the degree of hydrolysis and enzyme type. Food Chem 102:1317–1327
Lertittikul W, Benjakul S, Tanaka M (2007) Characteristics and antioxidative activity of Maillard reaction products from a porcine plasma protein–glucose model system as influenced by pH. Food Chem 100:669–677
Liu R, Wang M, Duan J, Guo J, Tang Y (2010) Purification and identification of three novel antioxidant peptides from Cornu bubali (water buffalo horn). Peptides 31(5):786–793
McCord JM (2000) The evolution of free radicals and oxidative stress. Am J Med 108:652–659
Mitsuta H, Yasumoto K, Iwami K (1996) Antioxidative action of indole compounds during the autoxidation of linoleic acid. Eiyo to Shokuryo 29:238–244
Nanjo F, Goto K, Seto R, Suzuki M, Sakai M, Hara Y (1996) Scavenging effects of tea catechins and their derivatives on 1,1,-diphenyl-2-picrylhydrazyl radical. Free Radic Biol Med 21:895–902
Osawa T, Namiki M (1985) Natural antioxidants isolated from eucalyptus leaf waxes. J Agric Food Chem 33:777–780
Peng X, Xiong YL, Kong B (2009) Antioxidant activity of peptide fractions from whey protein hydrolysates as measured by electron spin resonance. Food Chem 113:196–201
Pihlanto Leppala A (2001) Bioactive peptides derived from bovine whey proteins: opioid and ACE-inhibitory peptides. Trends Food Sci Technol 11:347–356
Pihlanto-Leppala A (2006) Antioxidative peptides derived from milk proteins. Int Dairy J 16:1306–1314
Pryor WA, Ann NY (1982) Free radical biology: xenobiotics, cancer, and aging. Acad Sci 393:1–22
Qian ZJ, Jung WK, Kim SK (2008) Free radical scavenging activity of a novel antioxidative peptide purified from hydrolysate of bull frog skin Rana catesbeiana Shaw. Bioresour Technol 99:1690–1698
Rajapakse N, Mendis E, Byun HG, Kim SK (2005) Purification and in vitro antioxidative effects of giant squid muscle peptides on free radical-mediated oxidative systems. J Nutr Biochem 16:562–569
Ranathung S, Rajapakse N, Kim SK (2006) Purification and characterization of antioxidative peptide derived from muscle of conger eel (Conger myriaster). Eur Food Res Technol 222:310–315
Roberts PR, Burney JD, Black KW, Zaloga GP (1999) Effect of chain length on absorption of biologically active peptides from the gastrointestinal tract. Digestion 60:332–337
Rosen GM, Rauckman EJ (1984) Spin trapping of superoxide and hydroxyl radicals. Methods Enzymol 105:198–209
Shabeena YN, Nazeer RA (2010) Antioxidant activity of hydrolysates and peptide fractions of Nemipterus japonicas and Exocoetus volitans muscle. J Aquat Food Prod Technol 19:182–192
Sherwin ER (1990) Antioxidant. In: Branen AL, Davidson PM, Salminen S (eds) Food additives. Marcel Dekker, New York, USA, pp 139–193
Suetsuna K (1998) Isolation and characterization of angiotensin I-converting enzyme inhibitor dipeptides derived from Allium sativum L. (garlic). J Nutr Biochem 9:415–419
Tang CH, Wang XS, Yang XQ (2009) Enzymatic hydrolysis of hemp (Cannabis sativa L) protein isolate by various proteases and antioxidant properties of the resulting hydrolysates. Food Chem 114:1484–1490
Thiansilakul Y, Benjakul S, Shahidi F (2007) Compositions, functional properties and antioxidative activity of protein hydrolysates prepared from round scad (Decapterus maruadsi). Food Chem 103:1385–1394
Wiseman H, Halliwell B (1996) Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J 313:17–29
Wu HC, Chen HM, Shiau CY (2003) Free amino acids and peptides as related to antioxidant properties in protein hydrolysates of markerel (Scomber austriasicus). Food Res Int 36:949–957
Xie Z, Huang J, Xu X, Jin Z (2008) Antioxidant activity of peptides isolated from alfalfa leaf protein hydrolysate. Food Chem 111:370–376
Yen GC, Chen HY (1995) Antioxidant activity of various tea extracts in relation to their antimutagenicity. J Agric Food Chem 43:27–32
Yildirim A, Mavi A, Oktay M, Kara AA, Algur OF, Bilaloglu V (2000) Comparison of antioxidant and antimicrobial activities of tilia (Tilia argentea Desf Ex DC), sage (Salvia triloba L.) and black tea (Camellia sinensis) extracts. J Agric Food Chem 48:5030–5034
Zhu K, Zhou H, Qian H (2006) Antioxidant and free radical-scavenging activities of wheat germ protein hydrolysates (WGPH) prepared with alcalase. Process Biochem 41:1296–1302
Acknowledgments
We gratefully acknowledge Dr. K. Ramasamy, Dean, School of Bioengineering, SRM University, for his support throughout the work. We extend our gratitude to the management, SRM University for providing the facilities.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sampath Kumar, N.S., Nazeer, R.A. & Jaiganesh, R. Purification and identification of antioxidant peptides from the skin protein hydrolysate of two marine fishes, horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber). Amino Acids 42, 1641–1649 (2012). https://doi.org/10.1007/s00726-011-0858-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-011-0858-6