
Abstract
The filamentous fungus Botrytis cinerea is an important agricultural pathogen affecting a wide range of cultivated plants. Since World War II, chemical fungicides have been the go-to method for agricultural pathogen control. However, the potential adverse environmental and health effects of these chemicals have led to an increasing demand for alternative methods of pathogen control, including biological control agents. In this study, we identified a bacterial isolate with strong antagonistic activity against B. cinerea. An analysis of the 16S rRNA gene sequence for this isolate identified it as a novel strain of Bacillus subtilis. Culture media from this isolate were harvested and fractionated using ion exchange and gel filtration chromatography. The fraction exhibiting the highest level of antifungal activity was identified, and its sequence determined by electrospray tandem mass spectrometry had significant similarity to flagellin. This flagellin-like protein was exogenously expressed in Escherichia coli, and screened for antifungal activity against B. cinerea. This flagellin-like protein demonstrated clear antifungal activity of inhibiting B. cinerea growth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The filamentous fungus Botrytis cinerea is an important agricultural pathogen affecting a wide range of cultivated plants, and can cause significant economic losses both before and after harvest (Guinebretiere et al. 2000). Chemical and biological approaches are commonly used to control B. cinerea (Daniel et al. 2006). Since World War II, broad-spectrum synthetic chemical fungicides have been used to control fungal populations, but the use of these fungicides is being phased out in many countries due to increasing resistance (Rosenberger and Meyer 1981; Spotts and Cervantes 1986). Furthermore, many governments have begun to restrict the use of chemical fungicides due to environmental concerns, echoing the demands of consumers’ concern on safety of chemical pesticides usage (Caia et al. 1988). Combined with the emergence of iatrogenic diseases caused by fungicides (Jordan 1973; Hislop 1976; Griffiths 1981), these concerns have increased interest in alternative methods of pathogen control (Mari et al. 1996). As a result, the use of natural fungicides, as a component of integrated pest management, has been gaining acceptance throughout the world (Estibaliz et al. 2010).
Numerous biocontrol methods have been successfully developed in recent years (Guinebretiere et al. 2000; Montealegre et al. 2003; Daniel et al. 2006). One such example is Bacillus thuringiensis, a Gram-positive, soil-dwelling bacterium that has been successfully used as a biopesticides in agriculture and forestry. Its advantages lie in its specific toxicity against target insects, lack of polluting residues, and safety to non-target organisms such as mammals, birds, amphibians, and reptiles (Estibaliz et al. 2010).
Microorganisms that grow in the soil are ideal for use as biocontrol agents, as many possess defense mechanisms that directly inhibit plant pathogens. Several mechanisms have been proposed to explain pathogen suppression (Thomashow and Weller 1996), including competition for nutrients (e.g., via the production of siderophores) and the production of toxic metabolites such as antibiotics and HCN (O’Sullivan and O’Gara 1992; Thomashow and Weller 1996). Considering the potential environmental and health risks of chemical fungicides, we attempted to identify bacterial isolates exhibiting antagonistic activity against fungal pathogens. In this paper, we describe the isolation and characterization of a novel strain of Bacillus subtilis exhibiting significant potential for use as an effective biocontrol agent. We also describe the identification of an antifungal compound isolated from B. subtilis culture filtrates, along with the gene responsible for its production. The in vitro antagonistic effects of this compound against B. cinerea were also investigated.
Materials and methods
Reagents and chemicals
All fungal strains (Fusarium moniliforme, Colletotrichum gloeosporioides, Verticillium dahliae Kleb., Fusarium solani, Ralstonia solanacearum, Polyporus hirsutus, B. cinerea, and Monilinia laxa) were isolated from different hosts and identified by the Department of Plant Science and Technology, Beijing University of Agriculture (Beijing, China). All reagents and solvents used in this study were analytical grade.
Isolation of bacteria for studies of antagonism
A total of 200 soil samples were collected from 50 different agricultural sites in a suburban area of Beijing. All locations were selected at random; the study sites consisted of public areas free of environmental or other restrictions governing scientific research. At each site, four plots were formed, each approximately 15 m × 5 m. A total of 20 individual samples were randomly collected from the surface layer (0–15 cm) of each plot using a sterile auger, and mixed to yield one composite sample per plot; a total of 4 composite soil samples were collected from each site. Mixing and root debris removal was achieved by passage through a 2-mm mesh sieve. Bacterial cultures were obtained by dissolving 5 g of soil into 100 mL of phosphate buffer solution (pH 7.4). The cultures were incubated at 37 °C on an orbital shaker for 15 min at 160 rpm. The supernatants were then diluted 10–1,000 fold. A total of 100 μL of each dilution was spread on an LB agar plate and incubated at 37 °C until most of the bacterial colonies became visible (Maria and Ruiz de Valladares 2003). A total of 103 isolates were selected for further analysis; bacterial isolates were purified by streaking on agar plates, and stored at −80 °C (Adesina et al. 2007). Based on in vitro dual plate cultures and potato dextrose agar (PDA) assays, isolate RN-061 was selected for further testing as an in vivo biological control agent.
Determination of the antagonistic activity of the isolated bacteria
To determine the potential antifungal activity of each bacterial isolate in vitro, isolates were co-cultured alongside the pathogenic fungus B. cinerea. The fungal strains were cultured on PDA plates for 5 days at 28 °C; bacterial strains were grown in LB liquid medium for 24 h at 37 °C (Suárez-Estrella et al. 2007). Competitive interactions between the bacterial isolates and B. cinerea were evaluated in dual-culture experiments on 90-mm Petri dishes containing 20 mL of PDA. Mycelial disks (5 mm in diameter) from fungal colonies and 2 μL of bacterial suspension from the LB cultures were placed on the agar surface, 30 mm apart. Control cultures were inoculated with 2 μL of distilled water instead of bacteria. Immediately after inoculation, the plates were sealed with plastic film and incubated at 28 °C in the dark for 3–5 days. Colony growth and inter-specific interactions were examined daily under a stereomicroscope (Gloria et al. 2003); inhibition was defined as the absence of contact between the bacterial and fungal strains (Hernández-Rodrígueza et al. 2008). From these analyses, isolate RN-061 was identified as exhibiting the strongest antifungal activity. This isolate was then tested against a range of fungi to determine its spectrum of antifungal activity.
Antifungal activity of culture filtrates from the antagonistic bacteria
Bacteria showing antagonistic activity against B. cinerea were cultured at 37 °C for 48 h at 160 rpm in LB media. Bacterial culture filtrates were concentrated by rotary evaporation and passed through sterile 0.45-μm Millipore filters (Millipore, Billerica, MA, USA). The inhibitory effects of these concentrated extracts toward B. cinerea were assessed using the method of Hastings and Kirby (1966) with modifications. Briefly, 10 mL of extract was mixed with 90 mL of molten PDA culture medium (45 °C) and spread on 90-mm plates. Next, mycelial disks (5 mm in diameter) from 5-day-old fungal cultures were placed in the center of each plate. Control cultures were inoculated using mycelial disks on standard PDA media. The plates were incubated for 3–5 days at 28 °C. Two perpendicular directions of radial growth of the fungal colony were measured using a stereo zoom microscope (Ah et al. 2009). The percentage of inhibition was calculated through a comparison with the control plate, as described in Bashan et al. (1996). The experiment was repeated three times with five replicates per treatment (Hernández-Rodrígueza et al. 2008).
Amplification of the 16S rRNA gene of isolate RN-061 by PCR
Strain RN-061 was further characterized using standard biochemical methods combined with 16S rRNA gene sequence analysis. Chromosomal DNA was isolated from bacterial cells using a bacterial genomic DNA preparation kit (Tiangen, Beijing, China). The 16S rRNA gene of RN-061 was amplified using the universal primers 27f (5′-AGAGTTTGATCCTGGCTCAG-3′) (Invitrogen, Carlsbad, CA, USA) and 1492r (5′-GGTTACCTTGTTACGACTT-3′) (Invitrogen), which are complementary to the 5′ and 3′ ends of the prokaryotic 16S rRNA gene, respectively (Lane 1991). All PCRs were performed using a MyCycler S1000 (Bio-Rad, Hercules, CA, USA). Amplification was carried out in a 25-μL reaction volume using a PCR master mix kit (Invitrogen) according to the manufacturer’s instructions, under the following conditions: 95 °C for a 5-min initial denaturation step, followed by 32 cycles of denaturation at 95 °C for 30 s, annealing at 53 °C for 30 s, and extension at 72 °C for 90 s, with a final extension at 72 °C for 5 min. The products were purified using an AxyPrep PCR Clean-Up Kit (Axygen, Union City, CA, USA) according to the manufacturer’s instructions, and analyzed by 1 % agarose gel electrophoresis. The purified products were sequenced using the same primers with an Applied Biosystems 3730XL genetic analyzer (Foster City, CA, USA) according to the manufacturer’s instructions. The sequences were analyzed for similarity to other known sequences found in the GenBank database using BLAST.
Ion exchange chromatography and gel filtration chromatography (GFC)
A single colony of isolate RN-061 was streaked onto LB agar plates and incubated at 37 °C for 12 h. Following incubation, the plates were washed with 1 mL of sterile water, and the suspension used to inoculate 30 mL of fermentation broth. The broth was incubated at 37 °C on an orbital shaker at 160 rpm for 48 h, followed by centrifugation at 10,000 rpm at 4 °C for 15 min. The supernatant was transferred to a new tube, saturated with 80 % (NH4)2SO4, and incubated at 4 °C for 48 h. The precipitate was collected by centrifugation at 10,000 rpm at 4 °C for 15 min, dissolved in distilled water, and dialyzed using a 25-kDa filter (Biomed Instruments Inc., Fullerton, CA, USA) to remove the (NH4)2SO4. Crude proteins were dissolved in Tris–HCl buffer (pH 8.0) at a concentration of 20 mg/mL, and loaded for fast protein liquid chromatography on a DEAE anion exchange column (CL-6B; Sigma, St. Louis, MO, USA) equilibrated with Tris–HCl buffer (pH 8.0). Elution was performed using a linear gradient of NaCl (0–1.5 M) in the same buffer at a flow rate of 1 mL/min. Each fraction, collected at a volume of 5 mL, was monitored at 280 nm. Next, pooled fractions were concentrated with a rotary evaporator, and the antifungal activity was investigated. Fractions exhibiting the strongest antifungal properties were lyophilized and subjected to further separation.
Those protein fractions exhibiting the highest levels of antifungal activity were further purified after dissolution in distilled water by loading onto a Sephadex G-25 gel filtration column (2.5 cm × 75 cm) previously equilibrated with distilled water. The column was eluted with distilled water at a flow rate of 1 mL/min. Each fraction was collected at a volume of 3 mL and monitored at 280 nm. Pooled fractions were concentrated using a rotary evaporator, and the antifungal activity was investigated. Those fractions exhibiting strong antifungal properties were lyophilized according to the method of Laemmli and Favre (1973). Lyophilized proteins were resolved by SDS-PAGE and stained with Coomassie blue. The molecular mass of the antifungal protein was determined by comparing its electrophoretic mobility to those of molecular mass marker proteins from Fermentas (Burlington, ON, Canada), followed by electrospray tandem mass spectrometry (ESI–MS/MS) to determine the sequence of the purified protein (Kumar et al. 2012).
Identification of the proteins by ESI–MS/MS
The protein bands were excised from the polyacrylamide gel and purified using an SDS-PAGE Clean-Up Kit (Axygen). The samples were dissolved in 75 % acetonitrile/25 % water (HPLC grade), and loaded into an FIA type 3200 QTRAP mass spectrometer (Applied Biosystems). The sample was passed at a flow rate of 20 μL/min via the electrospray interface, which was operated in the positive electrospray ionization (ESI + ve) mode. High-purity nitrogen was used for drying (35 psi) and ESI nebulization (45 psi). Spectra were recorded over the mass/charge (m/z) ratio. Three spectra were averaged in the MS and multiple MS (MS/MS) analyses. Protein sequencing was performed by manual calculation.
PCR amplification and sequence analysis of the flagellin gene
Specific primer pairs (FLA-f and -r) were manually designed based on the flagellin gene of B. subtilis subsp. subtilis str. 168 to amplify the complete sequence of the flagellin gene from the RN-061 genome. In this experiment, Flagellin-f (5′-CATGCCATGGATGAGAATTAACCACAATATTGC-3′) and Flagellin-r (5′-CCGCTCGAGACGTAATAATTGAAGTACGT-3′) were used as upstream and downstream primers, respectively. Amplification was carried out in a 50-μL reaction volume containing 10 μL of Taq polymerase buffer (5×), 1 μL of dNTPs (10 μM), 2 μL of MgCl2 buffer (25 mM), 0.5 μL of each primer (10 μL), 0.125 μL of Taq polymerase (5 U/μL), and 1–5 μL of template. The reaction conditions were as follows: denaturation for 5 min at 94 °C, followed by 25 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 45 s, with a final extension at 72 °C for 5 min. The products were resolved by electrophoresis on 1 % agarose gels and compared to a 4.5-kb DNA ladder. The purified products were sequenced using the same primers and analyzed for similarity to other known sequences found in the GenBank database using BLAST.
Identification of the antifungal function of the novel flagellin gene
The resulting PCR fragment was cloned using Phusion polymerase to introduce XhoI and NcoI restriction sites at the 5′ and 3′ ends, respectively. The PCR products were digested with XhoI and NcoI, and cloned into pET-28a(+) to produce pET-FLA061. pET-FLA061 was then transformed into Escherichia coli Rosetta competent cells, and grown on LB medium containing kanamycin (20 μg/mL) at 37 °C until individual colonies arose. Individual colonies were harvested and grown overnight at 37 °C in liquid LB medium again until the optical density at 600 nm reached 0.8–1.0. Expression of the flagellin protein (FLA) was induced using isopropyl-1-thio-β-d-galactopyranoside (0.5 mM) followed by incubation at 37 °C overnight. Transformed colonies lacking the target gene were used as controls. The antifungal activity of the transgenic E. coli strain was analyzed using the method described above.
Statistical analysis
All assays for antifungal activity were conducted in three replicates. The data are expressed as the mean ± standard deviation. Statistical analyses were performed using SPSS 10.0 software (SPSS Inc., Chicago, IL, USA). Significant differences were determined with 95 % confidence intervals (P < 0.05).
Results
Isolation, characterization, and screening of antagonistic bacterial strains
A total of 103 colonies were chosen at random from plates containing a wide array of divergent colonies (Thirumala et al. 2010). All isolates were screened for their ability to inhibit the growth of B. cinerea. As shown in Table 1, in the dual culture plate assay, only 14 of the 103 strains were found to be antagonistic to B. cinerea on PDA. The antagonistic activity of the strains eventually stabilized, with isolate RN-061 exhibiting the most potent suppression of fungal development (Table 1). The zone of inhibition between isolate RN-061 and B. cinerea was 7.12 mm after 5 days compared with −6.81 mm for the B. cinerea-inoculated control, indicating significant inhibition of fungal growth (P < 0.05; Fig. 1). The antifungal activity of isolate RN-061 was not limited to B. cinerea; it possessed activity against different fungi representing multiple genera (Table 2). As a result, this isolate was selected for further study.

The antagonistic activity of RN-061 on the growth of B. cinerea (after 5 days). The control plate is shown at the left; the treatment plate is shown at the right. The small white colony inoculated on both sides of the pathogen fungi is strain RN-061
PCR amplification and sequence analysis of the 16S rRNA gene of RN-061
The 16S rRNA gene from isolate RN-061 was amplified by PCR and compared against the GenBank database by BLAST. Our sequence analysis revealed 99 % similarity to the 16S rRNA gene of B. subtilis subsp. subtilis str. 168 (GenBank accession no. NC_000964.3), B. subtilis BEST7003 DNA (GenBank accession no. AP012496.1), B. subtilis QB928 (GenBank accession no. NC_018520.1), and B. subtilis BEST7613 (GenBank accession no. AP012495.1). Isolate RN-061 was therefore considered to represent a novel strain belonging to the B. subtilis family and has been named B. subtilis RN-061. The 16S rRNA sequence of this strain has been deposited in the GenBank database (accession no. KC840668).
Isolation of antifungal metabolites from B. subtilis RN-061
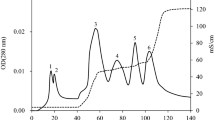
Media from B. subtilis RN-061 cultures were precipitated in vitro by 80 % (NH4)2SO4 saturation. Following dialysis and lyophilization, the cultures were fractionated using an anion exchange column at pH 8.0 with a linear gradient concentration of NaCl. All fractions were tested for antifungal activity against B. cinerea. As shown in Table 3, fractions 2 and 3 showed the highest overall activity with inhibition rates of 40.94 and 34.06 %, respectively (P < 0.05). The inhibition rates decreased over time for all fractions, similar to the results of our dual-culture experiments. Fraction 2 was further purified using a gel filtration column. As shown in Fig. 2, three peaks were eluted from this fraction. Of these peaks, fraction B showed the highest level of activity, with an inhibition rate of 50.00 % after 2 days, 45.86 % after 3 days, and 40.94 % after 4 days (P < 0.05; Table 4). The protein fractions were further resolved by SDS-PAGE, revealing a single 32-kDa band (Fig. 3).

Separation of an antifungal peptide from B. subtilis RN-061 by GFC. Following purification using DEAE anion exchange chromatography, the fraction with high activity was further purified by Sephadex G-25 GFC. Peak B exhibited the highest efficacy against B. cinerea

SDS-PAGE analysis of fraction B from Sephadex G-25 GFC. Left lane molecular mass marker. Right lane fraction B (purified antifungal protein)
Identification of an antifungal protein from B. subtilis RN-061
To identify the protein described above, the protein band was purified using a Sephadex G-25 gel filtration column and analyzed by ESI–MS/MS. The spectrum pattern of separated products (Fig. 4) showed five main relative abundance peaks, consistent with the molecular form of the purified protein. The resulting protein sequence was blasted against the NCBI database with the best overall match showing 44 % identity with the flagellin (FLA) protein of B. subtilis subsp. subtilis str. 168 (accession no. NP_391416.1). The specific alignment is shown in Fig. 5. These results are consistent with previous reports suggesting that the formation of the purified protein was mainly via flagellin activity (Zhao et al. 2013). As a result, we predicted that this protein was either flagellin or a member of the flagellin family, and that this protein was responsible for the antifungal activity exhibited by the isolate.

Mass spectra of the purified protein

Aligned amino acid sequence (in red, bold font) of the novel protein with sequences from the amino acid database of Mascot. (Color figure online)
PCR amplification and complete sequence analysis of the flagellin gene
To further confirm that the high level of antifungal activity of strain RN-061 was due to flagellin, we designed PCR primers using the flagellin gene of B. subtilis subsp. subtilis str. 168 as template. Primers FLA-f and FLA-r successfully amplified the complete flagellin gene from B. subtilis RN-061, producing an amplicon of 800 bp (Fig. 6). The resulting product was purified, sequenced, and the translated amino acid sequence (Fig. 7) blasted against the NCBI protein database. The sequence exhibiting the highest degree of identity (91 %) to our putative flagellin gene was flagellin-2 of Bacillus sp. JS (accession no. YP_006233347.1). This gene also showed significant similarity to flagellin hypothetical protein BSNT_05346 (90 %) (accession no. YP_005562843.1), as well as flagellin genes from Bacillus licheniformis WX-02 (89 %; accession no. ZP_17658917.1), Bacillus sp. M 2-6 (82 %; accession no. ZP_10164049.1), and B. subtilis subsp. subtilis str. 168 (69 %; accession no. NP_391416.1; Fig. 8). This isolated PCR product had no identical sequence in the protein database; we are therefore reporting the cloning of a new antifungal gene from the antifungal bacterium B. subtilis RN-061, to which the name FLA-RN061 was given. Figures 7 and 8 show the predicted amino acid sequence of this new flagellin and multiple alignment of FLA-RN061 to related proteins.

Agarose gel electrophoresis of the amplified FLA gene of B. subtilis RN-061. The FLA on the left side is the PCR product; on the right side is the DNA molecular weight marker

Deduced amino acid sequence of the new FLA from B. subtilis RN-061

Multiple alignment of the deduced amino acid sequence of flagellin RN061 (FLA) from B. subtilis RN061 and other closely related strains. The third branch is flagellin from our new isolated strain
Exogenous expression and antifungal activity of FLA-RN061
To further confirm the antifungal effect of this protein, the exogenous expression of FLA-RN061 was performed in E. coli Rosetta 2 competent cells using the plasmid pET-FLA061. Culture filtrates from cells transformed with this plasmid showed clear antifungal activity; no activity was seen for vector controls. The level of antifungal activity was determined by co-culture with B. cinerea. The growth of B. cinerea was greatly diminished in the presence of E. coli expressing FLA-RN061 compared to vector controls (Fig. 9). The inhibition rate of the metabolites from the E. coli strain expressing FLA-RN061 was 47.06 % after 48 h, 41.22 % after 72 h, and 36.72 % after 96 h; no antifungal effect was observed for control strains (Table 5). Together, these result clearly demonstrates that the antifungal activity of isolate RN061 attribute to its FLA-RN061 gene.

The antifungal effect of E. coli expressing FLA-RN061 compared to an unmodified E. coli control. a The control plate (shown on the left) contains the filtered cultures from an unmodified plasmid E. coli strain; the plate on the right contains the filtered cultures from the gene-modified E. coli strain
Discussion
Botrytis cinerea is one of the most serious pathogens of horticultural plants in China. We isolated a novel B. subtilis strain exhibiting a potent inhibitory effect on the growth of B. cinerea, consistent with previous reports. Fiddaman and Rossall (1993) described a strain of B. subtilis that produced an antibiotic metabolite with antifungal activity against Rhizoctonia solani and Pythium ultimum; Gueldner et al. (1988) reported that iturins isolated from B. subtilis may be useful for the biological control of peach brown rot. A third substance, fengycin, was isolated from B. subtilis strain F-29-3. This compound inhibited filamentous fungi, but was not effective against yeast or bacteria (Vanittanakom et al. 1986).
In the present study, we identified a novel protein, flagellin RN-061, which is capable of inhibiting the growth of B. cinerea. Exogenous expression of this protein in E. coli resulted in a 36.72 % decrease in the growth of B. cinerea after 96 h (Table 4); this effect was greater than that of some chemical fungicides.
The flagellum is an important virulence factor for many bacteria pathogenic to both animals and plants, and is a potent stimulator of innate immune responses. Plants have a highly sensitive chemoperception system for eubacterial flagellins, which specifically targets the most highly conserved domains within its N-terminus (Georg et al. 1999). Bacterial flagellins have well-conserved N- and C-termini, but they contain hypervariable regions in the center of the protein (Komoriya et al. 1999), consistent with the flagellin described here.
Flagellin proteins are expressed in B. subtilis, as well as many other genera. However, the antifungal activities described here are found primarily in B. subtilis; further study will be necessary to identify the mechanism by which these proteins inhibit fungal growth. Although most secreted flagellins are assembled in a flagellum, flagellin can also accumulate in the bacterial environment as a result of leaks and spillover during the construction of flagella (Komoriya et al. 1999). Such FLAs may be easily transported to the extracellular space. Accordingly, we hypothesize that the antifungal effect of FLA-RN061 is derived from the variable center region of the protein, and that this effect depends on the transportation of flagellin to the extracellular space.
Flagellin is an important pathogen-associated molecular pattern capable of inducing innate immune responses in both plants and animals. The detection of flagellin by plants induces a wide range of defense mechanisms, including oxidative bursts, callose deposition, and ethylene production, leading to the induction of defense-related genes such as PR1 and PR5 (Felix et al. 1999; Lemaitre et al. 1997; McDermott et al. 2000; Eaves-Pyles et al. 2001; Zipfel et al. 2004).
Despite the well-described role of flagellin in plants and animals, little is known about the molecular mechanisms linking flagellin receptor activation to intracellular signal transduction in fungi. Based on conserved mechanisms in plants and animals, in conjunction with our study on the antifungal effect of flagellin, we speculate that flagellin may trigger pattern-recognition receptors (PRRs) in fungi. The leucine-rich repeat receptor kinase flagellin-sensitive 2 (FLS2) in Arabidopsis thaliana acts as a PRR for bacterial flagellin, and contributes to resistance against bacterial pathogens (Chinchilla et al. 2007). This gene may also function as a PRR in fungi, though more work is necessary to test this hypothesis. The identification of PRRs in fungi may be helpful to determine the mechanism by which flagellin inhibits fungal growth.
Conclusions
Our results indicate that B. subtilis RN-061 is a promising candidate as a biocontrol agent in the prevention and cure of plant diseases. A complete flagellin gene with 91 % sequence homology with other known flagellins was characterized from this new strain. Its encoded protein expressed in E. coli was capable of inhibiting the growth of B. cinerea. Research on the antifungal mechanism of RN061 flagellin is underway.
References
Adesina MF, Lembke A, Costa R, Speksnijder A, Smalla K (2007) Screening of bacterial isolates from various European soils for in vitro antagonistic activity towards Rhizoctonia solani and Fusarium oxysporum: site-dependent composition and diversity revealed. Soil Biol Biochem 39:2818–2828
Ah RJ, Marimuthu J, Kyoung ML, Won IS, Jung SK, In WK (2009) Purification, characterization and synergistic activity of -1,3-glucanase and antibiotic extract from an antagonistic B. subtilis NSRS 89–24 against rice blast and sheath blight. Microbiol Biotechnol 83:285–294
Bashan Y, Holguı′n G, Ferrera-Cerrato R (1996) Interacciones entre plantas y microorganismos bene′ficos. Terra 14:159–192
Caia G, Bertoluzza A, Foschi F (1988) Fitofarmaci e legislazione. In: Goida`nich G, Pratella GC (eds) Fitofarmaci, Igiene e Ambiente. Maggioli, Rimini, pp 236–250
Chinchilla D, Zipfel C, Robatzek S, Kemmerling B, Nürnberger T, Jones JDG, Felix G, Boller T (2007) A flagellin-induced complex of the receptor FLS2 and BAK1 initiates plant defence. Nature 448:497–500
Daniel AN, Ursula K, Lukas S, Wilfried S (2006) Dual antagonism of aldehydes and epiphytic bacteria from strawberry leaf surfaces against the pathogenic fungus Botrytis cinerea in vitro. Biocontrol 51:279–291
Eaves-Pyles T, Murthy K, Liaudet L (2001) Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: IkBa degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J Immunol 166:1248–1260
Estibaliz S, Candelario V, Aurelio O (2010) Genetic manipulation in Bacillus thuringiensis for strain improvement. Biotechnol Lett 32:1549–1557
Felix G, Duran JD, Volko S, Boller T (1999) Plants recognize bacteria through the most conserved domain of flagellin. Plant J 18:265–276
Fiddaman PJ, Rossall S (1993) The production of antifungal volatiles by B. subtilis. J Appl Microbiol 74:119–126
Georg F, Duran JD, Sigrid V, Thomas B (1999) Plants have a sensitive perception system for the most conserved domain of bacterial flagellin. Plant J 18:265–276
Gloria I, Roberta R, Matteo M, Eva Z (2003) Efficacy of microorganisms antagonistic to Rhizoctonia cerealis and their cell wall degrading enzymatic activities. Mycol Res 107:421–427
Griffiths E (1981) Iatrogenic plant diseases. Annu Rev Phytopathol 19:69–82
Gueldner RC, Reilly CC, Pusey PL, Costello CE, Arrendale RF, Cox RH, Himmelsbach DS, Crumley FG, Cutler HG (1988) Isolation and identification of iturins as antifungal peptides in biological control of peach brown rot with B. subtilis. J Agric Food Chem 36:366–370
Guinebretiere MH, Nguyen-The C, Morrison N, Reich M, Nicot P (2000) Isolation and characterization of antagonists for the control of the post harvest wound pathogen Botrytis cinerea on strawberry fruits. Food Prot 63:386–394
Hastings JR, Kirby KS (1966) The nucleic acids of Drosophila melanogaster. Biochem J 100:532–539
Hernández-Rodrígueza A, Heydrich-Péreza M, Acebo-Guerreroa Y, Velazquez-del MJV, Hernández-Lauzardob AN (2008) Antagonistic activity of Cuban native rhizobacteria against Fusarium verticillioides (Sacc.) Nirenb. In maize (Zea mays L.). Appl Soil Ecol 39:180–186
Hislop EC (1976) Some effects of fungicides and other agrochemicals on the microbiology of the aerial surfaces of plants. In: Dickinson CH, Preece TF (eds) Microbiology of aerial plant surface. Academic Press, New York, pp 41–74
Jordan VWL (1973) The effects of prophylactic spray programs on the control of pre- and postharvest diseases of strawberry. Plant Pathol 22:67–70
Komoriya K, Shibano N, Higano T, Azuma N, Yamaguchi S, Aizawa SI (1999) Flagellar proteins and type III-exported virulence factors are the predominant proteins secreted into the culture media of Salmonella typhimurium. Mol Microbiol 34(4):767–779
Kumar NSS, Nazeer RA, Jaiganesh R (2012) Purification and identification of antioxidant peptides from the skin protein hydrolysate of two marine fishes, horse mackerel (Magalaspis cordyla) and croaker (Otolithes ruber). Amino Acids 42:1641–1649
Laemmli UK, Favre M (1973) Maturation of the head of bacteriophage T4.I.DNA packaging events. J Mol Biol 80:575–599
Lane DJ (1991) 16S/23S rRNA sequencing. In: Nucleic acid techniques in bacterial systematics, Wiley, Chichester, UK, pp 115–175
Lemaitre B et al (1997) Drosophila host defense: differential induction of antimicrobial peptide genes after infection by various classes of microorganisms. Proc Natl Acad Sci USA 94:14614–14619
Mari M, Guizzardi M, Pratella GC (1996) Biological control of gray mold in pears by antagonistic bacteria. Biol Control 7:30–37
Maria MT, Ruiz de Valladares R (2003) Antagonistic interactions among Fusobacterium nucleatum and Prevotella intermedia with oral Lactobacilli. Res Microbiol 154:669–675
McDermott PF et al (2000) High-affinity interaction between gram-negative flagellin and a cell surface polypeptide results in human monocyte activation. Infect Immun 68:5525–5529
Montealegre JR, Reyes R, Perez LM, Herrera R, Silvia P, Besoain X (2003) Selection of bioantagonistic bacteria to be used in biological control of Rhizoctonia solani in tomato. Environ Biotechnol 6:1–8
O’Sullivan DJ, O’Gara F (1992) Traits of fluorescent Pseudomonas spp. involved in suppression of plant root pathogens. Microbiol Rev 56:662–676
Rosenberger DA, Meyer FW (1981) Postharvest fungicides for apples: development of resistance to benomyl, vinclozolin, and iprodione. Plant Dis 65:1010–1013
Spotts RA, Cervantes LA (1986) Populations, pathogenicity and benomyl resistance of Botrytis spp., Penicillium spp. and Mucor piriformis in packinghouses. Plant Dis 70:106–108
Suárez-Estrella F, Vargas-García C, Lópeza MJ, Capel C, Moreno J (2007) Antagonistic activity of bacteria and fungi from horticultural compost against Fusarium oxysporum f.sp. Melonis. Crop Prot 26:46–53
Thirumala M, Sultanpuram VR, Mahmood SK (2010) Production and characterization of PHB from two novel strains of Bacillus spp. isolated from soil and activated sludge. J Ind Microbiol Biotechnol 37:271–278
Thomashow LS, Weller DM (1996) Current concepts in the use of introduction bacteria for biological disease control: mechanisms and antifungal metabolites. Plant-Microbe Interact 1:187–235
Vanittanakom N, Loeffler W, Koch U, Jung G (1986) Fengycin—a novel antifungal lipopeptide antibiotic produced by Bacillus subtilis F-29-3. J Antibiot 39:888–901
Zhao X, Zhou ZJ, Han Y, Wang ZZ, Fan J, Xiao HZ (2013) Isolation and identification of antifungal peptides from Bacillus BH072, a novel bacterium isolated from honey. Microbiol Res 3:23–26
Zipfel C, Robatzek S, Navarro L, Oakeley EJ, Jones JDG, Felix G, Boller T (2004) Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428:764–767
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (30872029, 31200483), the Platform Construction Project of Beijing Education Committee (PXM2012_014207_000014, PXM2012_014207_000028, PXM2012_014207_000016), Science and Technology Achievement Transformation and Industrialization of Beijing Education Committee (PXM2011_014207_000026), and the Beijing Municipal Science and Technology Commission Research Project (Z111100066111009).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ren, J.J., Shi, G.L., Wang, X.Q. et al. Identification and characterization of a novel Bacillus subtilis strain with potent antifungal activity of a flagellin-like protein. World J Microbiol Biotechnol 29, 2343–2352 (2013). https://doi.org/10.1007/s11274-013-1401-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-013-1401-6