
Abstract
Mercury (Hg) dynamics was evaluated in contaminated sediments and overlying waters from Tagus estuary, in two sites with different Hg anthropogenic sources: Cala Norte (CNOR) and Barreiro (BRR). Environmental factors affecting methylmercury (MMHg) production and Hg and MMHg fluxes across sediment/water interface were reported. [THg] and [MMHg] in solids (0.31–125 μg g−1 and 0.76–201 ng g−1, respectively) showed high variability with higher values in BRR. Porewater [MMHg] (0.1–63 ng L−1, 0.5–86% of THg) varied local and seasonally; higher contents were observed in the summer campaign, thus increasing sediment toxicity affecting the sediment/water Hg (and MMHg) fluxes. In CNOR and BRR sediments, Hg availability and organic carbon were the main factors controlling MMHg production. Noteworthy, an upward MMHg diffusive flux was observed in winter that was inverted in summer. Although MMHg production increases in warmer month, the MMHg concentrations in overlying water increase in a higher proportion compared to the levels in porewaters. This opposite trend could be explained by different extension of MMHg demethylation in the water column. The high concentrations of Hg and MMHg and their dynamics in sediments are of major concern since they can cause an exportation of Hg from the contaminated areas up to ca. 14,600 mg year−1 and an MMHg deposition of up to ca. 6000 mg year−1. The results suggest that sediments from contaminated areas of Tagus estuary should be considered as a primary source of Hg for the water column and a sink of MMHg to the sedimentary column.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Environmental contamination by mercury (Hg) is a worldwide problem. As such, it is important to assess the possible effects of increasing levels of mercury pollution on human health and the environment. In general, contaminated sites are places where high accumulation of toxic substances affects the soil, groundwater, sediments, and even air posing serious risks to the environment and ultimately to human health. The Minamata Convention on Mercury recognized Hg as a chemical of global concern due to its long-range atmospheric transport, its persistence in the environment once anthropogenically introduced, and especially to its ability to bioaccumulate in ecosystems, causing significant negative effects on human health and on the environment. After the Minamata Bay disaster (1950–1970), Hg has subsequently become one of the most critical trace metals examined in environmental studies (Tomiyasu et al. 2006). Mercury is released and mobilized through natural processes and by anthropogenic activities (Fang and Chen 2010). It exists in different chemical species with a wide range of properties and its ecotoxicological effects are strongly dependent on its chemical form (Logar et al. 2002; Ullrich et al. 2001). Inorganic mercury (Hg2+) is the main form of Hg in natural waters and sediments and can be naturally converted to its most toxic organic form methylmercury (MMHg), by biotic or abiotic processes (Celo et al. 2006). Methylmercury is then accumulated by aquatic organisms and is known to be biomagnified along the food chain, posing a threat to humans by fish consumption, driving exposure of this potent toxin with neurological, immunological, and cardiovascular effects (Karagas et al. 2012; Mahaffey et al. 2011; Mergler et al. 2007; Nyland et al. 2011). It is widely accepted that microorganisms can affect the mobility of Hg in a variety of metabolic pathways (Barkay and Wagner-Döbler 2005). Methylmercury has been thought to be produced predominantly by sulfate-reducing bacteria in anoxic sediments (Benoit et al. 2001; Ullrich et al. 2001); however, iron-reducing bacteria (Fleming et al. 2006; Kerin et al. 2006) and methanogens (Gilmour et al. 2013; Hamelin et al. 2011; Podar et al. 2015) are also capable of Hg methylation. In fact, recent studies demonstrated a diversity of Hg-methylating organisms other than sulfate-reducing bacteria (SRB) and iron-reducing bacteria (FeRB) and methanogens, including syntrophic acetogenic and fermentative Firmicutes (Gilmour et al. 2013; Podar et al. 2015). In sediments, microbial community is influenced by the extent of anoxic conditions, the quality and quantity of organic matter (Ullrich et al. 2001), and also by the concentration of electron acceptors such as sulfate and ferric ions (Hsu-Kim et al. 2013; Kerin et al. 2006). It was also been shown that microbial activity increased with temperature (e.g., King et al. 1999), which may be partially responsible for the seasonal variations in MMHg concentrations observed in many systems (e.g., Hammerschmidt and Fitzgerald 2004). However, the extent to which Hg is methylated is also highly dependent on the bioavailability of the inorganic Hg to the methylating organisms. Graham et al. (2012) reported that, in sediments under sulfidic conditions, dissolved organic matter (DOM) could play an important role affecting the uptake of inorganic Hg into methylating organisms. In fact, only a small amount (typically 5%) of the total amount of inorganic Hg in aquatic systems is in a “reactive” form that is considered available for microbial uptake and methylation (Domagalski 2001; Marvin-Dipasquale et al. 2009; Singer et al. 2016). The bioavailability of inorganic Hg in sediments is associated with the partitioning of Hg between solid and dissolved fractions, being related with the sediment-porewater distribution coefficient (K D) (Marvin-Dipasquale et al. 2009; Schartup et al. 2014). Sediment-porewater Hg partitioning is controlled by the amounts of Hg binding ligands, such as sulfide and thiols, in the solid and dissolved phases. These ligands are influenced by several factors such as sediment organic matter, redox conditions, and pH (Hammerschmidt and Fitzgerald 2004; Schartup et al. 2014). Overall, partitioning of Hg between solid and dissolved fractions can be an important factor controlling MMHg production and transport.
Extensive waste inputs from industrial and/or agricultural and/or urban sources have contributed to a significant increase of pollution in rivers and coastal areas. This is also the case of the Tagus estuary, where industrial activities are considered to be the main sources of mercury pollution (Canário et al. 2005; Figuères et al. 1985). As such, the study of the Hg dynamics in the sedimentary column of contaminated sites seems to be of great importance concerning the anthropogenic effects and changes in this aquatic ecosystem. Moreover, this estuarine ecosystem is of great importance due to artisanal fish and bivalve “fishing” that are further used for human consumption.
Sediment cores are often used as proxies to study temporal trends of Hg inputs into estuarine environments, including all natural and anthropogenic sources. In order to understand the biogeochemistry of Hg in contaminated sediments, this paper examines the local Hg patterns along contaminated zones of the Tagus estuary, investigating the factors that influence (1) the dynamics of total Hg and MMHg concentrations in the sediment column in two opposite seasons, (2) evaluating the processes responsible for the MMHg production, (3) quantifying the potential of Hg and MMHg diffusion within the sediment column by measuring Hg and MMHg in porewaters and finally, and (4) estimating the mobility and assessment of Hg species with respect to exchange between the sediment and water column. This study complements an ongoing investigation of Hg in non-contaminated zones of this estuary (Cesário et al. 2016), to understand the influence of different sources of Hg and MMHg in this estuarine ecosystem.
2 Material and Methods
2.1 Study Area and Sampling Dates
Tagus estuary is considered a contaminated aquatic system mainly due to the presence of two distinguished point sources of contaminant species: a chlor-alkali plant in Cala Norte (CNOR) and a smelting mineral plant in Barreiro (BRR) (Canário et al. 2003, 2005, 2007a, 2008a). The CNOR is a narrow navigation channel (max. 400 m width, 2 m depth) located in the north shore of the estuary. This area is surrounded by salt-marsh areas colonized by halophyte vegetation and receives the discharge of the chlor-alkali industry still in operation. Sediments of this channel incorporate anthropogenic materials associated with suspended particulate matter that settles (ca. 0.8 cm year−1) (Ferreira 1995) due to the low hydrodynamics of the channel. Consequently, the water residence time inside the channel is relatively high (8.6 days) (Hidroprojecto 1991), particularly in summer. Although the change in the electrolytic process has largely eliminated the use of Hg in the chlor-alkali plant of CNOR, historic emissions deposited in the Tagus estuary and those emanating from other sources still contributed to high contamination levels of Hg in the northward sediments of this channel, having thus an impact on water, sediment, and biota of the region (Canário et al. 2003, 2005, 2007a, 2008a). BRR site is a mudflat area without any vegetation around, located in the south shore of the estuary, near the channel used in the past as a navigation channel to fluvial transport of goods for and to the industrial complex (CUF channel). The industrial plants in BRR are no longer in operation (stopped in the end of 70s); however, high concentrations of mercury have been observed in sediment deposition areas, such as the CUF channel (Canário et al. 2005, 2007a), due to an intensive industrial activities between 1950 and 1960s. Besides, domestic discharges may still contribute to the high levels of Hg previously observed in this area (Canário et al. 2007a).
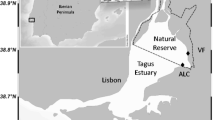
Sediment and water samples were collected in CNOR (38° 51′ 21.21″ N, 9° 3′ 40.51″ W) and in BRR (38° 40′ 45.40″ N, 9° 3′ 1.70″ W) (Fig. 1) in two sampling campaigns performed in summer and winter seasons, respectively, in July 2010 and January 2011. GPS coordinates were used to ensure that the sediment cores collected at both season periods were approximately in the same area (±3 m), therefore minimizing spatial variations within each site.

Sampling sites in Tagus Estuary: Cala Norte (CNOR) and Barreiro (BRR). Sampling was performed in two different periods: in summer (July 2010) and in winter (January 2011)
2.2 Sampling and Sample Processing
Sediment cores were collected for porewater extraction and analysis of both the solid and the dissolved phase. Sediment samples were collected using a 7-cm-diameter PVC core. Sediment cores were kept at 4 °C until arrival to the laboratory and, after overlying water was collected with a syringe, were extruded and sectioned into slices in a N2-filled glove box to preserve the original redox conditions. A total of 37 samples were collected in CNOR (summer core, 40-cm long, 19 sections; winter core, 35-cm long, 18 sections) and of 33 samples in BRR (summer core, 30-cm long, 17 sections; winter core, 25-cm long, 16 sections). Cores were sliced into 1-cm sections for the top 10 cm and at 2-cm sections between 10 and 20 cm. Deeper sediments beyond 25 cm were sliced in 5-cm sections until the end of the core. Porewaters were extracted from the bulk sediments by centrifugation at 8000 rpm at 10,160 rcf×g, for 45 min at +4 °C and immediately filtered inside a glovebox using 0.45-μm cellulose acetate syringe filters (Whatman). Filtrate subsamples were stored in glass flasks, Teflon flasks, and polypropylene tubes and acidified to pH <2, respectively, for reactive dissolved mercury (RHgD) analysis (HNO3, Merck, Hg free), total dissolved mercury (THgD) and methylmercury (MMHgD) analysis (HCl, Merck, Hg free), and total dissolved metal, sulfate and sulfide, determinations (bi-distilled HNO3). The remaining solid phase samples were stored at −20 °C for solid phase analysis. For each layer, pH and redox potential (E H (mV)) were also measured in situ. The pH was measured using a portable Metrohm 704 pH meter with temperature control and glass combined electrode, calibrated with two buffer solutions (4.00 ± 0.02 and 7.00 ± 0.02 at 25 °C, Crison Instruments SA). Redox potential (E H) was also measured with a combined electrode Ag/AgCl-platinum, calibrated with a buffer for 220 ± 0.05 mV vs. Ag/AgCl at 25 °C (Mettler Toledo). Values were corrected to the hydrogen reference electrode. Values of pH and EH were presented in Tables SI1 and SI2 (Supporting Information).
2.3 Chemical Analysis
2.3.1 Solid Phase
Sediment porosity (Φ) was calculated by measuring the weight loss of sediments dried overnight at 110 °C (Loring and Rantala 1992) to constant weight using the fallowing equation (Covelli et al. 1999): \( \varPhi =\left[\frac{\left(\raisebox{1ex}{${M}_w$}\!\left/ \!\raisebox{-1ex}{${\rho}_w$}\right.\right)}{\left(\raisebox{1ex}{${M}_w$}\!\left/ \!\raisebox{-1ex}{${\rho}_w$}\right.\right)+\left(\raisebox{1ex}{${M}_w$}\!\left/ \!\raisebox{-1ex}{${\rho}_w$}\right.\right)}\right] \), where M w is the weight of water lost on drying, M s is the weight of dry sediment, and ρ w = 1.025 g cm−3 is the water density, whereas ρ s = 2.65 g cm−3 is the sediment density. Water content and porosity values were presented in Tables SI1 and SI2. Sediments were oven dried at 40 °C, disaggregated, and homogenized with a mortar. Total determinations of Si, Al, Fe, and Mn were performed by mineralization of sediment samples with a mixture of acids (HF, HNO3, HCl) (Rantala and Loring 1975), and concentrations were obtained by flame-atomic absorption spectrometry (F-AAS) in a PerkinElmer AAnalyst 100, using direct aspiration into N2O–acetylene flame (Si and Al) or air–acetylene flame (Fe and Mn). Total carbon (TC) and total nitrogen (TN) were determined using a CHN Fisons NA 1500 Analyzer, calibrated with sulfanilamide standards. Organic carbon (OC) was estimated by the difference between total carbon and inorganic carbon. Total carbon was determined directly from dry sediments and the inorganic was measured after heating samples at 450 °C for 2 h, in order to remove the organic fraction (Verardo et al. 1990).
Total mercury (THgs) concentrations were determined in dried sediments by atomic absorption spectrometry (AAS) using a silicon UV diode detector LECO AMA-254 (Costley et al. 2000). Results obtained were in good agreement with the certified values from the Certified Reference Materials (CRMs) used (MESS-3, PACS-1, and PACS-2, marine sediments from National Research Council Canada (NRCC)) (Table SI3) and the precision, as relative standard deviation (%RSD, n = 5), was better than 5%. The limit of detection (LOD) was based on three standard deviations (3σ) of the blank and was 0.02 μg g−1. Methylmercury concentration in sediments (MMHgs) was determined by alkaline digestion (KOH/MeOH), organic extraction with dichloromethane (DCM) preconcentration in aqueous sulfide solution, back-extraction into DCM, and quantification by atomic fluorescence spectrometry coupled with gaseous chromatography (GC-AFS) using an Agilent Chromatograph and a Millennium Merlin PSA detector (Canário et al. 2004). To control the level of accuracy, two CRMs (BCR 580 and IAEA 405, both estuarine sediments from Institute for Reference Material and Measurements (IRMM) and International Atomic Energy Agency (IAEA), respectively) were also analyzed in each batch of samples (Table SI3). The precision in CRMs (% RSD, n = 3) was better than 5.0% and the LOD (3σ of the blanks) was 0.1 ng g−1.
2.3.2 Dissolved Phase
In overlying waters and porewaters, sulfate (SO4 2−) concentrations were determined by turbidimetry method using a Hitachi U-2000 spectrophotometer with a light path of 5-cm length (APHA 1995). The dissolved sulfide concentrations (HS−) were determined by differential pulse polarography (DPP) using a Metrohm apparatus equipped with a 693 VA Processor and a 694 VA Stand (O’Driscoll et al. 2011). Dissolved total Fe and Mn were determined by F-AAS, using air–acetylene flame, in a PerkinElmer AAnalyst 100. Dissolved organic carbon (DOC) analysis was performed by high temperature catalytic oxidation (HTCO) using a commercial Shimadzu TOC-5000A analyzer (Benner and Strom 1993).
THgD was analyzed by cold vapor-atomic fluorescence spectroscopy (CV-AFS) applying the EPA method 1631B (USEPA 2002), using a PSA Merlin mercury system. The CRM for THgD was BCR 579 (coastal seawater from Institute for Reference Materials and Measurements (IRMM)), and the obtained concentrations (2.17 ± 0.3 ng L−1 Hg) were in agreement with the certified ones (1.9 ± 0.5 ng L−1 Hg). RHgD was also determined by CV-AFS on a PSA Merlin mercury system (Beckvar et al. 1996). Recoveries of Hg ranged from 90 to 109% and the LOD, as 3σ of the blanks, was 0.7 ng L−1. MMHgD was determined following the EPA method 1630 (USEPA 2001), by CV-AFS in a Brooks Rand Model III system. Concentrations were determined applying the standard addition method using a fresh MMHg-Cl solution (prepared daily from CH3HgCl, Aldrich) in ethanol (Canário et al. 2006, 2008b). Recoveries of MMHg ranged from 92 to 103% (MMHg-Cl spikes). Replicate samples were also used to assess variability of the data. Blanks were repeated every 20 samples in order to evaluate cross-contaminations and to ensure that equipment is operating in the same conditions. The LOD (3σ of the blanks) was 0.02 ng L−1, and precision (% RSD, n = 8) was better than 6.0%. Detailed information can be found in Supporting Information.
2.4 Estimation of Sediment-Water Fluxes of Hg Species
The diffusive fluxes across the sediment-water interface (SWI) were estimated according to Cesário et al. (2016) using Fick’s first law of diffusion:
J = −(ϕDw/θ 2)(∂C/∂z), where J is the diffusion flux (ng m−2 day−1) of solute with concentration C (ng dm−3) at depth z (cm); ϕ and θ, respectively, are the porosity and tortuosity; Dw is the ionic/molecular diffusion coefficient in water; and (∂C/∂z) is the concentration gradient (C ow − C pw). For all cores, we used z = 1 cm (i.e., the average depth of the uppermost porewater sample). Tortuosity was estimated from porosity using Boudreau’s formulation: θ 2 = 1 − ln(ϕ 2) (Boudreau 1996).
According to Cesário et al. (2016 and references therein), we used Dw, 25 °C = 1.2 × 10−5 cm2 s−1 for MMHgD and 2 × 10−6 cm2 s−1 for HgD (assuming that inorganic Hg is bound to macromolecules in the colloidal size range), corrected for temperature (T, °C) and expressed as follows: Dw, 25 °C = Dt (1 + 0.048 × (25 − t)) (Gill et al. 1999). Detailed information can be found in Supporting Information.
The water temperatures used for the fluxes calculations were the ones measured in the field during the campaigns, being those of 24 °C in summer period (July 2010) and 12 °C in winter period (January 2011).
2.5 Statistical Analysis
Statistical tests were performed using the computer software Statistica 7.0. The normality of all data was assessed by a Shapiro-Wilks test. Since some variables did not present a normal distribution, concentrations of several parameters in solid and dissolved fractions were compared using the non-parametric Wilcoxon paired-sample (T) test. Kruskal–Wallis ANOVA by ranks (K–W) were used to compare partition coefficients for THg and MMHg between sediment cores from CNOR and BRR sites in both sampling periods. Correlations were determined with the Spearman correlation coefficient (r) and were considered significant at p < 0.05.
3 Results and Discussion
3.1 Sediment Characteristics
Concentrations of Si normalized to Al content (Si/Al) were used as a proxy of the terrigenous fraction (sand or coarser silt) in sediments (Loring 1991). An increase of Si/Al ratio is generally associated with an increase of coarser grained quartz, feldspar, and other components containing little or even no trace metals (Singh et al. 1999). Sediments from CNOR appear to be mostly constituted by a mixture of sand and mud particles with Si/Al ratios ranging from 2 to 4 (Fig. 2a). On the other hand, sediments from BRR varied in a wider range (Si/Al = 2–32) showing an irregular distribution with depth (Fig. 2b) with topmost sediments and bottom layers constituted by coarser particles with higher Si/Al ratios. These results were in line with those reported by Franz et al. (2014), who observed that the estuary channels are composed mostly of sand and the intertidal zones are composed mainly of cohesive sediments, which consist of inorganic (clays, silts, and very fine sands) and organic particles (e.g., detritus) (Hayter and Mehta 1986). Also Caçador and Duarte (2012) reported that silt and clay were the main constituents of the estuary salt marshes, with sand accounting for less than 5%.

Si/Al ratios (a, b), organic carbon content (OC, %) (c, d), and total nitrogen (N tot , %) (e, f) concentrations in sediments from Cala Norte (CNOR) and Barreiro (BRR) collected in summer (s) and winter periods (w)
Sediments from CNOR are also characterized by relatively similar OC content (Fig. 2c) (1.3–2.3%). In BRR, OC vertical distributions were irregular (Fig. 2d) and between the two sampling periods suggesting a large heterogeneity. Total N (Ntot) values in CNOR and BRR cores presented similar distribution to OC, slightly decreasing in depth in both campaigns (Fig. 2e, f). In BRR cores, OC and Ntot showed similar patterns, with slightly lower values. In both locations and periods, the Ntot concentrations paralleled the OC content behavior as evidenced by the strong positive correlation (r = 0.85, p < 0.05) between them (Fig. SI1—Supporting Information). However, CNOR sediments in general presented higher contents of organic matter (OC = 1.9 ± 0.2%, Ntot = 0.11 ± 0.03% in summer; OC = 1.8 ± 0.2%; Ntot = 0.10 ± 0.01% in winter) compared to BRR sediments (OC = 1.3 ± 1.2%, Ntot = 0.07 ± 0.05% in summer; OC = 0.9 ± 0.4%; Ntot = 0.05 ± 0.03% in winter). In fact, OC (%) and Ntot (%) contents were statistically different (p < 0.05) between sites. This may be due to higher levels of riverine inputs from Tagus and Sorraia rivers and by the presence of the industrial activity in the margins of CNOR. The broad peak of OC observed in the intermediate sediment layers of BRR in summer season suggested the existence of additional diffuse OC sources in this period, since this peak was not observed in BRR winter sediments. The observed differences in OC content in depth are probably due to a capping with sand layers, which were applied in the past 10 years to prevent the resuspension of the extremely contaminated sediments (personal communication). The C/N (OC/Ntot) molar ratio is used to differentiate between terrestrial and aquatic inputs (Goñi et al. 2003; Tolun et al. 2002) and the values obtained in this study, above 12 at both sites and sampling periods, pointing to a terrestrial source of organic matter. Similar results were obtained by Goñi et al. (2003) in estuarine sediments from Winyah Bay, SC, USA.
3.2 Total Hg and MMHg in Sediments
Sediments from CNOR presented similar vertical distribution of THgs concentrations in both sampling periods. Lower THgs concentrations were observed at the topmost layers, increasing with depth (Fig. 3a). In CNOR, the THgs levels observed in both periods were in the same order of magnitude (summer = 0.99–18 μg THgs g−1; winter = 6.9–29 μg THgs g−1), with similar average values (summer = 12 ± 5.1 μg THgs g−1, n = 17; winter = 15 ± 8.1 μg THgs g−1, n = 16). BRR sediments also presented the same THgs patterns between periods although in different ranges (THgs = 0.30–126 μg g−1 in summer and 1.4–41 μg g−1 in winter) (Fig. 3b) and with higher depth variations due to the broad peaks observed between 10- and 20 cm depth. These variations in THgs concentrations with depth may be related to particle heterogeneity in those cores with more or less Hg (ad)sorbing capacity (Ravichandran 2004). In fact, the THgs maximums were found at depths with fine grained sediments, while coarser material at upper and bottom layers retained less THgs. Negative correlations obtained between THgs and Si/Al (summer: above 8 cm depth: r = 0.78, p < 0.05; below 9 cm depth: r = 0.94, p < 0.05; winter: above 5 cm depth: r = 0.74, p < 0.05; below 6 cm depth: r = 0.73, p < 0.05, Fig. SI2) in BRR sediments supports the above hypothesis. In the first 5-cm of BRR sediments, higher Si/Al ratios were determined, probably due to a capping with sand layers, reported before.

Total mercury (THg s , μg g−1) (a, b), methylmercury (MMHg S , ng g−1) (c, d) concentrations, and methylmercury proportions of the total mercury (MMHg s /THg s ) (e, f) in sediments from Cala Norte (CNOR) and Barreiro (BRR) collected in summer (s) and winter (w) periods
The relationship between THgs and OC content may also play an important role in metal retention because of the capacity of organic matter, particularly some functional groups of organic carbon, to efficiently bind Hg (Ravichandran 2004). In our samples, this was only observed in the CNOR sediments collected in winter period (r = 0.75, p < 0.05, Fig. SI3a) and in BRR sediments from summer period (r = 0.86, p < 0.05, Fig. SI3b). In the other two campaigns (CNOR in summer and BRR in winter periods), no significant correlations (p > 0.05) were observed suggesting that other processes such as sorption or coprecipitation with Fe and Mn oxyhydroxides may also be relevant. Depth variation of Fes and Mns in sediments (Fig. SI4) in our sediments suggested that in BRR summer sediments, Hg possibly coprecipitate with Fes and Mns oxyhydroxides. This can be corroborated by the strong positive correlations between both metals and THgs (r = 0.91, p < 0.05 for THgs vs Fes, Fig. SI5a and r = 0.92, p < 0.05 for THgs vs Mns, Fig. SI6a). The same pattern was observed in BRR and CNOR winter sediments but only between THgs and Fes (r = 0.84, p < 0.05 for THgs vs Fes, in BRR, Fig. SI5b; r = 83, p < 0.05 for THgs vs Fes, in CNOR, Fig. SI6b) where a possible Hg coprecipitation only with oxyhydroxides of Fes may have occurred.
The observed differences at CNOR and BRR between periods were considered a consequence of sediment heterogeneity. The THgs concentrations observed in this study are extremely high, being 1 to 2 orders (in CNOR) and almost 3 orders (in BRR) of magnitude higher than the Mediterranean background values (0.04–0.17 μg g−1) (Ogrinc et al. 2007), the values in Guadiana estuary, Portugal (0.16–1.3 μg g−1) (Canário et al. 2007b) or in Vigo Ria, Spain (0.02–1.8 μg g−1) (Canário et al. 2007c) but comparable with the ones (4.06 and 79.0 μg g−1) reported for sediments of other coastal areas affected by relevant industrial or mining activities (Acquavita et al. 2012; Bratkič et al. 2013; Covelli et al. 2011; Emili et al. 2016; Oliveri et al. 2016; Wang et al. 2009). Comparing these results with the previous work done by the same authors in the RAMSAR areas of Tagus estuary (Cesário et al. 2016), these estuarine areas can be considered as highly Hg contaminated (Canário et al. 2005, 2007a, b).
MMHgs present a similar vertical distribution in both campaigns for each site. The higher MMHgs concentrations were observed in warmer periods, which is in line with previous studies in the Tagus estuary (Canário et al. 2007a; Cesário et al. 2016) and also in other international estuarine environments (Bratkič et al. 2013; Hammerschmidt and Fitzgerald 2008; Hines et al. 2012; Hollweg et al. 2009; Mitchell and Gilmour 2008). In CNOR sediments, MMHgs concentrations ranged between 1.0 and 87 ng g−1 in summer and from 1.3 to 25 ng g−1 in winter, with maximum values observed between 5 and 10 cm depth (Fig. 3c). The same pattern was observed in BRR sediments, although in deeper sediment layers. In the summer period, MMHgs concentrations ranged from 0.76 to 201 ng g−1, with a broad peak observed between 7 and 13 cm depth, decreasing in winter period to values ranging from 1.2 to 61 ng g−1 (Fig. 3d). The concentrations of MMHg obtained in this study were 10 to 25 times higher than values reported in other contaminated areas such as in Deûle and Lys Rivers, Northern France (1.6–7.6 ng g−1) (Daye et al. 2015), in Soca/Isonzo River, Northern Adriatic Sea (0.2–6.3 ng g−1) (Bratkič et al. 2013), and in Marano and Grado Lagoon, Italy (1.03–2.39 ng g−1) (Hines et al. 2012). The results obtained from CNOR in summer and BRR in both campaigns showed positive correlations between THgs and MMHgs concentrations in sediments [r = 0.95, p < 0.05 in CNOR (above 10 cm depth, Fig. SI7); r = 0.88, p < 0.05 in BRR (above 13 cm depth, Fig. SI8a) in summer period and r = 0.94, p < 0.05 in BRR (all depths, Fig. SI8b) in winter period], suggesting co-deposition of both Hg forms and/or MMHgS production as result of higher THgS concentration. This last observation was also reported in estuarine sediments of the Patuxent River (USA) (Benoit et al. 1998) and in deeper sediments from polluted sediments of the Deûle River (France) (Kadlecová et al. 2012). However, in the deeper sediments from CNOR and BRR cores collected in summer, no correlations were found between THgs and MMHgs, which suggest that MMHgs production/retention is not Hg bioavailability dependent. As an example, BRR sediments collected in summer showed a positive correlation between MMHgs concentrations and OC content (r = 0.85, p < 0.05, Fig. SI9), indicating that in addition to Hg bioavailability, organic matter also plays an important role in the sedimentary Hg methylation process (Ullrich et al. 2001). However, in CNOR sediments collected in winter period, in situ MMHg production seems to be mediated by other factors, such as (i) the presence of organic and/or inorganic complexing agents (Mikac et al. 1999; Ullrich et al. 2001); (ii) sorption/desorption MMHg mechanisms (Canário et al. 2008a); and (iii) availability of labile organic substrates, a fraction of the total OC (Ravichandran 2004), rather than overall OC content, or hypothetically, MMHg might be exported from others sources rather than in situ produced, probably by influencing of Tagus River discharge during a rainy period in winter season. The observed seasonal variations in MMHgs concentrations suggest that methylation processes were more effective in summer period, when sediments were up to 12 °C warmer than in winter period. This is in agreement with other studies that showed a maximum methylation activity in mid or late summer (Hintelmann and Wilken 1995), presumably due to the increase of temperature that directly induces microbial activity (e.g., King et al. 1999). In addition, the increment of the primary production and biological activity during the warmer periods (Gameiro et al. 2011) seems to stimulate the methylation process, providing to methylating bacteria nutrients and a greater abundance of relatively Hg-rich organic matter at the sediment-water interface (Hintelmann and Wilken 1995). At both sites, the MMHgs concentrations observed in this study accounted for higher proportions to THgs in the warmer month, with values above 1% in CNOR and 1.5% in BRR (Fig. 3e, f). In colder months, these values decrease to below 0.29 and 0.37% MMHgs in CNOR and BRR sediments, respectively. The proportion of MMHgs at the CNOR and BRR sites from Tagus estuary is relatively low but is comparable to other highly polluted estuaries impacted by historical discharges of ore minerals (Table 1).
3.3 Porewater Mercury Distribution and Its Controls
Concentrations of THgD, RHgD, and MMHgD in porewaters varied between sites and sampling campaigns. In BRR site, concentrations of THgD ranged between 71 and 365 ng L−1 (maximum at 3 cm depth) in summer (Fig. 4a) and from 11 to 140 ng L−1 in winter (Fig. 4b). In the CNOR site, THgD concentrations were lower ranging from 6.8 to 166 ng L−1 (maximum at 13 cm depth) in summer (Fig. 5a) and between 5.3 and 96 ng L−1 in winter (Fig. 5b). A different pattern was observed between seasons; once in winter, the higher concentrations were observed in deeper sediments at both sites (140 ng L−1 at 10 cm in BRR (Fig. 4b) and 96 ng L−1 at 17 cm in CNOR) (Fig. 5b). Total HgD concentrations found in CNOR and BRR sediments were 10 to 20 times higher than others found in sediments affected by point source pollution in Deûle and Lys Rivers, Northern France (16 ± 3.2 and 11 ± 3.5 ng L−1) (Daye et al. 2015) but were in the same order of magnitude than those found in Soca/Isonzo River, Northern Adriatic Sea (2–220 ng L−1) (Bratkič et al. 2013) and in sediment cores from Augusta Bay, Southern Italy (8.4–226 ng L−1) (Oliveri et al. 2016). Vertical profiles of RHgD were similar to those from THgD in both periods for BRR (Fig. 4c, d) and CNOR (Fig. 5c, d). This similarity suggests that these two Hg fractions were in equilibrium with each other (correlation between both species in Figs. SI10 and SI11). The higher values of RHgD were determined in BRR porewaters in warmer periods, as it was observed in THgD levels. Accordingly, higher concentrations were determined in summer, varying between 1.2 and 28 ng L−1 in CNOR and between 4.9 and 98 ng L−1 in BRR. Lower RHgD levels in winter were observed in CNOR (0.2–10 ng L−1), when compared with BRR (0.1–26 ng L−1).

Total dissolved mercury (THg D ) (a, b), reactive dissolved mercury (RHg D ) (c, d), and dissolved methylmercury (MMHgD) (e, f) concentrations (ng L−1) in porewaters from sediments collected in Barreiro (BRR) in summer (s) and winter (w) periods. The bare point corresponds to the overlying water and the full points correspond to porewaters

Total dissolved mercury (THg D ) (a, b), reactive dissolved mercury (RHg D ) (c, d), and dissolved methylmercury (MMHgD) (e, f) concentrations (ng L−1) in porewaters from sediments collected in Cala Norte (CNOR) in summer (s) and winter (w) periods. The bare point corresponds to the overlying water and the full points correspond to porewaters
MMHgD concentrations in porewaters varied between 0.8 and 63 ng L−1 in summer (Fig. 4e) and between 0.4 and 5.3 ng L−1 in winter (Fig. 4f) in BRR porewaters. However, MMHgD in CNOR porewaters were slightly lower ranging from 0.9 to 26 ng L−1 in summer (Fig. 5e) and from 0.1 to 4.1 ng L−1 in winter (Fig. 5f). The exhibited seasonal variation on MMHgD concentrations was also observed in a study conducted by Covelli et al. (2008) in Grado Lagoon, Italy, who reported higher porewater MMHg contents in the warmer period. MMHgD concentrations observed in the colder period were in the same order of magnitude of those reported in contaminated sediments from Soca/Isonzo River, Northern Adriatic Sea (0.12–8.55 ng L−1) (Bratkič et al. 2013) but were generally higher than those reported in porewaters of the San Francisco Bay Delta, USA (0–13 ng L−1; Choe et al. (2004)), the Marano Lagoon, Italy (0.1–6.25 ng L−1; Emili et al. (2012)), the Grado Lagoon, Italy (0.9–7.9 ng L−1; Covelli et al. (2008)), and the St. Lawrence River, Canada (<0.01–1.94 ng L−1; Goulet et al. (2007)).
MMHgD vertical profiles showed, in general, a subsurface maximum with the highest values in summer period. In this period, higher levels at CNOR were observed between 6.5 and 9.5 cm depth (26 ng L−1), while in BRR, the maximum of 63 ng L−1 was at 7.5 cm. In winter period, MMHgD values decreased at both sites. Although in BRR maximum MMHgD concentrations (5.3 ng L−1) were observed more near the surface (4.5 cm), in CNOR, the higher MMHgD concentrations were observed deeper (17 cm) and were six times lower (4.1 ng L−1) compared to the summer maximum. In addition to the positive correlation (r = 0.88, p < 0.05) observed between THgD and RHgD in porewaters from CNOR in colder period (Fig. SI10b), also positive correlations between these dissolved Hg species and MMHgD (r = 0.84, p < 0.05 for THgD and MMHgD; r = 0.86, p < 0.05 for RHgD and MMHgD, Fig. SI12) were observed suggesting equilibriums between the three dissolved Hg species. Therefore, MMHgD production was presumably controlled by the bioavailability of the other dissolved Hg species. A different pattern was observed in the CNOR sediments collected in summer, which showed a broad peak between 7 and 10 cm depth, with MMHgD maximum concentration (26 ng L−1) at 9 cm depth (Fig. 5e). These results suggest that RHgD was mobilized in the sedimentary column between those two depths and is now possibly available for methylation. Either RHgD was mobilized in the sedimentary column by downward diffusion or it was consumed during methylation thereby decreasing its concentrations and consequently increasing MMHgD concentrations. This pattern was also observed in BRR sediments from winter period. Although in BRR summer sediments the methyl peak (7.5 cm depth) was observed below the RHgD peak (2.5 cm depth), suggesting that below this depth, all RHgD was consumed during methylation. The obtained results indicated that distribution of Hg species in porewater is not solely governed by exchange equilibrium between solid and dissolved phases. The decoupling between THgD and MMHgD profiles of summer sediments from CNOR and BRR suggested that Hg availability was not the main driver for methylation in these sediments. Moreover, several other processes may affect MMHgD concentrations in porewaters like in situ production by methylating bacteria (Fleming et al. 2006; Kerin et al. 2006), dissolution of authigenic Fe and Mn oxyhydroxides (Bloom et al. 1999; Gagnon et al. 1997), and quantity and quality of DOM in anoxic sediments (Lambertsson and Nilssons 2006; Randall and Chattopadhyay 2013). Despite the removal of Hg from the dissolved fraction can be attributed to the formation of Fe/Mn oxyhydroxides that sorb Hg onto solids (Bloom et al. 1999; Gagnon et al. 1996), the lack of correlation between Fe/Mn and THg concentrations in porewaters (Table SI4) at both sites and periods suggested that the diagenetic reactions involving Hg and Fe/Mn oxyhydroxides were not a key factor in the dynamic of Hg in these sediments. It is also important to evaluate the influence of sulfur chemistry in the Hg and MMHg dynamics in sediments. Measurements of porewater sulfide (HS−) and sulfate (SO4 2−) concentrations were limited to the warmer period. Overall, SO4 2− concentrations were higher in top layers in both sediment cores and inversely correlated with lower HS− levels in the corresponding layers. The anoxic layer of these sediments [below 6 cm depth in CNOR (Fig. 6a) and below 3 cm depth in BRR (Fig. 6b)] indicated that sulfate was, in fact, reduced to HS−. Concentrations of MMHgD increased with the HS− levels in porewaters of CNOR, while RHgD concentrations decreased with the increments in HS−. This points to the apparent activity of SRBs on the Hg cycle, suggesting that these bacteria are methylating inorganic Hg. If SO4 2− and RHgD are necessary for SRBs to methylate Hg, HS− may contribute to diminish MMHgD concentrations due to HgS precipitation (Du Laing et al. 2009; Ullrich et al. 2001) and thereby reducing the available RHgD and/or possibly producing dimethylmercury (Gagnon et al. 1996; King et al. 2000). In fact, MMHgD concentrations in CNOR sediments generally parallel HS− concentrations and a significant positive correlation (r = 0.89, p < 0.05, Fig. SI13) was observed, indicating that Hg available for methylation is not limited by HgS precipitation (Ullrich et al. 2001). A different pattern was observed in BRR sediments, where HS− and MMHgD concentrations were not correlated (p > 0.05), suggesting that the formation of HgS by precipitation may be a limiting factor in Hg methylation. Moreover, above 5 cm depth, a small HS− peak was observed with a parallel increase in RHgD and decrease of SO4 2− concentrations, pointing to the reduction of SO4 2− to HS− probably due to the activity of SRBs. Otherwise, below 10 cm depth, HS− seems to contribute to the decrease in MMHgD concentrations either by controlling HgS precipitation or reducing the available RHgD.

Concentrations of sulfate (SO 4 2−, mM) and sulfide (HS −, μM) in porewaters from sediments collected in Cala Norte (CNOR) (a) and Barreiro (BRR) (b) in summer period
The MMHgD concentrations accounted from 1.0 to 43% of the THgD in CNOR and from 0.22 to 86% in BRR porewaters in warmer period (Fig. 7a, b). However, these proportions were lower in winter, decreasing to values between 2.1 and 20% in CNOR and from 0.5 to 15% in BRR (Fig. 7c, d). The %MMHgD values determined in this study were higher than those found in contaminated sediments from Soca/Isonzo River estuary (Northern Adriatic Sea), (MMHgD <25%) (Bratkič et al. 2013) but in the same range to those reported in Truckee River (67 ± 26%) (Pizarro-Barraza et al. 2014) and in Carson River (20–80%) (Bonzongo et al. 1996), both located in the USA.

Dissolved methylmercury (MMHg D ) proportions of the total dissolved mercury (THg D ) (MMHgD/THgD, %) in porewaters from sediments collected in summer (s) (a, b) and in winter (w) (c, d) periods in Cala Norte (CNOR) and in Barreiro (BRR)
The decrease of MMHg production in winter may be controlled not only by a decrease of microbial activity in colder months but also by the Hg availability and the concentration of DOC in the porewaters. Likewise, DOC can promote MMHg production by stimulating microbial activity and thus increasing methylation rates (Graham et al. 2012). In fact, our data suggests that Hg availability and porewater DOC are also important drivers for biomethylation in porewaters from CNOR in the colder month. The strong positive correlations observed between MMHgD and HgD species (r = 0.84, p < 0.05 for MMHgD vs THgD, Fig. SI12a, and r = 0.86, p < 0.05 for MMHgD vs RHgD, Fig. SI12b) suggest that inorganic Hg dissolved species may be a limiting factor for MMHg production. However, the absence of positive correlations (p > 0.05) between DOC concentrations (Fig. SI14) and %MMHgD, in CNOR (in summer) and in BRR (in both periods), suggested that different relationships between %MMHgD and DOC occurred. Whereas in BRR sediments the increases of %MMHgD and DOC concentrations with temperature are coupled, in CNOR sediments, the opposite trend was observed with %MMHgD increasing with the decrease of DOC (Fig. SI15). Similar results were attributed to (i) different nature of DOC (Ravichandran 2004), (ii) to the character (e.g., aromaticity) of DOC, which induced strong binding sites to inorganic Hg and MMHg attenuating the partitioning of dissolved MMHg (Tsui and Finlay 2011), and also to (iii) molecular composition of DOC constituents (Mitchell and Gilmour 2008). The positive relationship between MMHg production and DOC composition was also observed by Hollweg et al. (2009), in sediments from Chesapeake Bay and mid-Atlantic continental margin. Accordingly, the nature and chemical composition of DOC and presumably their binding sites are driving factors that need to be considered in the methylation process. In fact, CNOR and BRR have different morphological characteristics, while salt-marsh areas, colonized by different halophyte species, surround CNOR. BRR site has no influence of vegetation, thus providing DOC with different characteristics. Moreover, in CNOR winter sediments, an increasing tendency of %MMHgD with DOC concentrations (r = 0.85, p < 0.05, Fig. SI16) was observed, which is in line with results published for San Francisco Bay, California (Conaway et al. 2003) and New Jersey/New York Harbor (Hammerschmidt et al. 2008), both located in the USA. As such, further research on the speciation/composition of DOC will be necessary to provide a better knowledge about the mechanisms involving these mercury species, when concerning Hg methylation/MMHg demethylation processes.
3.4 Mercury Partition Between Solids and Porewaters
The Hg and MMHg distribution between solid and dissolved phases can be expressed in terms of logK D value, with K D coefficient expressed as ([THgs]/[THgD]), for THg and as ([MMHgs]/[MMHgD]) for MMHg (Bratkič et al. 2013; Daye et al. 2015), both in liters per kilogram. In sediments, this distribution has considerable effect on the bioavailability of Hg for methylation, as well as the availability of MMHg to organisms at the base of the food web (Rothenberg et al. 2008). The strong positive correlation (r = 0.89, p < 0.05, Fig. SI17a) observed between MMHgs and MMHgD in sediments from CNOR core collected in summer evidenced that levels of MMHgs in the solids are reflected in high MMHgD values in porewaters. This trend was also observed between THgS and THgD (r = 0.97, p < 0.05, Fig. SI17b) in the first 10 cm depth of CNOR sediments from the colder period, indicating that total Hg in solid and dissolved fractions was in equilibrium. However, THgD concentrations in porewaters of CNOR from summer and of BRR from both periods are not significantly correlated to THgs, reflecting the absence of equilibrium’s between the dissolved and solid phases. In fact, in these cases, THgD and THgs distributions were mirror images from each other. The same pattern was also observed for MMHg between solid and dissolved fractions from BRR sediments in both periods. LogK D values for THg (Table 2) were slightly higher in winter for both sites (5.7 ± 0.3 in CNOR and 5.2 ± 0.5 in BRR) compared to summer values (5.2 ± 0.5 for CNOR and 4.9 ± 0.9 for BRR), without statistical differences (p > 0.05), reflecting lower Hg remobilization from solid sediments to porewaters in the colder season. Since the remobilization of Hg from the solid fraction is frequently associated to the microbial diagenetic reactions occurring in the sediment, with the consequent release of solute to porewaters (Canário et al. 2003; Schulz and Zabel 2000), it is expected that this remobilization should be lower when the microbial activity is also lower. Similarly, higher logK D (THg) values were reported in contaminated sediments from Augusta Bay, Southern Italy (4.8–5.8) (Oliveri et al. 2016), from Olt River, Romania (5.1–6.7) (Bravo et al. 2014), from Deûle and Lys Rivers, Northern France (4.1–6.0) (Daye et al. 2015), from Soca/Izonzo River, Northern Adriatic Sea (4.6–6.7) (Bratkič et al. 2013), and from Marano and Grado Lagoon, Italy (3.6–5.6) (Emili et al. 2012).
Seasonal average logK D values for MMHg (Table 2) present the same trend that was seen for THg, being also slightly higher in the colder period. However, the differences between sites were minimal, and, in fact, no statistical differences (p > 0.05) were observed in logKD (MMHg) values between sites and sampling periods (Fig. SI18). It has been pointed out that Hg availability in sediments is dependent on its release/scavenging by three major bulk phases: sulfur, iron, and manganese and organic matter (Daye et al. 2015). Considering the latter, our results pointed to the relevant influence of the organic carbon content (%OC) in the Hg binding, transport, and transformation processes in sediments from BRR collected in summer. Moreover, the relevant associations between logK D of THg and MMHg and %OC were observed only in sediment from BRR during the warmer period (r = 0.80, p < 0.05 for logK D (THg) and %OC, Fig. SI19a, and r = 0.71, p < 0.05 for logK D (MMHg) and %OC, Fig. SI19b). These results indicate that the OC content influences the partitioning of both species proportionally in this specific case. Similar correlations between logK D of Hg species (THg and MMHg) and sedimentary organic content were also reported in two independent sites in Deûle and Lys Rivers (Northern France) (Daye et al. 2015), in Lavaca Bay (Texas) (Gill et al. 1999), and in New Jersey/New York Harbor (USA) (Hammerschmidt et al. 2008). However, the same patterns were not observed in BRR sediments from winter and in CNOR sediments from both periods, as was shown earlier by the lack of correlation between THg, MMHg, and %OC contents, suggesting that other factors such as dissolution and precipitation of hydroxides and sulfides and/or quality of organic matter are also important in controlling sediment-water partitioning and furthermore influenced the Hg dynamics in sediments.
3.5 Estimated Sediment-Water Diffusive Fluxes of THg and MMHg
Sediments can be a potential source of Hg and MMHg to the water column through molecular diffusion or advection of dissolved Hg species (Abi-Ghanem et al. 2011; Bratkič et al. 2013; Daye et al. 2015; Rothenberg et al. 2008). Based on the THgD and MMHgD concentration gradients between the uppermost porewaters (0–1 cm) and overlying surface waters, we estimate the transport of Hg across the sediment-water interface due to diffusion. In general, during colder periods, sediments were always the principal source of THgD and MMHgD to the water column. In warmer period, the same pattern was observed for the THgD diffusive flux, but the opposite process—influx (from overlying water to porewater) of MMHgD—was observed in CNOR and BRR sediments (Table 3). This indicates that these sediments were always a source of THgD but only a source of MMHgD to the water column in colder periods. In summer, sediments acted as a sink of MMHgD at both sampling sites. During this period, the higher MMHgD fluxes were found for BRR sediments (−45 ng m−2 day−1) compared to CNOR (−10 ng m−2 day−1). This difference can be due to a more active exchange between sediment and overlying water, which tends to create higher concentration gradients during warmer temperatures. Due to the lower hydrodynamics in CNOR, the concentration gradients in the sediment-water interfaces are lower. Also, sediment resuspension in CNOR is less pronounced (low hydrodynamics), limiting the upward transport of Hg species by advection mechanisms. Since the Tagus River (upstream of both sites) is the main source of dissolved oxygen to estuary (Canário et al. 2008a) and considering that in warmer period the river flow is lower, anoxic conditions are frequently observed in the water column, which may contribute to high MMHg levels in bottom water and therefore inverted fluxes during the warmer months. Estimations of diffusive THgD fluxes from our sediments (Table 3) were in the same range than those reported in contaminated sediments from Soca/Isonzo River (Northern Adriatic Sea) (2.48–55.1 ng m−2 day−1) (Bratkič et al. 2013) but were lower than those reported by Gray et al. (2004), in sediments from Almaden mining district, Valdeazognes River, Spain (0.66–6236 ng m−2 day−1), and by Daye et al. (2015) in sediments from Deûle and Lys Rivers, France (1493 and 13–353 ng m−2 day−1, respectively). However, our values were higher than those reported in sediments from Lavaca Bay, Texas, USA (0.4–16 ng m−2 day−1) (Gill et al. 1999). Conversely, MMHgD fluxes estimated in this study were generally higher than those reported in Marano and Grado Lagoons, Italy (4.2 ng m−2 day−1 in summer and of 4.0 ng m−2 day−1 in winter seasons) (Emili et al. 2012) than those estimated in Lebanese Coast, Eastern Mediterranean (0.38–0.80 ng m−2 day−1) (Abi-Ghanem et al. 2011), also influenced by industrial activities. Considering the total area of each site (CNOR = 2.71 × 106 m2 and BRR = 1.02 × 106 m2), assuming that each period (summer and winter) is representative of half of the year and sediments are covered by tidal water 12 h a day (semidiurnal tidal cycle), we estimated the amount of HgD exchanged between sediment and the water column on an annual basis. A positive flux of approximately 11.5 and 3.15 g of HgD per year was estimated for CNOR and BRR sediments, respectively. The annual balance is positive indicating that sediments from both contaminated sites are sources of Hg to the water column of the Tagus estuary. Although the MMHgD diffusive fluxes presented opposite behavior between summer and winter periods, the efflux of the methyl form in CNOR and BRR only accounts for 8.2 and 10%, respectively, of the annual MMHgD flux meaning that MMHg depositions in surface sediments at both sites are up to 90% higher than exportation to the water column. Thus, sediments from both sites act mainly as sinks of MMHg with an average “deposition” of ca. 2330 mg year−1 for CNOR and 3740 mg year−1 for BRR. However, it must be noted that this tentative budget is probably very much underestimated once benthic fluxes are currently reported to be up two orders of magnitude higher than the estimated diffusive fluxes for both Hg and MMHg (Choe et al. 2004; Covelli et al. 1999, 2008; Gill et al. 1999; Point et al. 2007). This discrepancy could be due to inappropriate resolution of porewater concentration gradients near the sediment-water interface. Thus, the impact of these two contaminated areas in the estuarine ecosystem can be much higher than the predicted in this study. Moreover, we should also consider other transport processes like tidal advection (Cesário et al. 2017) or bioturbation/bioirrigation activity (Fanjul et al. 2011; Zagar et al. 2007) to evaluate the total Hg and MMHg budget.
4 Conclusions
Our study clearly confirms the continuous permanence of two Hg hotspots in the Tagus estuary with Hg and MMHg concentrations in sediments reaching 125 μg Hg g−1 and 200 ng MMHg g−1, reflecting the degree of contamination that the Tagus Estuary has been exposed to over the years. This study reports the impact of seasonal variation in the several processes involved in the Hg methylation and in the fluxes of Hg and MMHg between sediment-water interfaces. Higher proportions of MMHg in both fractions of sediments were found in summer period, thus increasing sediment toxicity, affecting the sediment/water MMHg (and Hg) fluxes and increasing the risk of contamination to the water column. The positive correlations between MMHg, THg, and OC contents and DOC concentrations in BRR and CNOR sediments corroborate that MMHg production is mainly mediated by Hg availability and organic carbon, although in different phases. The sulfur chemistry seems to influence Hg methylation in both sites, being this more pronounced in CNOR in warmer period. Levels of Hg and MMHg in CNOR and BRR sites were considered of major concern once his dynamics in sediments cause an exportation of ca. 11,500 and 3100 mg year−1 of Hg and a “deposition” of 2300 and 3700 mg year−1 of MMHg, respectively. However, these annual budgets of both Hg species were probably underestimated if we consider that benthic fluxes can be up to orders of magnitude higher than the estimated diffusive fluxes. Among these major findings, this work also provides updated data of Hg and MMHg in surface and depth sediments in Tagus estuary, indicating that not only the contamination still persist (even after the end of Hg inputs in the two industrial areas) but also that this contamination spreads along the estuary reaching protected areas, without anthropogenic influences of this metal (Cesário et al. 2016). However, understanding the major factors controlling methylation and demethylation is essential to control the concentrations and the accumulation of MMHg in sediments. Therefore, further studies are needed to better understand the Hg reactivity in highly contaminated sediments from Tagus estuary. From an ecological point of view and considering the complexity of factors that affect Hg methylation in estuarine environments, such as the Tagus estuary, it is necessary to keep track of Hg levels and the processes responsible for the spread of this contamination to other areas in the estuary.
References
Abi-Ghanem, C., Nakhlé, K., Khalaf, G., & Cossa, D. (2011). Mercury distribution and methylmercury mobility in the sediments of three sites on the Lebanese coast, eastern Mediterranean. Archives of Environmental Contamination and Toxicology, 60(3), 394–405. doi:10.1007/s00244-010-9555-9.
Acquavita, A., Covelli, S., Emili, A., Berto, D., Faganeli, J., Giani, M., et al. (2012). Mercury in the sediments of the Marano and Grado Lagoon (northern Adriatic Sea): sources, distribution and speciation. Estuarine, Coastal and Shelf Science, 113, 20–31. doi:10.1016/j.ecss.2012.02.012.
APHA. (1995). In A. E. Greenberg (Ed.), Standard methods for the examination of water and wastewater (19th ed.). Washington, DC: American Public Health Association, American Water Works Association, and Water Environment Federation.
Barkay, T., & Wagner-Döbler, I. (2005). Microbial transformations of mercury: potentials, challenges, and achievements in controlling mercury toxicity in the environment. Advances Applied Microbiology, 57, 1–52. doi:10.1016/S0065-2164(05)57001-1.
Beckvar, N., Field, J., Salazar, S., & Hoff, R. (1996). Contaminants in aquatic habitats at hazardous waste sites: mercury. Technical Memorandum NOS ORCA 100, (December), 80 pp.
Benner, R., & Strom, M. (1993). A critical evaluation of the analytical blank associated with DOC measurements by high-temperature catalytic oxidation. Marine Chemistry, 41(1–3), 153–160. doi:10.1016/0304-4203(93)90113-3.
Benoit, J. M., Gilmour, C. C., & Mason, R. P. (2001). Aspects of bioavailability of mercury for methylation in pure cultures of Desulfobulbus propionicus (1pr3). Applied and Environmental Microbiology, 67(1), 51–58. doi:10.1128/AEM.67.1.51-58.2001.
Benoit, J. M., Gilmour, C. C., Mason, R. P., Riedel, G. S., & Riedel, G. F. (1998). Behavior of mercury in the Patuxent River estuary. Biogeochemistry, 40(2–3), 249–265. doi:10.1023/a:1005905700864.
Bloom, N. S., Gill, G. A., Cappellino, S., Dobbs, C., McShea, L., Driscoll, C., et al. (1999). Speciation and cycling of mercury in Lavaca Bay, Texas, sediments. Environmental Science & Technology, 33(1), 7–13. doi:10.1021/es980379d.
Bonzongo, J. C. J., Heim, K. J., Chen, Y. A., Lyons, W. B., Warwick, J. J., Miller, G. C., & Lechler, P. J. (1996). Mercury pathways in the Carson River-Lahontan reservoir system, Nevada, USA. Environmental Toxicology and Chemistry, 15(5), 677–683. doi:10.1897/1551-5028(1996)015<0677:MPITCR>2.3.CO;2.
Boudreau, B. P. (1996). The diffusive tortuosity of fine-grained unlithified sediments. Geochimica et Cosmochimica Acta, 60(16), 3139–3142. doi:10.1016/0016-7037(96)00158-5.
Bratkič, A., Ogrinc, N., Kotnik, J., Faganeli, J., Žagar, D., Yano, S., et al. (2013). Mercury speciation driven by seasonal changes in a contaminated estuarine environment. Environmental Research, 125, 171–178. doi:10.1016/j.envres.2013.01.004.
Bravo, A. G., Cosio, C., Amouroux, D., Zopfi, J., Chevalley, P.-A., Spangenberg, J. E., et al. (2014). Extremely elevated methyl mercury levels in water, sediment and organisms in a Romanian reservoir affected by release of mercury from a chlor-alkali plant. Water Research, 49, 391–405. doi:10.1016/j.watres.2013.10.024.
Caçador, I., & Duarte, B. (2012). Tagus estuary salt marsh structure and dynamics: a historical perspective. Estuaries: Classification, Ecology and Human Impacts, (January), 41–56.
Canário, J., Antunes, P., Lavrado, J., & Vale, C. (2004). Simple method for monomethylmercury determination in estuarine sediments. TrAC - Trends in Analytical Chemistry, 23(10–11), 799–806. doi:10.1016/j.trac.2004.08.009.
Canário, J., Branco, V., & Vale, C. (2007a). Seasonal variation of monomethylmercury concentrations in surface sediments of the Tagus Estuary (Portugal). Environmental Pollution, 148(1), 380–383. doi:10.1016/j.envpol.2006.11.023.
Canário, J., Caetano, M., & Vale, C. (2006). Validation and application of an analytical method for monomethylmercury quantification in aquatic plant tissues. Analytica Chimica Acta, 580(2), 258–262. doi:10.1016/j.aca.2006.07.055.
Canário, J., Caetano, M., Vale, C., & Cesário, R. (2007b). Evidence for elevated production of methylmercury in salt marshes. Environmental Science and Technology, 41(21), 7376–7382. doi:10.1021/es071078j.
Canário, J., Vale, C., & Nogueira, M. (2008a). The pathway of mercury in contaminated waters determined by association with organic carbon (Tagus Estuary, Portugal). Applied Geochemistry, 23(3), 519–528. doi:10.1016/j.apgeochem.2007.12.019.
Canário, J., Poissant, L., O’Driscoll, N., Ridal, J., Delongchamp, T., Pilote, M., et al. (2008b). Mercury partitioning in surface sediments of the Upper St. Lawrence River (Canada): evidence of the importance of the sulphur chemistry. Water, Air, and Soil Pollution, 187(1–4), 219–231. doi:10.1007/s11270-007-9510-1.
Canario, J., Prego, R., Vale, C., & Branco, V. (2007c). Distribution of mercury and monomethylmercury in sediments of Vigo Ria, NW Iberian Peninsula. Water, Air, and Soil Pollution, 182(1–4), 21–29. doi:10.1007/s11270-006-9317-5.
Canário, J., Vale, C., & Caetano, M. (2005). Distribution of monomethylmercury and mercury in surface sediments of the Tagus Estuary (Portugal). Marine Pollution Bulletin, 50(10), 1142–1145. doi:10.1016/j.marpolbul.2005.06.052.
Canário, J., Vale, C., Caetano, M., & Madureira, M. J. (2003). Mercury in contaminated sediments and pore waters enriched in sulphate (Tagus Estuary, Portugal). Environmental Pollution, 126(3), 425–433. doi:10.1016/S0269-7491(03)00234-3.
Celo, V., Lean, D. R. S., & Scott, S. L. (2006). Abiotic methylation of mercury in the aquatic environment. Science of the Total Environment, 368(1), 126–137. doi:10.1016/j.scitotenv.2005.09.043.
Cesário, R., Monteiro, C. E., Nogueira, M., O’Driscoll, N. J., Caetano, M., Hintelmann, H., et al. (2016). Mercury and methylmercury dynamics in sediments on a protected area of Tagus Estuary (Portugal). Water, Air, & Soil Pollution, 227(12), 475. doi:10.1007/s11270-016-3179-2.
Cesário, R., Poissant, L., Pilote, M., O’Driscoll, N. J., Mota, A. M., & Canário, J. (2017). Dissolved gaseous mercury formation and mercury volatilization in intertidal sediments. Science of the Total Environment. doi:10.1016/j.scitotenv.2017.06.093.
Choe, K.-Y., Gill, G. A., Lehman, R. D., Han, S., Heim, W. A., & Coale, K. H. (2004). Sediment-water exchange of total mercury and monomethyl mercury in the San Francisco Bay Delta. Limnology and Oceanography, 49(5), 1512–1527. doi:10.4319/lo.2004.49.5.1512.
Conaway, C. H., Squire, S., Mason, R. P., & Flegal, A. R. (2003). Mercury speciation in the San Francisco Bay estuary. Marine Chemistry, 80(2–3), 199–225. doi:10.1016/S0304-4203(02)00135-4.
Costley, C. T., Mossop, K. F., Dean, J. R., Garden, L. M., Marshall, J., & Carroll, J. (2000). Determination of mercury in environmental and biological samples using pyrolysis atomic absorption spectrometry with gold amalgamation. Analytica Chimica Acta, 405(1–2), 179–183. doi:10.1016/S0003-2670(99)00742-4.
Covelli, S., Emili, A., Acquavita, A., Koron, N., & Faganeli, J. (2011). Benthic biogeochemical cycling of mercury in two contaminated northern Adriatic coastal lagoons. Continental Shelf Research, 31(16), 1777–1789. doi:10.1016/j.csr.2011.08.005.
Covelli, S., Faganeli, J., De Vittor, C., Predonzani, S., Acquavita, A., & Horvat, M. (2008). Benthic fluxes of mercury species in a lagoon environment (Grado Lagoon, Northern Adriatic Sea, Italy). Applied Geochemistry, 23(3), 529–546. doi:10.1016/j.apgeochem.2007.12.011.
Covelli, S., Faganeli, J., Horvat, M., & Brambati, A. (1999). Porewater distribution and benthic flux measurements of mercury and methylmercury in the Gulf of Trieste (Northern Adriatic Sea). Estuarine, Coastal and Shelf Science, 48(4), 415–428. doi:10.1006/ecss.1999.0466.
Daye, M., Kadlecova, M., & Ouddane, B. (2015). Biogeochemical factors affecting the distribution, speciation, and transport of Hg species in the Deule and Lys Rivers (Northern France). Environmental Science and Pollution Research, 22(4), 2708–2720. doi:10.1007/s11356-014-3528-x.
Domagalski, J. (2001). Mercury and methylmercury in water and sediment of the Sacramento River Basin, California. Applied Geochemistry, 16(15), 1677–1691. doi:10.1016/s0883-2927(01)00068-3.
Du Laing, G., Rinklebe, J., Vandecasteele, B., Meers, E., & Tack, F. M. G. (2009). Trace metal behaviour in estuarine and riverine floodplain soils and sediments: a review. Science of the Total Environment, 407(13), 3972–3985. doi:10.1016/j.scitotenv.2008.07.025.
Emili, A., Acquavita, A., Covelli, S., Spada, L., Di Leo, A., Giandomenico, S., & Cardellicchio, N. (2016). Mobility of heavy metals from polluted sediments of a semi-enclosed basin: in situ benthic chamber experiments in Taranto’s Mar Piccolo (Ionian Sea, Southern Italy). Environmental Science and Pollution Research, 23(13), 12582–12595. doi:10.1007/s11356-015-5281-1.
Emili, A., Acquavita, A., Koron, N., Covelli, S., Faganeli, J., Horvat, M., et al. (2012). Benthic flux measurements of Hg species in a northern Adriatic lagoon environment (Marano and Grado Lagoon, Italy). Estuarine, Coastal and Shelf Science, 113, 71–84. doi:10.1016/j.ecss.2012.05.018.
Fang, T., & Chen, R. (2010). Mercury contamination and accumulation in sediments of the East China Sea. Journal of Environmental Sciences, 22(8), 1164–1170. doi:10.1016/S1001-0742(09)60233-3.
Fanjul, E., Bazterrica, M. C., Escapa, M., Grela, M. A., & Iribarne, O. (2011). Impact of crab bioturbation on benthic flux and nitrogen dynamics of Southwest Atlantic intertidal marshes and mudflats. Estuarine, Coastal and Shelf Science, 92(4), 629–638. doi:10.1016/j.ecss.2011.03.002.
Ferreira, J. G. (1995). ECOWIN—an object-oriented ecological model for aquatic ecosystems. Ecological Modelling, 79, 21–34.
Figuères, G., Martin, J. M., Meybeck, M., & Seyler, P. (1985). A comparative study of mercury contamination in the Tagus estuary (Portugal) and major French estuaries (Gironde, Loire, Rhône). Estuarine, Coastal and Shelf Science, 20(2), 183–203. doi:10.1016/0272-7714(85)90037-X.
Fleming, E. J., Mack, E. E., Green, P. G., & Nelson, D. C. (2006). Mercury methylation from unexpected sources: molybdate-inhibited freshwater sediments and an iron-reducing bacterium. Applied and Environmental Microbiology, 72(1), 457–464. doi:10.1128/AEM.72.1.457-464.2006.
Franz, G., Pinto, L., Ascione, I., Mateus, M., Fernandes, R., Leitão, P., & Neves, R. (2014). Modelling of cohesive sediment dynamics in tidal estuarine systems: case study of Tagus estuary, Portugal. Estuarine, Coastal and Shelf Science, 151, 34–44. doi:10.1016/j.ecss.2014.09.017.
Gagnon, C., Pelletier, É., & Mucci, A. (1997). Behaviour of anthropogenic mercury in coastal marine sediments. Marine Chemistry, 59(1–2), 159–176. doi:10.1016/S0304-4203(97)00071-6.
Gagnon, C., Pelletier, E., Mucci, A., & Fitzgerald, W. F. (1996). Diagenetic behavior of methylmercury in organic-rich coastal sediments. Limnology and Oceanography, 41(3), 428–434. doi:10.2307/2838577.
Gameiro, C., Zwolinski, J., & Brotas, V. (2011). Light control on phytoplankton production in a shallow and turbid estuarine system. Hydrobiologia, 669(1), 249–263. doi:10.1007/s10750-011-0695-3.
Gill, G. A., Bloom, N. S., & Driscoll, C. T. (1999). Sediment–water fluxes of mercury in Lavaca Bay, Texas. Environmental Science & Technology, 33, 663–669.
Gilmour, C. C., Podar, M., Bullock, A. L., Graham, A. M., Brown, S. D., Somenahally, A. C., et al. (2013). Mercury methylation by novel microorganisms from new environments. Environmental Science and Technology, 47(20), 11810–11820. doi:10.1021/es403075t.
Goñi, M. A., Teixeira, M. J., & Perkeya, D. W. (2003). Sources and distribution of organic matter in a river-dominated estuary (Winyah Bay, SC, USA). Estuarine, Coastal and Shelf Science, 57(5–6), 1023–1048. doi:10.1016/S0272-7714(03)00008-8.
Goulet, R. R., Holmes, J., Page, B., Poissant, L., Siciliano, S. D., Lean, D. R. S., et al. (2007). Mercury transformations and fluxes in sediments of a riverine wetland. Geochimica et Cosmochimica Acta, 71(14), 3393–3406. doi:10.1016/j.gca.2007.04.032.
Graham, A. M., Aiken, G. R., & Gilmour, C. C. (2012). Dissolved organic matter enhances microbial mercury methylation under sulfidic conditions. Environmental Science & Technology, 46(5), 2715–2723. doi:10.1021/es203658f.
Gray, J. E., Hines, M. E., Higueras, P. L., Adatto, I., & Lasorsa, B. K. (2004). Mercury speciation and microbial transformations in mine wastes, stream sediments, and surface waters at the Almadén mining district, Spain. Environmental Science & Technology, 38(16), 4285–4292. doi:10.1021/es040359d.
Hamelin, S., Amyot, M., Barkay, T., Wang, Y., & Planas, D. (2011). Methanogens: principal methylators of mercury in lake periphyton. Environmental Science & Technology, 45(18), 7693–7700. doi:10.1021/es2010072.
Hammerschmidt, C. R., & Fitzgerald, W. F. (2004). Geochemical controls on the production and distribution of methylmercury in near-shore marine sediments. Environmental Science and Technology, 38(5), 1487–1495. doi:10.1021/es034528q.
Hammerschmidt, C. R., & Fitzgerald, W. F. (2008). Sediment–water exchange of methylmercury determined from shipboard benthic flux chambers. Marine Chemistry, 109(1–2), 86–97. doi:10.1016/j.marchem.2007.12.006.
Hammerschmidt, C. R., Fitzgerald, W. F., Balcom, P. H., & Visscher, P. T. (2008). Organic matter and sulfide inhibit methylmercury production in sediments of New York/New Jersey Harbor. Marine Chemistry, 109(1–2), 165–182. doi:10.1016/j.marchem.2008.01.007.
Hayter, E. J., & Mehta, A. J. (1986). Modelling cohesive sediment transport in estuarial waters. Applied Mathematical Modelling, 10(4), 294–303. doi:10.1016/0307-904X(86)90061-2.
Hidroprojecto (1991). Melhoria das condições de navegabilidade do Rio Tejo (Cala do Norte). Vol. I. Análise do estado de referência. Lisnoa. 191pp.
Hines, M. E., Poitras, E. N., Covelli, S., Faganeli, J., Emili, A., Žižek, S., & Horvat, M. (2012). Mercury methylation and demethylation in Hg-contaminated lagoon sediments (Marano and Grado Lagoon, Italy). Estuarine, Coastal and Shelf Science, 113, 85–95. doi:10.1016/j.ecss.2011.12.021.
Hintelmann, H., & Wilken, R. D. (1995). Levels of total mercury and methylmercury compounds in sediments of the polluted Elbe River: influence of seasonally and spatially varying environmental factors. Science of the Total Environment, 166(1–3), 1–10. doi:10.1016/0048-9697(95)04506-V.
Hollweg, T. A., Gilmour, C. C., & Mason, R. P. (2009). Methylmercury production in sediments of Chesapeake Bay and the mid-Atlantic continental margin. Marine Chemistry, 114(3–4), 86–101. doi:10.1016/j.marchem.2009.04.004.
Hsu-Kim, H., Kucharzyk, K. H., Zhang, T., & Deshusses, M. A. (2013). Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: a critical review. doi:10.1021/es304370g.
Kadlecová, M., Ouddane, B., & Dočekalová, H. (2012). Speciation of mercury in the strongly polluted sediments of the Deûle River (France). Journal of Environmental Monitoring, 14, 961. doi:10.1039/c2em10561f.
Karagas, M. R., Choi, A. L., Oken, E., Horvat, M., Schoeny, R., Kamai, E., et al. (2012). Evidence on the human health effects of low-level methylmercury exposure. Environmental Health Perspectives, 120(6), 799–806. doi:10.1289/ehp.1104494.
Kerin, E. J., Gilmour, C. C., Roden, E., Suzuki, M. T., Coates, J. D., & Mason, R. P. (2006). Mercury methylation by dissimilatory iron-reducing bacteria. Applied and Environmental Microbiology, 72(12), 7919–7921. doi:10.1128/AEM.01602-06.
King, J. K., Kostka, J. E., & Frischer, M. E. (2000). Sulfate-reducing bacteria methylate mercury at variable rates in pure culture and in marine sediments, 66(6), 2430–2437. doi:10.1128/AEM.66.6.2430-2437.2000.
King, J. K., Saunders, F. M., Lee, R. F., & Jahnke, R. A. (1999). Coupling mercury methylation rates to sulfate reduction rates in marine sediments. Environmental Toxicology and Chemistry, 18(7), 1362–1369. doi:10.1002/etc.5620180704.
Lambertsson, L., & Nilssons, M. (2006). Organic material: the primary control on mercury methylation and ambient methyl mercury concentrations in estuarine sediments. Environmental Science and Technology, 40(6), 1822–1829. doi:10.1021/es051785h.
Logar, M., Horvat, M., Akagi, H., & Pihlar, B. (2002). Simultaneous determination of inorganic mercury and methylmercury compounds in natural waters. Analytical and Bioanalytical Chemistry, 374(6), 1015–1021. doi:10.1007/s00216-002-1501-x.
Loring, D. H. (1991). Normalization of heavy-metal data from estuarine and coastal sediments. ICES Journal of Marine Science: Journal du Conseil, 48(1), 101–115. doi:10.1093/icesjms/48.1.101.
Loring, D. H., & Rantala, R. T. T. (1992). Manual for the geochemical analyses of marine sediments and suspended particulate matter. Earth-Science Reviews, 32(4), 235–283.
Mahaffey, K. R., Sunderland, E. M., Chan, H. M., Choi, A. L., Grandjean, P., Mariën, K., et al. (2011). Balancing the benefits of n-3 polyunsaturated fatty acids and the risks of methylmercury exposure from fish consumption. Nutrition Reviews, 69(9), 493–508. doi:10.1111/j.1753-4887.2011.00415.x.
Marvin-Dipasquale, M., Lutz, M. A., Brigham, M. E., Krabbenhoft, D. P., Aiken, G. R., Orem, W. H., & Hall, B. D. (2009). Mercury cycling in stream ecosystems. 2. Benthic methylmercury production and bed sediment-pore water partitioning. Environmental Science and Technology, 43(8), 2726–2732. doi:10.1021/es802698v.
Mergler, D., Anderson, H. A., Chan, L. H. M., Mahaffey, K. R., Murray, M., Sakamoto, M., & Stern, A. H. (2007). Methylmercury exposure and health effects in humans: a worldwide concern. Ambio: A Journal of the Human Environment, 36(1), 3–11.
Mikac, N., Niesse, S., & Ouddane, B. (1999). Speciation of mercury in sediments of the Seine Estuary (France). Applied Organometallic Chemistry, 13(April 2016), 715–725. doi:10.1002/(SICI)1099-0739(199910)13.
Mitchell, C. P. J., & Gilmour, C. C. (2008). Methylmercury production in a Chesapeake Bay salt marsh. Journal of Geophysical Research: Biogeosciences, 113(G2), 1–14. doi:10.1029/2008JG000765.
Nyland, J. F., Wang, S. B., Shirley, D. L., Santos, E. O., Ventura, A. M., de Souza, J. M., & Silbergeld, E. K. (2011). Fetal and maternal immune responses to methylmercury exposure: a cross-sectional study. Environmental Research, 111(4), 584–589. doi:10.1016/j.envres.2011.02.010.
O’Driscoll, N. J., Canário, J., Crowell, N., & Webster, T. (2011). Mercury speciation and distribution in coastal wetlands and tidal mudflats: relationships with sulphur speciation and organic carbon. Water, Air, and Soil Pollution, 220(1–4), 313–326. doi:10.1007/s11270-011-0756-2.
Ogrinc, N., Monperrus, M., Kotnik, J., Fajon, V., Vidimova, K., Amouroux, D., et al. (2007). Distribution of mercury and methylmercury in deep-sea surficial sediments of the Mediterranean Sea. Marine Chemistry, 107(1), 31–48. doi:10.1016/j.marchem.2007.01.019.
Oliveri, E., Salvagio Manta, D., Bonsignore, M., Cappello, S., Tranchida, G., Bagnato, E., et al. (2016). Mobility of mercury in contaminated marine sediments: biogeochemical pathways. Marine Chemistry, 186, 1–10. doi:10.1016/j.marchem.2016.07.002.
Pizarro-Barraza, C., Gustin, M. S., Peacock, M., & Miller, M. (2014). Evidence for sites of methylmercury formation in a flowing water system: impact of anthropogenic barriers and water management. Science of the Total Environment, 478, 58–69. doi:10.1016/j.scitotenv.2014.01.081.
Podar, M., Gilmour, C. C., Brandt, C. C., Soren, A., Brown, S. D., Crable, B. R., et al. (2015). Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Science Advances, 1(9), 1–13. doi:10.1126/sciadv.1500675.
Point, D., Monperrus, M., Tessier, E., Amouroux, D., Chauvaud, L., Thouzeau, G., et al. (2007). Biological control of trace metal and organometal benthic fluxes in a eutrophic lagoon (Thau Lagoon, Mediterranean Sea, France). Estuarine, Coastal and Shelf Science, 72(3), 457–471. doi:10.1016/j.ecss.2006.11.013.
Randall, P. M., & Chattopadhyay, S. (2013). Mercury contaminated sediment sites—an evaluation of remedial options. Environmental Research, 125, 131–149. doi:10.1016/j.envres.2013.01.007.
Rantala, R. T. T., & Loring, D. H. (1975). Multi-element analysis of silicate rocks and marine sediments by atomic absorption spectrophotometry. Atomic absorption Newsletter, 14, 117–120.
Ravichandran, M. (2004). Interactions between mercury and dissolved organic matter––a review. Chemosphere, 55(3), 319–331. doi:10.1016/j.chemosphere.2003.11.011.
Rimondi, V., Gray, J. E., Costagliola, P., Vaselli, O., & Lattanzi, P. (2012). Concentration, distribution, and translocation of mercury and methylmercury in mine-waste, sediment, soil, water, and fish collected near the Abbadia San Salvatore mercury mine, Monte Amiata district, Italy. Science of the Total Environment, 414, 318–327. doi:10.1016/j.scitotenv.2011.10.065.
Rothenberg, S. E., Ambrose, R. F., & Jay, J. A. (2008). Mercury cycling in surface water, pore water and sediments of Mugu Lagoon, CA, USA. Environmental Pollution, 154(1), 32–45. doi:10.1016/j.envpol.2007.12.013.
Schartup, A. T., Balcom, P. H., & Mason, R. P. (2014). Sediment-porewater partitioning, total sulfur, and methylmercury production in estuaries. Environmental Science and Technology, 48(2), 954–960. doi:10.1021/es403030d.
Schulz, H. D., & Zabel, M. (2000). In H. D. Schulz & M. Zabel (Eds.), Marine geochemistry (1st ed.). Heidelberg: Springer Berlin Heidelberg.
Singer, M. B., Harrison, L. R., Donovan, P. M., Blum, J. D., & Marvin-DiPasquale, M. (2016). Hydrologic indicators of hot spots and hot moments of mercury methylation potential along river corridors. Science of the Total Environment, 568, 697–711. doi:10.1016/j.scitotenv.2016.03.005.
Singh, A. K., Hasnain, S. I., & Banerjee, D. K. (1999). Grain size and geochemical partitioning of heavy metals in sediments of the Damodar River—a tributary of the lower Ganga, India. Environmental Geology, 39(1), 90–98. doi:10.1007/s002540050439.
Tolun, L., Çağatay, M. N., & Carrigan, W. J. (2002). Organic geochemistry and origin of Late Glacial–Holocene sapropelic layers and associated sediments in Marmara Sea. Marine Geology, 190(1–2), 47–60. doi:10.1016/S0025-3227(02)00342-0.
Tomiyasu, T., Matsuyama, A., Eguchi, T., Fuchigami, Y., Oki, K., Horvat, M., et al. (2006). Spatial variations of mercury in sediment of Minamata Bay, Japan. Science of the Total Environment, 368(1), 283–290. doi:10.1016/j.scitotenv.2005.09.090.
Tsui, M. T. K., & Finlay, J. C. (2011). Influence of dissolved organic carbon on methylmercury bioavailability across Minnesota stream ecosystems. Environmental Science & Technology, 45(14), 5981–5987. doi:10.1021/es200332f.
Ullrich, S. M., Tanton, T. W., & Abdrashitova, S. A. (2001). Mercury in the aquatic environment: a review of factors affecting methylation. Critical Reviews in Environmental Science and Technology, 31(3), 241–293. doi:10.1080/20016491089226.
US-EPA (2001). Method 1630: methyl mercury in water by distillation, aqueous ethylation, purge and trap, and cold vapor atomic fluorescence spectrometry (CV-AFS) United States, EPA-821-R-01-020, January, 1–49.
US-EPA (2002). Method 1631, revision E: mercury in water by oxidation, purge and trap, and cold vapor atomic fluorescence spectrometry (CV-AFS). United States, EPA 821-R-02-019. US EPA, August, 1–46.
Verardo, D. J., Froelich, P. N., & McIntyre, A. (1990). Determination of organic carbon and nitrogen in marine sediments using the Carlo Erba NA-1500 analyzer. Deep Sea Research Part A. Oceanographic Research Papers, 37(1), 157–165. doi:10.1016/0198-0149(90)90034-S.
Wang, S., Jia, Y., Wang, S., Wang, X., Wang, H., Zhao, Z., & Liu, B. (2009). Total mercury and monomethylmercury in water, sediments, and hydrophytes from the rivers, estuary, and bay along the Bohai Sea coast, northeastern China. Applied Geochemistry, 24(9), 1702–1711. doi:10.1016/j.apgeochem.2009.04.037.
Zagar, D., Petkovsek, G., Rajar, R., Sirnik, N., Horvat, M., Voudouri, A., et al. (2007). Modelling of mercury transport and transformations in the water compartment of the Mediterranean Sea. Marine Chemistry, 107(1), 64–88. doi:10.1016/j.marchem.2007.02.007.
Acknowledgements
This work was performed under the projects PROFLUX—Processes and Fluxes of Mercury and Methylmercury in a Contaminated Coastal Ecosystem, Tagus Estuary, Portugal (PTDC/MAR/102748/2008) PLANTA—Effect of salt-marsh plants on mercury methylation, transport and volatilization to the atmosphere (PTDC/AAC-AMB/115798/2009), Project UID/QUI/00100/2013, and by the Rute Cesário PhD grant (SFRH/BD/86441/2012), all funded by the Portuguese Foundation for Science and Technology (FCT). The authors would also like to thank to Joana Raimundo for the help in the statistical analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
ESM 1
(DOCX 1263 kb)
Rights and permissions
About this article

Cite this article
Cesário, R., Hintelmann, H., O’Driscoll, N.J. et al. Biogeochemical Cycle of Mercury and Methylmercury in Two Highly Contaminated Areas of Tagus Estuary (Portugal). Water Air Soil Pollut 228, 257 (2017). https://doi.org/10.1007/s11270-017-3442-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11270-017-3442-1