
Abstract
Venous thromboembolism, which is common in cancer patients and accompanies or even precedes malignant tumors, is known as cancer-related thrombosis and is an important cause of cancer- associated death. At present, the exact etiology of the elevated incidence of venous thrombosis in cancer patients remains elusive. Platelets play a crucial role in blood coagulation, which is intimately linked to the development of arterial thrombosis. Additionally, platelets contribute to tumor progression and facilitate immune evasion by tumors. Tumor cells can interact with the coagulation system through various mechanisms, such as producing hemostatic proteins, activating platelets, and directly adhering to normal cells. The relationship between platelets and malignant tumors is also significant. In this review article, we will explore these connections.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer and cardiovascular disease are diseases with intricate pathophysiology and the most common causes of death; prevalence is steadily increasing worldwide [1, 2]. Although venous thromboembolism (VTE), including deep venous thrombosis (DVT) and pulmonary embolism (PE), is common in the general population, it is especially common in cancer patients, and is accompanied by or can occur prior to malignant tumors [3]. Research indicates that the annual incidence rate of venous thromboembolism (VTE) in cancer patients is 0.5%, compared to 0.1% in the general population[4]. Active cancer accounts for 20% of the total VTE incidence [5, 6]. Furthermore, cancer-associated thrombosis (CAT) is a significant contributor to mortality in cancer patients [7, 8]. Compared with non-malignant tumor patients, cancer patients have a nine-fold increased risk of developing VTE [9]. The high incidence of VTE in cancer patients arises from a complex interplay between acquired and genetic factors that perturb the delicate balance of hemostasis, ultimately culminating in thrombosis [10]. In the general population, numerous genetic factors associated with VTE have been identified, including mutations in genes related to anticoagulation (SERPINC1, PROC, and PROS1), as well as various genetic polymorphisms [11]. Common factors contributing to the high incidence of VTE in cancer patients include advanced age, reduced mobility, and various cancer treatments such as surgery, radiotherapy, and chemotherapy. During the development of cancer, tumor cells release inflammatory cytokines and pro-angiogenic/procoagulant factors that stimulate stromal cells to express prothrombotic components, thereby promoting blood clotting [12, 13]. Table 1 provides a comprehensive overview of the factors that predisposed patients to thrombosis. Furthermore, cancer patients undergoing immune checkpoint therapy are highly susceptible to developing CAT and exhibit elevated mortality rates [14,15,16]. The inherent heterogeneity and biological characteristics of tumors may also contribute to an increased susceptibility to VTE [17], such as glioblastoma multiforme (GBM), pancreatic cancer, gastric cancer, and lung cancer carry a significantly higher risk of VTE due to the unique tumor mechanism that induces a hypercoagulable state in the body [18]. VTE is an independent predictor of reduced survival in cancer patients, meaning that the presence of VTE may indicate tumor progression, tumor treatment failure, and some underlying cancers [14, 19,20,21]. Gene mutations in tumor cells, particularly those affecting TP53, KRAS, EGFR, PTEN and IDH1 genes, can have an impact on hemostasis by inducing the expression of tumor tissue factor (TF) and vascular endothelial growth factor (VEGF), as well as promoting thrombosis through the release of pro-inflammatory cytokines and extracellular vesicles [13].
Platelets are enucleated cells released from the membrane processes of mature megakaryocytes (pro-platelets), which contribute to the formation of clots and, if maladjusted, may lead to thrombosis [33]. With the development of platelet biology, we have a new understanding of platelet formation, function, and signaling (Fig. 1). Despite numerous hypotheses, the etiology underlying the high prevalence of venous thrombosis in cancer patients remains uncertain. Tumor cells can interact with the hemostatic system in a myriad of ways, including the production of hemostatic proteins such as TF and thrombin, activation of platelets, and direct adhesion to normal cells like endothelial cells, monocytes, and platelets [34, 35]. In this review, our objective is to provide a concise overview of the pivotal role that platelets play in hemostasis and thrombosis, as well as to examine the intricate interplay between tumors and platelets, the current state of research, and the possibility of future treatment of tumors.

Main signal events and responses in the process of platelet activation (a) The release of ADP and thromboxane A2(TXA2) activates purine receptors P2Y12, P2Y1, TXA2 receptors (TP) and platelet activation. P2Y12 inhibits AC but stimulates phosphoinositide 3 kinase (PI3Ks) via GαI protein. The P2Y1 and TP signals activate the GαQ protein, which stimulates phospholipase C β (PLC β) to release Ca2 + into the cytoplasm, leading to protein kinase C (PKC) activation and downstream signaling events. The TP receptor also activates Gα12 and 13 proteins, leading to the activation of rho-associated protein kinase (ROCK) and contributing to platelet shape change and diffusion.thrombin activates platelets by binding to GαQ-coupled receptors PAR1 and PAR4. b Platelet inhibition is achieved through nitric oxide (NO) and prostaglandin I2 (PGI2) IP receptor-mediated activation of guanylate cyclase (GC) and adenylate cyclase (AC), leading to protein kinase G (PKG) and protein kinase A (PKA) activation. c Enhance platelet activation. GPVI and type c lectin-like receptor 2 (CLEC2) induce strong signals through the protein tyrosine kinase pathway, resulting in the release of Ca2 + into the cytoplasm. d Platelet activation is initiated by the interaction between adhesion receptors (integrin α6β1, α2β1, αIIbβ3 and glycoprotein Ib-V-IX complex) and their ligands, such as collagen and von Willebrand factor. This involves signal transduction through small G protein regulators (SGRs), src family kinases (SFKs) and serine/threonine protein kinases (STKs).The conformational changes of integrins, such as αIIbβ3, involve a pathway downstream of PLC, PKC and PI3K that leads from low to high affinity. This pathway includes CalDAG-GEFI (a guanosine nucleotide exchange factor regulated by Ca2 + and diacylglycerol), SGRs (RAS3A and RAP1B), as well as signaling molecules connected to the cytoskeleton (kindlin and talin subtypes). e Platelet membrane swelling and exposure of phosphatidylserine (PtdSer) occur through high Ca2 + mobilization agonists mediated by the ion channel anooctamin6 (ANO6), while calpain-2 mediates intracellular protein degradation. f The activation of Ca2 + -dependent and protein kinase-dependent cytoplasmic phospholipase A2 (cPLA2) and cyclooxygenase 1 (COX1) mediates the release of TXA2
Tumor progression with platelet aggregation and activation
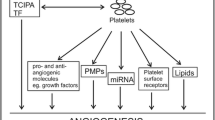
While platelets can have a positive impact on the behavior of cancer cells, the physiology and phenotype of platelets are also subject to influence by tumor cells [36]. In fact, several studies have revealed that tumor cells can regulate the RNA profile, number, and functionality of platelets. Cancer patients often exhibit activated clotting pathways that lead to a four-fold increased risk of thrombosis [37]. Professors Levin and Conley found that at least 40% of their hospitalized cancer patients had thrombocytosis [38]. Since then, a growing number of studies have reported a significant link between thrombocytosis and solid tumors, with a prevalence of 4–55% at the time of initial diagnosis [39,40,41,42]. As described in a review study of 3,654 patients with stage I-III breast cancer, 6.5% of these patients were diagnosed with inflammatory breast cancer (IBC), this study concluded that thrombocytosis, which is more prevalent in IBC patients, was associated with lower overall survival in these subjects, but not in non-IBC patients [43]. Tumor cells have the ability to increase thrombin production through direct and indirect mechanisms. To proliferate and foster metastasis, these cells concurrently manipulate platelet behavior by enhancing the synthesis and/or secretion of various compounds that incite platelet activation and aggregation [44,45,46]. The initial observation that tumor cells can induce platelet activation is based on a co-culture system, wherein tumor-induced platelet aggregation (TCIPA) is mitigated following the introduction of direct thrombin inhibitors[47]. Despite TCIPA being characterized by a heightened platelet count and conventional platelet activation, contemporary studies have demonstrated alterations in platelet derivatives within cancer patients. It's been discovered that EGFRvIII-mRNA can be isolated from the platelets of glioma patients, and PCA3-mRNA resides in the platelets of those with prostate cancer [48]. Furthermore, aberrant fusions of specific EML4-ALK genes have been detected in the platelets of patients suffering from non-small cell lung cancer [49]. Dysexpression of platelet protein can also be used as a diagnostic marker and prognostic factor. It has been reported that platelet concentrations of vascular endothelial growth factor (VEGF) are higher in cancer patients than in healthy individuals, including lung cancer patients [50, 51], Liver Cancer [52, 53] and colorectal cancer [54]. Furthermore, malignant tumor cells exhibit heightened thrombin production compared to benign tumors, underscoring thrombin's role as a potent platelet activator with significant pro-coagulant attributes [55]. Previous research has also indicated that the rise in thrombin levels is related to the tumor's location, with lung cancer demonstrating a more significant increase compared to brain and pancreatic cancers [56]. Previous research has demonstrated that TF plays a crucial role in initiating platelet activation and aggregation, with TF being expressed on the cell membranes of various cancer cells [57]. Extracellular vesicles (EVs), a diverse collection of cell-derived membrane structures including exosomes, microvesicles, and apoptotic bodies, are released by cells and significantly contribute to blood coagulation, thereby facilitating the direct generation of thrombin in cancer patients. Numerous studies have identified that EVs can express TF, linking it to thrombin production across different types of cancer [58]. Furthermore, activated host cells, such as monocytes and cancer cells, can release EVs that bear transferrin on their surface, further enhancing platelet activation [59,60,61]. Infact, it is reported that transferrin potentiates thrombin/FXIIa and blocks AT's inactivation effect on coagulation proteases inducing activation coagulation and platelet[62]. EVs are also exposed to negatively charged phospholipids, such as phosphatidyl serine (PS), on adventitia lobules [63], is an effective substance to promote coagulation. Circulating TF and EVs are good biomarkers of thrombosis and disseminated intravascular coagulation (DIC) in different diseases [64, 65].
Mechanisms of cancer-induced coagulation
Platelets are closely related to the blood hypercoagulable state, which is an important cause of cancer-related venous thrombosis and and tumors may affect the blood hypercoagulable state in many ways [66]. Existing studies have found that, tumor-derived G-CSF induces neutrophilia, triggering NET release and promoting thrombosis in murine model[67] and tumor-derived IL-6 induces thrombopoietin (TPO) expression in hepatocytes, and cancer cells have the ability to generate thrombin as well, promoting thrombosis and increasing the risk of thrombotic events in cancer patients[68,69,70]. Studies have found that about 5% of patients with idiopathic venous thromboembolism show latent malignant tumors, which are found within one year after the diagnosis of venous thromboembolism [71]. Previous studies have shown that 10%–57% of cancer patients have a high platelet count, which is associated with a poor prognosis [72]. Many cancer types, such as adenocarcinoma, ovarian cancer, brain cancer, gastric cancer, colon cancer, and lung cancer, have a higher risk of developing VTE, while in cancer types such as breast and prostate cancer, the risk of VTE is lower, indicating that there may be specific VTE pathways in different cancer types (Table 2). Additionally, cancer patients often exhibit a hypercoagulable state at the time of diagnosis, with a higher prevalence of certain prothrombotic gene mutations compared to the general population [73, 74]. This suggests a strong correlation between blood status and tumor growth, with potential implications for the role of blood coagulation gene polymorphisms in cancer development. Upon activation of prothrombin, thrombin is generated, which subsequently triggers fibrinogenesis and platelet activation to maintain hemostasis. In addition to its hemostatic function, thrombin, especially through its interaction with the receptor PAR1, is also thought to affect key mechanisms of tumor initiation, including cell survival, proliferation, and adhesion [75, 76]. Previous studies have found that F7rs510317A alleles are associated with an increased risk of breast cancer [77]. This allele is also linked to an elevation in the levels of FVII, the activating enzyme in the exogenous coagulation pathway, within circulation [78, 79].
As mentioned earlier, cancer patients with VTE have a more aggressive underlying cancer phenotype, disease progression, and treatment failure, and tumors may affect the blood hypercoagulable state in many ways (Fig. 2). Platelets play a crucial role in coagulation, indicating their close association with tumor progression. Markers of platelet activation and coagulation have been identified in primary tumor tissues [97].

Partial pathways of tumor affecting thrombosis (a) Tumor-derived G-CSF induces neutrophilia, triggering NET release and promoting thrombosis. b Tumor cells release TF1+MVs, causing thrombosis in tumor patients through circulation. c Tumor-derived IL-6 induces TPO expression in hepatocytes, promoting thrombosis and increasing the risk of thrombotic events in cancer patients
FVII is structurally expressed in cancer cells under hypoxic conditions, and forms a complex with TF (TF/FVIIa) to enhance blood clotting activity. FX plays a pivotal role in both endogenous and exogenous coagulation pathways. Tumor cells produce a thrombin called a cancer coagulant, which activates FX [98]. Tissue factor pathway inhibitor (TFPI) is known to modulate the activity of transferrin, which triggers the exogenous coagulation pathway and represents the most extensively studied coagulation factor in the context of malignant tumors [99, 100]. TF is the center of current thrombosis models: exposed when injured. Following oncogene induction, expression of TF in tumor tissue is related to cell survival, proliferation, invasion, angiogenesis, and metastasis, and generally related to poor prognosis in different tumor types. As previously mentioned, TF interacts with plasma coagulation factors to facilitate the generation of thrombin, which exerts pleiotropic effects on various cellular processes. Thrombin contributes to thrombosis by cleaving fibrinogen and activating protease activated receptors (PARs), which mediate a variety of cellular effects. However, when considering the biological function of the thrombin-PAR axis, it remains challenging to determine how this pathway specifically contributes to both blood coagulation and tumor progression. Studies have demonstrated that thrombin activates human platelets via PAR-1 and PAR-4, leading to the secretion of a diverse array of molecules, which facilitates the formation of thrombus [101, 102].
In cancer, transferrin is often structurally overexpressed and can act locally or remotely, and is carried by tumor-derived extracellular vesicles and released into the circulation [18]. In tumor tissue, blood coagulation can be activated by destruction of the blood vessel wall, leading to bleeding and intravascular coagulation, or extravascular coagulation due to increased vascular permeability and plasma extravasation [103]. In addition, the entry of metastatic cancer cells into the circulation and the recruitment/activation of inflammatory cells (immune thrombosis) may further amplify these processes.
Some biomarkers have been shown to be associated with the occurrence of VTE in cancer (Table 3). Parameters such as increased white blood cell and platelet count and decreased hemoglobin have been shown to be good predictors of the risk of venous thromboembolism in cancer patients. In addition, the increased concentrations of prothrombin fragments 1–2, soluble P-selectin, coagulation factor VIII, and D-dimer are closely bound up with an increased incidence of cancer VTE. In an extensive synthesis of eighteen studies incorporating thirty-six biomarkers, D-dimer levels and epidermal growth factor receptor (EGFR) mutations emerged as the paramount predictors for thromboembolic incidents.[104].
Role of platelets in cancer-related thrombosis
The role of platelets in CAT is increasingly recognized. Platelet-cancer cell interactions are involved in the regulation of cancer-associated thrombosis [110]. A noteworthy clinical study has shown that patients with high-grade gliomas (HGGs) who exhibit a low platelet count — accounting for 25% of the participants — are at a heightened risk for VTE [86]. This discovery is particularly surprising when contrasted with the commonly observed phenomenon of elevated platelet counts in patients with solid tumors, suggesting a unique aspect of HGG biology. Moreover, the expression of P-selectin on the surface of activated platelets, and its release as soluble P-selectin into the circulation, has been identified as a contributory factor in thrombosis development [111,112,113]. Elevated levels of P-selectin might result in decreased platelet counts due to its role in augmenting platelet adhesion, potentially leading to enhanced platelet consumption [114, 115]. This theory is corroborated by the observation of increased soluble P-selectin levels in individuals with conditions that lead to platelet depletion before thrombotic events, such as disseminated intravascular coagulation, heparin-induced thrombocytopenia, and thrombotic thrombocytopenic purpura (TTP) [116]. Furthermore, the formation of nodules targeted by prothrombin activity is noted in specific pathological conditions characterized by extensive coagulation activity, including VTE in ovarian cancer [117], highlighting the involvement of podoplanin (PDPN) [87, 118]. PDPN induces platelet aggregation in a CLEC-2-dependent manner within human glioblastoma cells (LN319) [119], and PDPN expression is negatively correlated with platelet count, but positively correlated with D-dimer level [87]. These findings suggest that tumors may release PDPN1-containing microvesicles, which can activate circulating platelets and lead to an increased risk of venous thromboembolism in cancer patients (Fig. 2).
Activated platelets interact with the coagulation system directly or indirectly through MPs or secretory factors. Once activated, expression of anionic phospholipids on the procoagulant surface is involved in thrombin production, fibrin formation, and clotting [120]. In addition, the interaction between platelets and neutrophils leads to the formation of NETs. NETs can be formed without infection, such as in cancer and autoimmunity, and can promote thrombosis [121]. Activated platelets are involved in cancer-related thrombosis by providing procoagulant surfaces, releasing inflammatory molecules, and interacting with monocytes and neutrophils [120].
In cancer patients, platelets play a crucial role not only in the development of arterial thrombosis but also significantly contribute to venous thrombosis [122, 123]. Thrombocytosis is common in cancer patients, especially in gastrointestinal, lung, breast, and ovarian cancer [104]. Previous studies have found that individuals with elevated platelet counts prior to cancer diagnosis have a higher frequency of VTE compared to individuals with lower platelet counts [83], and similar results were observed when measuring platelet counts in cancer patients [84, 85]. The reduction of VTE in patients with multiple myeloma who are treated with thalidomide or lenalidomide and low-dose aspirin is comparable to that achieved by low molecular weight heparin, indicating the involvement of platelets in VTE development among these patients [124]. In addition, use of aspirin was associated with reduced edge of venous thromboembolism in patients with ovarian cancer, but not in patients with breast cancer [125, 126]. Many studies have measured the biomarkers of platelet activating factor [123]. In general, these biomarkers are increased in cancer patients, but only a limited number of studies have determined whether they can predict venous thromboembolism in this group of patients. A recent study failed to find a link between various platelet activation markers, including PF4 and VTE, in patients with various cancer types [127]. However, a pancreatic cancer study found that increased PF4 levels were associated with an approximately three-fold increase in VTE risk [128]. This disparity highlights the significance of examining distinct cancer categories.
The role of platelets in thrombosis has also been investigated in murine cancer models. A study employing a syngeneic orthotopic model of pancreatic cancer has demonstrated that clopidogrel can mitigate the binding of tumor-derived microvesicles to sites of thrombosis [129]. We have observed that TF1 microvesicles are capable of activating platelets via thrombin, while clopidogrel can effectively mitigate the augmented effect of exogenous TF1 microvesicles on thrombosis in murine models [130]. Collectively, these fundamental and clinical investigations propose that the administration of antiplatelet agents may serve as a preventive measure against venous thromboembolism in select cancer patients [123].
Numerous platelet receptors and signaling pathways, in addition to the release of platelet agonists by malignant cells, are implicated in TCIPA [131,132,133]. At the same time, the specific mechanism of platelet aggregation in malignant tumor cells is related to the type of cancer cell [132]. Several cancer cell lines, including melanoma, are capable of producing ADP [133], glioblastoma [134], breast cancer [135, 136], lung cancer [137], pancreatic ductal adenocarcinoma [138], and TCIPA induced by fibrosarcoma [137]. The activation of the P2Y12 receptor initiates a positive feedback mechanism in ADP-induced platelet activation, leading to the release of more ADP, ATP, and calcium from dense granules [139]. In human MCF-7 breast cancer cells, the reduction of ADP levels and the inhibition of the ADP receptor P2Y12 were found to diminish platelet activation and aggregation [136].
Platelets in the tumor microenvironment and interaction with PD-L1
Further clinical retrospective studies provide additional evidence that platelets present in the TME contribute to chemotherapy resistance and are associated with early disease recurrence and poor survival outcomes [140]. GPIbα platelet markers were detected in 59% of HER2-negative breast cancer biopsies and were significantly associated with reduced efficacy of neoadjuvant therapy. Circulating platelets are not mere pliable sacs summoned by VWF to impede damaged blood vessels and avert hemorrhage [141]. Platelets indeed express PD-L1, which facilitates direct interaction with T cells and may have an impact on the efficacy of immune checkpoint therapy [142]. Studies have shown that the proliferation and activity of T cells bound to platelets decreased [143]. This may be linked to the significantly reduced efficacy of immune checkpoint therapy utilizing PD-1 antibodies, indicating the direct involvement of platelets in cancer-related immune evasion. In addition to the aforementioned tumor-promoting effects, there is compelling evidence that thrombin cleaves platelet glycoprotein A (GARP) repeats, resulting in the liberation of TGF-β derived from platelets [144, 145]. Activated platelets can also trigger innate immune responses by recruiting neutrophils and inducing reticulocytosis, thereby unleashing a cascade of physiological events [146]. Neutrophils can block the function of cytotoxic T cells through the expression of inducible nitric oxide synthase (iNOS) and production of nitric oxide (NO) [147]. In addition, the presence of PD-L1 on neutrophils is associated with initiating T cell apoptosis through interference induced activation of T cells and subsequent interaction with PD-1 [148].
Tumor patients exhibiting a procoagulant state, who subsequently develop CAT, manifest significant tumor progression, limited treatment efficacy and poor overall survival. Current evidence from in vitro and in vivo studies suggests that the procoagulant environment not only facilitates tumor immune evasion but also impedes immunotherapeutic interventions. Revealing the intricate mechanisms by which endothelial cells orchestrate inflammation and thrombosis is pivotal in devising innovative strategies to augment the potency of immune checkpoint inhibition and targeted therapy (Fig. 3). A combined approach targeting both tumor-mediated coagulation and tumor-induced immune evasion has been postulated to potentially bestow clinical benefits upon cancer patients. In view of the intricacy of the tumor microenvironment, targeted therapies aimed at specific pathways may augment response rates and ameliorate adverse effects (Table 4). Inhibition of the hypercoagulable state can be accomplished by administering anti-VWF nanoparticles that obstruct the A1 domain, thereby impeding VWF-mediated platelet binding. Alternatively, recombinant disintegrin and metalloproteinase member 13 (ADAMTS13) with thrombin reactive protein type 1 motif can cleave and inactivate VWF to provide a therapeutic option for supporting immune checkpoint inhibition [149]. In addition, the investigation of platelet mRNA analysis or detection of cancer cell activation induced by platelets is also being explored for diagnostic and monitoring purposes related to tumor development [150].

Interaction between cancer-associated immune thrombosis and immune checkpoint inhibition Immune thrombosis associated with cancer is formed by the interaction of a variety of cells. Activation of plasma coagulation and complement factor, as well as TF released by endothelial cells, platelets, monocytes and neutrophils can modify T cell responses in relation to immune checkpoint inhibition
Therapeutic perspectives targeting tumor progression
Platelets are critical not only for hemostasis and coagulation but also, as extensive research has shown, in tumor progression, including aspects like invasion and exosmosis, closely linking them to CAT. This means that this property of platelets could be used for anti-tumor therapy. Integrin αIIbβ3, which is highly expressed in platelets and their progenitors, plays a pivotal role in platelet function, hemostasis, and arterial thrombosis, as well as in tumor progression, including cell proliferation and metastasis [158]. The binding of abciximab to integrin αIIbβ3 creates steric hindrance, preventing fibrinogen and other ligands from interacting with the integrin, thus interfering with platelet aggregation and thrombosis [159]. Caplacizumab, a humanized immunoglobulin fragment, specifically targets the A1 domain of vWF, disrupting the interaction between vWF and the platelet receptor GP1b- α [160]. This action prevents platelet adhesion, a critical step in the formation of microvascular thrombosis, and has been shown to normalize platelet counts in preclinical studies [160, 161]. Currently approved for the treatment of acquired TTP [162,163,164], but there is no relevant research in the field of cancer. The combination of antitumor drugs with Caplacizumab may be a potential treatment to reduce the occurrence of CAT and improve the prognosis of tumor patients, which needs further research to prove. Whilding et al. constructed αVβ3 specific CAR T cells and evaluated their antitumor function in vitro and in vivo preclinical models. These αVβ3-CAR T cells rapidly and specifically targeted αvβ3-positive tumor cells, secreting IFN-γ and IL-2. In a mouse xenograft model of metastatic A-375 melanoma, the intervention led to the complete eradication of melanoma lesions and long-term tumor-free survival. Integrins are also used to generate specific, controllable, and improved cytotoxicity of CAR-T therapies [165]. These studies illustrate the potential of the integrin family in anti-cancer and prevention of CAT.
Targeted drug delivery has become a new therapeutic strategy for cancer treatment. The tumor microenvironment is highly acidic compared to normal tissues, so Ph-sensitive nanosystems have been developed based on this difference. Drug release is activated in response to the acidic microenvironment and can enhance the therapeutic effects of cancer therapy. Crucially, platelets present promising avenues for targeted drug delivery. Nanocarriers can be engineered with peptides, enzymes, and antibodies [166]. For instance, a dual-targeted delivery system, consisting of paclitaxel-supported PEGylated-polylactic acid nanoparticles and cyclic peptides, is designed to selectively bind to the platelet-derived PDGF/PDGβ, offering improved treatment options for multiple myeloma [167].
Liposomes have been extensively studied as drug delivery systems, and many liposomal nanomedicines have been approved for clinical use [168]. Exosomes, which are biological extracellular vesicles, transmit signals through ligands or adhesion molecules on their membranes and have shown significant potential for targeted cancer drug delivery due to their origin from tumor cells [169]. Additionally, virus simulation systems are being developed for cancer diagnosis and targeted therapy. These systems mimic viruses in structure but lack genetic material, allowing them to be ingested by host cells without causing infection. Importantly, nanocarriers can be functionalized with small molecules, aptamers, and antibodies that possess high specificity and affinity [168]. There's growing evidence that these platelet-targeted drug delivery systems can enhance the efficacy of cancer treatments while minimizing potential side effects.
Conclusion and future prospects
Cancer-related venous thromboembolism is a well-known disease, but little is known about the potential biological mechanism of linking tumors to thrombosis [170]. Platelets play an important role in the process of blood coagulation. From a clinical point of view, platelet therapy may reduce the incidence of CAT and the rate of tumor progression. The use of some antiplatelet drugs may have a certain effect on the treatment of tumors (Table 5). Patient-related factors (including acquired and genetic determinants), cancer biology, and antineoplastic therapy are thought to play a synergistic role in venous thrombosis [11]. It is worth noting that the hemostatic system in cancer patients not only causes thrombosis, but also contributes to the growth and spread of tumors, suggesting cancer-related coagulation disorders may not be an accidental phenomenon [171].
In view of the adverse effects of venous thromboembolization on the prognosis of patients, disease prevention in cancer patients is important. It has been difficult to identify coagulation and fibrinolysis biomarkers. There is a strong correlation between TF and primary tumor types, while fibrinolysis is more closely related to the cellular composition of the TME [178], which requires exploration of the kinetics of coagulation and fibrinolysis simultaneously [8]. It was recently reported that longitudinal evaluation of D-dimer significantly improved the estimation of VTE risk in cancer patients [179]. Functional analysis of thrombin production is also used in the early diagnosis of postoperative recurrence of breast cancer [180]. Because studies in the general population have shown that venous thromboembolism is hereditary, genetic markers, especially genetic polymorphisms, are attractive candidates for predicting cancer-related venous thromboembolism. In addition, given the role of hemostatic components in cancer development, these genetic markers may also predict cancer susceptibility and/or progression [11].
Thrombosis in tumor patients may be different from VTE in the general population. According to tumor models, some biological mechanisms are more related than others. In terms of the susceptibility and progression of cancer, the few studies so far have mainly focused on oral squamous cell carcinoma, breast cancer, and gastrointestinal cancer. Twelve and nine genetic polymorphisms related to cancer susceptibility and progression have been reported. It is worth noting that most of these variants have not been confirmed as predictors of cancer-related venous thromboembolism [3]. However, according to rare data, hemostatic gene polymorphisms seem to have an impact on cancer occurrence, which seems to be specific to cancer models, but may also may be related to tumor staging. Taking into account thrombus prevention, cancer screening, prognostic assessment, and the development of potential antineoplastic therapy targeting coagulation, the scientific basis of these observations requires further investigation [11].
Blood coagulation is closely related to cancer, and platelets are an important player in blood clotting. The development of strategies that selectively target blood coagulation and simultaneously affect complement factors and the inflammatory response may promote anti-tumor effects. Further research is needed to fully understand the interaction of these pathways in the TME. Targeted coagulation, as a bridge between innate and acquired immune systems, may provide a new strategy to overcome drug resistance to checkpoint inhibition [181, 182]. Specifically, targeted platelets may provide a novel idea for the treatment of tumors and tumor-related embolism in the future.
Data availability
The authors declare that no new data were created for this narrative review.
References
Anderson E, Durstine JL (2019) Physical activity, exercise, and chronic diseases: a brief review. Sports Med Health Sci 1(1):3–10
Curigliano G, Lenihan D, Fradley M et al (2020) Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol: Off J Eur Soc Med Oncol 31(2):171–190
Tavares V, Neto BV, Vilas-Boas MI et al (2022) Impact of hereditary thrombophilia on cancer-associated thrombosis, tumour susceptibility and progression: a review of existing evidence. Biochim Biophys Acta 1877(5):188778
Elyamany G, Alzahrani AM, Bukhary E (2014) Cancer-associated thrombosis: an overview. Clin Med Insights Oncol 8:129–137
Heit JA, Spencer FA, White RH (2016) The epidemiology of venous thromboembolism. J Thromb Thrombolysis 41(1):3
Laporte S, Mismetti P, Décousus H et al (2008) Clinical predictors for fatal pulmonary embolism in 15,520 patients with venous thromboembolism: findings from the Registro Informatizado de la Enfermedad TromboEmbolica venosa (RIETE) Registry. Circulation 117(13):1711–1716
Farge D, Frere C, Connors JM et al (2019) 2019 international clinical practice guidelines for the treatment and prophylaxis of venous thromboembolism in patients with cancer. Lancet Oncol 20(10):e566–e581
Khorana AA, Cohen AT, Carrier M et al (2020) Prevention of venous thromboembolism in ambulatory patients with cancer. ESMO Open 5(6):e000948
Gran OV, Smith EN, Brækkan SK et al (2016) Joint effects of cancer and variants in the factor 5 gene on the risk of venous thromboembolism. Haematologica 101(9):1046–1053
Reitsma PH, Versteeg HH, Middeldorp S (2012) Mechanistic view of risk factors for venous thromboembolism. Arterioscler Thromb Vasc Biol 32(3):563–568
Tavares V, Pinto R, Assis J et al (2020) Venous thromboembolism GWAS reported genetic makeup and the hallmarks of cancer: linkage to ovarian tumour behaviour. Biochim Biophys Acta 1873(1):188331
Skille H, Paulsen B, Hveem K et al (2021) Prothrombotic genotypes and risk of venous thromboembolism in occult cancer. Thromb Res 205:17–23
Buijs JT, Versteeg HH (2020) Genes and proteins associated with the risk for cancer-associated thrombosis. Thromb Res 191:S43–S49
Moik F, Chan W-SE, Wiedemann S et al (2021) Incidence, risk factors, and outcomes of venous and arterial thromboembolism in immune checkpoint inhibitor therapy. Blood 137(12):1669–1678
Sussman TA, Li H, Hobbs B et al (2021) Incidence of thromboembolism in patients with melanoma on immune checkpoint inhibitor therapy and its adverse association with survival. J Immunother Cancer 9(1):e001719
Roopkumar J, Swaidani S, Kim AS et al (2021) Increased incidence of venous thromboembolism with cancer immunotherapy. Med (New York, NY) 2(4):423–434
Galmiche A, Rak J, Roumenina LT et al (2022) Coagulome and the tumor microenvironment: an actionable interplay. Trends Cancer 8(5):369–383
Falanga A, Schieppati F, Russo L (2019) Pathophysiology 1. Mechanisms of Thrombosis in Cancer Patients. Cancer Treat Res 179:11–36
Khorana AA (2010) Venous thromboembolism and prognosis in cancer. Thromb Res 125(6):490–493
Posch F, Riedl J, Reitter E-M et al (2016) Hypercoagulabilty, venous thromboembolism, and death in patients with cancer. A multi-state model. Thromb Haemost 115(4):817–826
Riedl JM, Schwarzenbacher E, Moik F et al (2022) Patterns of thromboembolism in patients with advanced pancreatic cancer undergoing first-line chemotherapy with FOLFIRINOX or gemcitabine/nab-paclitaxel. Thromb Haemost 122(4):633–645
Ay C, Pabinger I, Cohen AT (2017) Cancer-associated venous thromboembolism: Burden, mechanisms, and management. Thromb Haemost 117(2):219–230
Gao S, Ma J-J, Lu C (2014) Venous thromboembolism risk and erythropoiesis-stimulating agents for the treatment of cancer-associated anemia: a meta-analysis. Tumour Biol 35(1):603–613
Ashrani AA, Gullerud RE, Petterson TM et al (2016) Risk factors for incident venous thromboembolism in active cancer patients: a population based case-control study. Thromb Res 139:29–37
Cohen AT, Katholing A, Rietbrock S et al (2017) Epidemiology of first and recurrent venous thromboembolism in patients with active cancer. A population-based cohort study. Thromb Haemost 117(1):57–65
Königsbrügge O, Pabinger I, Ay C (2014) Risk factors for venous thromboembolism in cancer: novel findings from the Vienna cancer and thrombosis study (CATS). Thromb Res 133(Suppl 2):S39–S43
Fiedler T, Rabe M, Mundkowski RG et al (2018) Adipose-derived mesenchymal stem cells release microvesicles with procoagulant activity. Int J Biochem Cell Biol 100:49–53
Farge D, Le Maignan C, Doucet L et al (2019) Women, thrombosis, and cancer. Thromb Res 181(Suppl 1):S47–S53
Würtz M, Grove EL, Corraini P et al (2020) Comorbidity and risk of venous thromboembolism after hospitalization for first-time myocardial infarction: a population-based cohort study. J Thromb Haemost 18(8):1974–1985
Chaturvedi S, Braunstein EM, Yuan X et al (2020) Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood 135(4):239–251
Karasu A, Engbers MJ, Cushman M et al (2016) Genetic risk factors for venous thrombosis in the elderly in a case-control study. J Thromb Haemost 14(9):1759–1764
Ghouse J, Tragante V, Ahlberg G et al (2023) Genome-wide meta-analysis identifies 93 risk loci and enables risk prediction equivalent to monogenic forms of venous thromboembolism. Nat Genet 55(3):399–409
Raghunathan S, Rayes J, Sen Gupta A (2022) Platelet-inspired nanomedicine in hemostasis thrombosis and thromboinflammation. J Thromb Haemost 20(7):1535–1549
Levi M (2019) Disseminated intravascular coagulation in cancer: an update. Semin Thromb Hemost 45(4):342–347
Mitrugno A, Tassi Yunga S, Sylman JL et al (2019) The role of coagulation and platelets in colon cancer-associated thrombosis. Am J Physiol Cell Physiol 316(2):C264–C273
Ghoshal K, Bhattacharyya M (2014) Overview of platelet physiology: its hemostatic and nonhemostatic role in disease pathogenesis. Sci World J 2014:781857
Wang S, Li Z, Xu R (2018) Human cancer and platelet interaction, a potential therapeutic target. Int J Mol Sci 19(4):1246. https://doi.org/10.3390/ijms19041246
Levin J, Conley CL (1964) Thrombocytosis associated with malignant disease. Arch Intern Med 114:497–500
Wojtukiewicz MZ, Sierko E, Hempel D et al (2017) Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev 36(2):249–262
Zhang X, Ran Y (2015) Prognostic role of elevated platelet count in patients with lung cancer: a systematic review and meta-analysis. Int J Clin Exp Med 8(4):5379–5387
Gu D, Szallasi A (2017) Thrombocytosis portends adverse prognosis in colorectal cancer: a meta-analysis of 5,619 patients in 16 individual studies. Anticancer Res 37(9):4717–4726
Ji Y, Sheng L, Du X et al (2015) Elevated platelet count is a strong predictor of poor prognosis in stage I non-small cell lung cancer patients. Platelets 26(2):138–142
Harano K, Kogawa T, Wu J et al (2017) Thrombocytosis as a prognostic factor in inflammatory breast cancer. Breast Cancer Res Treat 166(3):819–832
Zarà M, Canobbio I, Visconte C et al (2018) Molecular mechanisms of platelet activation and aggregation induced by breast cancer cells. Cell Signal 48:45–53
Reddel CJ, Tan CW, Chen VM (2019) Thrombin generation and cancer: contributors and consequences. Cancers (Basel) 11(1):100. https://doi.org/10.3390/cancers11010100
Chang J, Jiang L, Wang Y et al (2015) 12/15 Lipoxygenase regulation of colorectal tumorigenesis is determined by the relative tumor levels of its metabolite 12-HETE and 13-HODE in animal models. Oncotarget 6(5):2879–2888
Heinmöller E, Weinel RJ, Heidtmann HH et al (1996) Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J Cancer Res Clin Oncol 122(12):735–744
Nilsson RJA, Balaj L, Hulleman E et al (2011) Blood platelets contain tumor-derived RNA biomarkers. Blood 118(13):3680–3683
Nilsson RJA, Karachaliou N, Berenguer J et al (2016) Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget 7(1):1066–1075
Sabrkhany S, Kuijpers MJE, van Kuijk SMJ et al (2017) A combination of platelet features allows detection of early-stage cancer. Eur J Cancer 80:5–13. https://doi.org/10.1016/j.ejca.2017.04.010
Yao L, Dong H, Luo Y et al (2014) Net platelet angiogenic activity (NPAA) correlates with progression and prognosis of non-small cell lung cancer. PLoS One 9(4):e96206
Alkozai EM, Porte RJ, Adelmeijer J et al (2015) Levels of angiogenic proteins in plasma and platelets are not different between patients with hepatitis B/C-related cirrhosis and patients with cirrhosis and hepatocellular carcinoma. Platelets 26(6):577–582
Kim SJ, Choi IK, Park KH et al (2004) Serum vascular endothelial growth factor per platelet count in hepatocellular carcinoma: correlations with clinical parameters and survival. Jpn J Clin Oncol 34(4):184–190
Peterson JE, Zurakowski D, Italiano JE et al (2012) VEGF, PF4 and PDGF are elevated in platelets of colorectal cancer patients. Angiogenesis 15(2):265–273
Duvernay MT, Temple KJ, Maeng JG et al (2017) Contributions of protease-activated receptors PAR1 and PAR4 to thrombin-induced GPIIbIIIa activation in human platelets. Mol Pharmacol 91(1):39–47
Reitter EM, Kaider A, Ay C et al (2016) Longitudinal analysis of hemostasis biomarkers in cancer patients during antitumor treatment. J Thromb Haemost 14(2):294–305
Arnout J, Hoylaerts MF, Lijnen HR (2006) Haemostasis. Handb Exp Pharmacol 176(Pt 2):1–41. https://doi.org/10.1007/3-540-36028-x_1
Gardiner C, Harrison P, Belting M et al (2015) Extracellular vesicles, tissue factor, cancer and thrombosis - discussion themes of the ISEV 2014 Educational Day. J Extracell Vesicles 4:26901
Mackman N, Tilley RE, Key NS (2007) Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol 27(8):1687–1693
Gregory SA, Morrissey JH, Edgington TS (1989) Regulation of tissue factor gene expression in the monocyte procoagulant response to endotoxin. Mol Cell Biol 9(6):2752–2755
Geddings JE, Mackman N (2013) Tumor-derived tissue factor-positive microparticles and venous thrombosis in cancer patients. Blood 122(11):1873–1880
Tang X, Zhang Z, Fang M et al (2020) Transferrin plays a central role in coagulation balance by interacting with clotting factors. Cell Res 30(2):119–132
György B, Szabó TG, Pásztói M et al (2011) Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci: CMLS 68(16):2667–2688
Reddy EC, Rand ML (2020) Procoagulant phosphatidylserine-exposing platelets and. Front Cardiovasc Med 7:15
Tesselaar MET, Romijn FPHTM, Van Der Linden IK et al (2007) Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost 5(3):520–527
Mulder FI, Horváth-Puhó E, van Es N et al (2021) Venous thromboembolism in cancer patients: a population-based cohort study. Blood 137(14):1959–1969
Gomes T, Várady CBS, Lourenço AL et al (2019) IL-1β blockade attenuates thrombosis in a neutrophil extracellular trap-dependent breast cancer model. Front Immunol 10:2088
Reusswig F, Fazel Modares N, Brechtenkamp M et al (2021) Efficiently restored thrombopoietin production by Ashwell-Morell receptor and IL-6R induced Janus Kinase 2/Signal transducer and activator of transcription signaling early after partial hepatectomy. Hepatology 74(1):411–427
Zucchella M, Dezza L, Pacchiarini L et al (1989) Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 74(6):541–545
Stone RL, Nick AM, McNeish IA et al (2012) Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med 366(7):610–618
van Es N, Le Gal G, Otten H-M et al (2017) Screening for occult cancer in patients with unprovoked venous thromboembolism: a systematic review and meta-analysis of individual patient data. Ann Intern Med 167(6):410–417
Sierko E, Wojtukiewicz MZ (2004) Platelets and angiogenesis in malignancy. Semin Thromb Hemost 30(1):95–108. https://doi.org/10.1055/s-2004-822974
Pihusch R, Danzl G, Scholz M et al (2002) Impact of thrombophilic gene mutations on thrombosis risk in patients with gastrointestinal carcinoma. Cancer 94(12):3120–3126
Tinholt M, Viken MK, Dahm AE et al (2014) Increased coagulation activity and genetic polymorphisms in the F5, F10 and EPCR genes are associated with breast cancer: a case-control study. BMC Cancer 14:845
Maragoudakis ME, Tsopanoglou NE, Andriopoulou P et al (2000) Effects of thrombin/thrombosis in angiogenesis and tumour progression. Matrix Biol: J Int Soc Matrix Biol 19(4):345–351
Green D (2010) Karpatkin S Role of thrombin as a tumor growth factor. Cell Cycle (Georgetown, Tex). 9(4):656–661
Eroğlu A, Oztürk A, Akar N (2011) Association between the -402GA, -401GT, and -323ins10-bp polymorphisms of factor VII gene and breast cancer. Breast Cancer (Tokyo, Japan) 18(4):282–285
Fang J, Yuan Q, Du Z et al (2021) Contribution of factor VII polymorphisms to coagulopathy in patients with isolated traumatic brain injury. Clin Neurol Neurosurg 208:106836
Ken-Dror G, Drenos F, Humphries SE et al (2010) Haplotype and genotype effects of the F7 gene on circulating factor VII, coagulation activation markers and incident coronary heart disease in UK men. J Thromb Haemost 8(11):2394–2403
DuPre SA, Hunter KW (2007) Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: association with tumor-derived growth factors. Exp Mol Pathol 82(1):12–24
Kowanetz M, Wu X, Lee J et al (2010) Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 107(50):21248-21255
Demers M, Krause DS, Schatzberg D et al (2012) Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A 109(32):13076-13081
Jensvoll H, Blix K, Brækkan SK et al (2014) Platelet count measured prior to cancer development is a risk factor for future symptomatic venous thromboembolism: the Tromsø Study. PLoS One 9(3):e92011
Khorana AA, Francis CW, Culakova E et al (2005) Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer 104(12):2822–2829
Simanek R, Vormittag R, Ay C et al (2010) High platelet count associated with venous thromboembolism in cancer patients: results from the Vienna Cancer and Thrombosis Study (CATS). J Thromb Haemost 8(1):114–120
Thaler J, Ay C, Kaider A et al (2014) Biomarkers predictive of venous thromboembolism in patients with newly diagnosed high-grade gliomas. Neuro-oncology 16(12):1645–1651
Riedl J, Preusser M, Nazari PMS et al (2017) Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 129(13):1831–1839
Khorana AA, Kuderer NM, Culakova E et al (2008) Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 111(10):4902–4907
Granger JM, Kontoyiannis DP (2009) Etiology and outcome of extreme leukocytosis in 758 nonhematologic cancer patients: a retrospective, single-institution study. Cancer 115(17):3919–3923
Kasuga I, Makino S, Kiyokawa H et al (2001) Tumor-related leukocytosis is linked with poor prognosis in patients with lung carcinoma. Cancer 92(9):2399–2405
Ruka W, Rutkowski P, Kaminska J et al (2001) Alterations of routine blood tests in adult patients with soft tissue sarcomas: relationships to cytokine serum levels and prognostic significance. Ann Oncol: Off J Eur Soc Med Oncol 12(10):1423–1432
Blix K, Jensvoll H, Brækkan SK et al (2013) White blood cell count measured prior to cancer development is associated with future risk of venous thromboembolism–the Tromsø study. PLoS One 8(9):e73447
Pabinger I, Posch F (2014) Flamethrowers: blood cells and cancer thrombosis risk. Hematol Am Soc Hematol Educ Program 2014(1):410–417
Khorana AA, Francis CW, Menzies KE et al (2008) Plasma tissue factor may be predictive of venous thromboembolism in pancreatic cancer. J Thromb Haemost 6(11):1983–1985
Thaler J, Ay C, Mackman N et al (2012) Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J Thromb Haemost 10(7):1363–1370
Bharthuar A, Khorana AA, Hutson A et al (2013) Circulating microparticle tissue factor, thromboembolism and survival in pancreaticobiliary cancers. Thromb Res 132(2):180–184
Verheul HM, Hoekman K, Lupu F et al (2000) Platelet and coagulation activation with vascular endothelial growth factor generation in soft tissue sarcomas. Clin Cancer Res: Off J Am Assoc Cancer Res 6(1):166–171
Gordon SG, Mielicki WP (1997) Cancer procoagulant: a factor X activator, tumor marker and growth factor from malignant tissue. Blood Coagul Fibrinolysis: Int J Haemost Thromb 8(2):73–86
Tinholt M, Vollan HKM, Sahlberg KK et al (2015) Tumor expression, plasma levels and genetic polymorphisms of the coagulation inhibitor TFPI are associated with clinicopathological parameters and survival in breast cancer, in contrast to the coagulation initiator TF. Breast Cancer Res: BCR 17(1):44
Bazzarelli AK, Scheer AS, Tai LH et al (2016) Tissue factor pathway inhibitor gene polymorphism -33T → C predicts improved disease-free survival in colorectal cancer. Ann Surg Oncol 23(7):2274–2280
Han N, Jin K, He K et al (2011) Protease-activated receptors in cancer: a systematic review. Oncol Lett 2(4):599–608
Ruf W (2007) Tissue factor and PAR signaling in tumor progression. Thromb Res 120(Suppl 2):S7-12
Tumors DHF (2019) Wounds that do not heal—a historical perspective with a focus on the fundamental roles of increased vascular permeability and clotting. Semin Thromb Hemost 45(06):576–592
Hisada Y, Mackman N (2017) Cancer-associated pathways and biomarkers of venous thrombosis. Blood 130(13):1499–1506
Lundbech M, Krag AE, Christensen TD et al (2020) Thrombin generation, thrombin-antithrombin complex, and prothrombin fragment F1+2 as biomarkers for hypercoagulability in cancer patients. Thromb Res 186:80–85
Orcutt SJ, Pietropaolo C, Krishnaswamy S (2002) Extended interactions with prothrombinase enforce affinity and specificity for its macromolecular substrate. J Biol Chem 277(48):46191–46196
Cosmi B, Legnani C, Libra A et al (2023) D-Dimers in diagnosis and prevention of venous thrombosis: recent advances and their practical implications. Pol Arch Intern Med 133(11):16604. https://doi.org/10.20452/pamw.16604
Ronchetti L, Terrenato I, Ferretti M et al (2023) Correction: circulating cell free DNA and citrullinated histone H3 as useful biomarkers of NETosis in endometrial cancer. J Exp Clin Cancer Res 42(1):278
Wurtzel JGT, Lazar S, Askari S et al (2024) Plasma growth factors maintain constitutive translation in platelets to regulate reactivity and thrombotic potential. Blood Adv 8(6):1550–1566. https://doi.org/10.1182/bloodadvances.2023011734
Palacios-Acedo A-L, Langiu M, Crescence L et al (2022) Platelet and cancer-cell interactions modulate cancer-associated thrombosis risk in different cancer types. Cancers (Basel) 14(3):730. https://doi.org/10.3390/cancers14030730
Kansas GS (1996) Selectins and their ligands: current concepts and controversies. Blood 88(9):3259–3287
Vandendries ER, Furie BC, Furie B (2004) Role of P-selectin and PSGL-1 in coagulation and thrombosis. Thromb Haemost 92(3):459–466
Michelson AD, Barnard MR, Hechtman HB et al (1996) In vivo tracking of platelets: circulating degranulated platelets rapidly lose surface P-selectin but continue to circulate and function. Proc Natl Acad Sci U S A 93(21):11877-11882
Palabrica T, Lobb R, Furie BC et al (1992) Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin on adherent platelets. Nature 359(6398):848–851
Frenette PS, Johnson RC, Hynes RO et al (1995) Platelets roll on stimulated endothelium in vivo: an interaction mediated by endothelial P-selectin. Proc Natl Acad Sci U S A 92(16):7450-7454
Chong BH, Murray B, Berndt MC et al (1994) Plasma P-selectin is increased in thrombotic consumptive platelet disorders. Blood 83(6):1535–1541
Glassman D, Bateman NW, Lee S et al (2022) Molecular Correlates of venous thromboembolism (VTE) in ovarian cancer. Cancers (Basel) 14(6):1496. https://doi.org/10.3390/cancers14061496
Tawil N, Bassawon R, Meehan B et al (2021) Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv 5(6):1682–1694
Suzuki-Inoue K, Kato Y, Inoue O et al (2007) Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J Biol Chem 282(36):25993–26001
Mege D, Aubert M, Lacroix R et al (2019) Involvement of platelets in cancers. Semin Thromb Hemost 45(6):569–575
Cedervall J, Hamidi A, Olsson A-K (2018) Platelets, NETs and cancer. Thromb Res 164(Suppl 1):S148–S152
Connolly GC, Phipps RP, Francis CW (2014) Platelets and cancer-associated thrombosis. Semin Oncol 41(3):302–310
Riedl J, Pabinger I, Ay C (2014) Platelets in cancer and thrombosis. Hamostaseologie 34(1):54–62
Larocca A, Cavallo F, Bringhen S et al (2012) Aspirin or enoxaparin thromboprophylaxis for patients with newly diagnosed multiple myeloma treated with lenalidomide. Blood 119(4):933–939
Shai A, Rennert HS, Rennert G et al (2014) Statins, aspirin and risk of thromboembolic events in ovarian cancer patients. Gynecol Oncol 133(2):304–308
Shai A, Rennert HS, Lavie O et al (2014) Statins, aspirin and risk of venous thromboembolic events in breast cancer patients. J Thromb Thrombolysis 38(1):32–38. https://doi.org/10.1007/s11239-013-1015-8
Riedl J, Hell L, Kaider A et al (2016) Association of platelet activation markers with cancer-associated venous thromboembolism. Platelets 27(1):80–85
Poruk KE, Firpo MA, Huerter LM et al (2010) Serum platelet factor 4 is an independent predictor of survival and venous thromboembolism in patients with pancreatic adenocarcinoma. Cancer Epidemiol Biomarkers Prev: Publ Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol 19(10):2605–2610
Mezouar S, Darbousset R, Dignat-George F et al (2015) Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int J Cancer 136(2):462–475
Geddings JE, Hisada Y, Boulaftali Y et al (2016) Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J Thromb Haemost 14(1):153–166
Medina C, Harmon S, Inkielewicz I et al (2012) Differential inhibition of tumour cell-induced platelet aggregation by the nicotinate aspirin prodrug (ST0702) and aspirin. Br J Pharmacol 166(3):938–949
Bastida E, Ordinas A, Jamieson GA (1981) Differing platelet aggregating effects by two tumor cell lines: absence of role for platelet-derived ADP. Am J Hematol 11(4):367–378
Boukerche H, Berthier-Vergnes O, Penin F et al (1994) Human melanoma cell lines differ in their capacity to release ADP and aggregate platelets. Br J Haematol 87(4):763–772
Bastida E, Escolar G, Almirall L et al (1986) Platelet activation induced by a human neuroblastoma tumor cell line is reduced by prior administration of ticlopidine. Thromb Haemost 55(3):333–337
Camez A, Dupuy E, Bellucci S et al (1986) Human platelet-tumor cell interactions vary with the tumor cell lines. Invasion Metastasis 6(6):321–334
Alonso-Escolano D, Strongin AY, Chung AW et al (2004) Membrane type-1 matrix metalloproteinase stimulates tumour cell-induced platelet aggregation: role of receptor glycoproteins. Br J Pharmacol 141(2):241–252
Jurasz P, Sawicki G, Duszyk M et al (2001) Matrix metalloproteinase 2 in tumor cell-induced platelet aggregation: regulation by nitric oxide. Can Res 61(1):376–382
Palacios-Acedo AL, Mezouar S, Mège D et al (2021) P2RY12-inhibitors reduce cancer-associated thrombosis and tumor growth in pancreatic cancers. Front Oncol 11:704945
Ballerini P, Dovizio M, Bruno A et al (2018) P2Y receptors in tumorigenesis and metastasis. Front Pharmacol 9:66. https://doi.org/10.3389/fphar.2018.00066
Ishikawa S, Miyashita T, Inokuchi M et al (2016) Platelets surrounding primary tumor cells are related to chemoresistance. Oncol Rep 36(2):787–794
Brass S (2001) Cardiovascular biology. Small cells, big issues. Nature 409(6817):145–147
Rolfes V, Idel C, Pries R et al (2018) PD-L1 is expressed on human platelets and is affected by immune checkpoint therapy. Oncotarget 9(44):27460–27470
Zamora C, Cantó E, Nieto JC et al (2017) Binding of platelets to lymphocytes: a potential anti-inflammatory therapy in rheumatoid arthritis. J Immunol (Baltimore, Md: 1950) 198(8):3099–3108
Metelli A, Wu BX, Riesenberg B et al (2020) Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-β. Sci Transl Med 12(525):eaay4860. https://doi.org/10.1126/scitranslmed.aay4860
Dahmani A, Delisle J-S (2018) TGF-β in T cell biology: implications for cancer immunotherapy. Cancers (Basel) 10(6):194. https://doi.org/10.3390/cancers10060194
Lisman T (2018) Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res 371(3):567–576
Coffelt SB, Kersten K, Doornebal CW et al (2015) IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522(7556):345–348
de Kleijn S, Langereis JD, Leentjens J et al (2013) IFN-γ-stimulated neutrophils suppress lymphocyte proliferation through expression of PD-L1. PLoS One 8(8):e72249
Scully M, Cataland SR, Peyvandi F et al (2019) Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med 380(4):335–346. https://doi.org/10.1056/NEJMoa1806311
Corvigno S, Johnson AM, Wong K-K et al (2022) Novel markers for liquid biopsies in cancer management: circulating platelets and extracellular vesicles. Mol Cancer Ther 21(7):1067–1075
Strauss J, Heery CR, Schlom J et al (2018) Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res: Off J Am Assoc Cancer Res 24(6):1287–1295
Goshua G, Sinha P, Hendrickson JE et al (2021) Cost effectiveness of caplacizumab in acquired thrombotic thrombocytopenic purpura. Blood 137(7):969–976
Loschi M, Porcher R, Barraco F et al (2016) Impact of eculizumab treatment on paroxysmal nocturnal hemoglobinuria: a treatment versus no-treatment study. Am J Hematol 91(4):366–370
Cofiell R, Kukreja A, Bedard K et al (2015) Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 125(21):3253–3262
Afshar-Kharghan V (2017) The role of the complement system in cancer. J Clin Invest 127(3):780–789. https://doi.org/10.1172/JCI90962
Scully M, Knöbl P, Kentouche K et al (2017) Recombinant ADAMTS-13: first-in-human pharmacokinetics and safety in congenital thrombotic thrombocytopenic purpura. Blood 130(19):2055–2063
Wang T-F, Li A, Garcia D (2018) Managing thrombosis in cancer patients. Res Pract Thromb Haemost 2(3):429–438
Huang J, Li X, Shi X et al (2019) Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting. J Hematol Oncol 12(1):26
Giordano A, Musumeci G, D’Angelillo A et al (2016) Effects of glycoprotein IIb/IIIa antagonists: anti platelet aggregation and beyond. Curr Drug Metab 17(2):194–203
Ulrichts H, Silence K, Schoolmeester A et al (2011) Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 118(3):757–765
Callewaert F, Roodt J, Ulrichts H et al (2012) Evaluation of efficacy and safety of the anti-VWF Nanobody ALX-0681 in a preclinical baboon model of acquired thrombotic thrombocytopenic purpura. Blood 120(17):3603–3610
Scully M, Cataland SR, Peyvandi F et al (2019) Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N Engl J Med 380(4):335–346
Völker LA, Kaufeld J, Balduin G et al (2023) Impact of first-line use of caplacizumab on treatment outcomes in immune thrombotic thrombocytopenic purpura. J Thromb Haemost 21(3):559–572
Peyvandi F, Scully M, Kremer Hovinga JA et al (2016) Caplacizumab for acquired thrombotic thrombocytopenic purpura. N Engl J Med 374(6):511–522
Whilding LM, Halim L, Draper B et al (2019) CAR T-cells targeting the integrin αvβ6 and co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel) 11(5):674. https://doi.org/10.3390/cancers11050674
Haider T, Sandha KK, Soni V et al (2020) Recent advances in tumor microenvironment associated therapeutic strategies and evaluation models. Mater Sci Eng C Mater Biol Appl 116:111229
Wang H, Liu H, Sun C et al (2021) Nanoparticles dual targeting both myeloma cells and cancer-associated fibroblasts simultaneously to improve multiple myeloma treatment. Pharm 13(2):274. https://doi.org/10.3390/pharmaceutics13020274
Liu Y, Castro Bravo KM, Liu J (2021) Targeted liposomal drug delivery: a nanoscience and biophysical perspective. Nanoscale Horiz 6(2):78–94
Shao J, Zaro J, Shen Y (2020) Advances in exosome-based drug delivery and tumor targeting: from tissue distribution to intracellular fate. Int J Nanomedicine 15:9355–9371
Roopkumar J, Swaidani S, Kim AS et al (2021) Increased incidence of venous thromboembolism with cancer immunotherapy - ScienceDirect. Med 2(4):423–434. https://doi.org/10.1016/j.medj.2021.02.002
Abe K, Yoshimura H, Tanaka H et al (2004) Comparison of conventional and diffusion-weighted MRI and proton MR spectroscopy in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like events. Neuroradiology 46(2):113–117
Crescente M, Mezzasoma AM, Del Pinto M et al (2011) Incomplete inhibition of platelet function as assessed by the platelet function analyzer (PFA-100) identifies a subset of cardiovascular patients with high residual platelet response while on aspirin. Platelets 22(3):179–187
Paniccia R, Antonucci E, Gori AM et al (2007) Different methodologies for evaluating the effect of clopidogrel on platelet function in high-risk coronary artery disease patients. J Thromb Haemost 5(9):1839–1847
Lele M, Sajid M, Wajih N et al (2001) Eptifibatide and 7E3, but not tirofiban, inhibit alpha(v)beta(3) integrin-mediated binding of smooth muscle cells to thrombospondin and prothrombin. Circulation 104(5):582–587
Kujovich JL Factor V (2011) Leiden thrombophilia. Genet Med 13(1):1–16. https://doi.org/10.1097/GIM.0b013e3181faa0f2
Mori J, Pearce AC, Spalton JC et al (2008) G6b-B inhibits constitutive and agonist-induced signaling by glycoprotein VI and CLEC-2. J Biol Chem 283(51):35419–35427
Perrault C, Mangin P, Santer M et al (2003) Role of the intracellular domains of GPIb in controlling the adhesive properties of the platelet GPIb/V/IX complex. Blood 101(9):3477–3484
Saidak Z, Soudet S, Lottin M et al (2021) A pan-cancer analysis of the human tumor coagulome and its link to the tumor immune microenvironment. Cancer Immunol Immunother: CII 70(4):923–933
Posch F, Riedl J, Reitter E-M et al (2020) Dynamic assessment of venous thromboembolism risk in patients with cancer by longitudinal D-Dimer analysis: a prospective study. J Thromb Haemost 18(6):1348–1356
Marchetti M, Giaccherini C, Masci G et al (2020) Thrombin generation predicts early recurrence in breast cancer patients. J Thromb Haemost 18(9):2220–2231
Pantel K, Alix-Panabières C (2019) Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol 16(7):409–424
Keller L, Pantel K (2019) Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat Rev Cancer 19(10):553–567
Funding
The present study was funded by the National Natural Science Foundation of China (82172776), Tianjin Science and Technology Plan Project (19ZXDBSY00060), Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-061B), and Diversified Input Project of Tianjin National Natural Science Foundation (21JCYBJC01770).
Author information
Authors and Affiliations
Contributions
Conceptualization, Y.-F.Z., J.-T.Z., B.Z., S.-H.B., X.-J.L., H.-Q.W., Y.C. and H.Z.; L.-L.Z., X.-H.X., S.X., Z.-Q.S. methodology, Y.-F.Z., J.-T.Z., B.Z., S.-H.B., X.-J.L., H.-Q.W., Y.C. and H.Z.; L.-L.Z., X.-H.X., S.X., Z.-Q.S.; writing—original draft preparation, Y.-F.Z.; writing—review and editing, Y.-F.Z. and J.-T.Z. contributed equally to this study. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics statement
Not applicable.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationship.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Authors Yifan Zhang, Jingtong Zeng, and Shihao Bao denotes as the first authors or first co-authors.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Y., Zeng, J., Bao, S. et al. Cancer progression and tumor hypercoagulability: a platelet perspective. J Thromb Thrombolysis 57, 959–972 (2024). https://doi.org/10.1007/s11239-024-02993-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-024-02993-0