
Abstract
Aims
In the present study, the effects of Cu (2.0 and 8.0 μM) on root growth of Allium cepa var. agrogarum L. were addressed and protein abundance levels were analyzed using the technology of proteomics combined with transcriptomics, in order to go deeper into the understanding of the mechanism of Cu toxicity on plant root systems at the protein level and to provide valuable information for monitoring and forecasting the effects of exposure to Cu in real scenarios conditions.
Methods
Protein extraction; Two-dimensional electrophoresis (2-DE) analysis; Mass spectrometry analysis; Establishment of the in-house database; Restriction enzyme map of the in-house database and protein identification.
Results
Root growth was dramatically inhibited after 12 h Cu treatment. By establishing an in-house database and using mass spectrometry analysis, 27 differentially abundant proteins were identified. These 27 proteins were involved in multiple biological processes including defensive response, transcription regulation and protein synthesis, cell wall synthesis, cell cycle and DNA replication, and other important functions.
Conclusions
Our results provide new insights at the proteomic level into the Cu-induced responses, defensive responses and toxic effects, and provide new molecular markers of the early events of plant responses to Cu toxicity. Moreover, the establishment of an in-house database provides a big improvement for proteomics research on non-model plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Copper (Cu) is an essential microelement required for plant growth and development. As one of the most crucial cofactors, Cu is responsible for the activity or structure of many metalloproteins (Burkhead et al. 2009; Yruela 2009). Cu plays an important role in photosynthesis, respiration, superoxide scavenging, cell wall metabolism and lignification (Andrés-Colás et al. 2010). Despite being essential, Cu is highly toxic to plants at elevated concentrations and interferes with numerous biochemical and physiological processes (Mostofa and Fujita 2013). Unfortunately, in the last decades, Cu contamination has accumulated in the environment due to anthropogenic activities, such as inappropriate agricultural practices, mining activities and manifold industrial uses, causing a potential threat to plants and plant consumers (Wang and Björn 2014; Mirlean et al. 2007; Saunders et al. 2013; Fujimori and Takigami 2014). The root systems of plants absorb Cu from the soil, and most plants are very sensitive to Cu toxicity.
A large number of physiological and biochemical analyses have been conducted on the responses of different plant species to Cu intoxication, but how Cu affects metabolism in plants is still not completely understood. Identification of the functional genes or proteins involved in the responses of plants to Cu is needed in order to understand the molecular mechanisms of Cu stress (Ahsan et al. 2009). Proteomics, which combines high-resolution protein separation by two-dimensional electrophoresis (2-DE) with matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry (MALDI-TOF/TOF MS), aided by improved databases, is recognized as a powerful approach for large-scale analysis and identification of proteins in many fields of plant biology (Song et al. 2013). The approach can therefore help to elucidate new aspects of plant heavy metal stress (Cvjetko et al. 2014; Hossain and Komatsu 2013). Using this method, the proteomic changes induced by Cu have been conducted in several species, including Cannabis sativa (Bona et al. 2007), Arabidopsis (Haensch and Mendel 2009), Elsholtzia splendens (Li et al. 2009), Triticum aestivum L. (Li et al. 2013), Oryza sativa L. (Song et al. 2013, 2014), cyanobacterium Cyanothece (Mota et al. 2015), Oryza sativa L. (Chen et al. 2015), and Sargassum fusiforme (Zou et al. 2015). Despite the literature available, knowledge of plant responses to Cu stress at the proteomic level is scarce, and little is known about the responses of sensitive plants to Cu stress at an early stage, especially, for non-model plants without genomic background information.
Although proteomics is a widely accepted technique, there are still some factors, primarily protein identification and functional annotation, which hinder proteomic investigation on non-model plants. Protein identification and functional annotation for non-model plants without a well-characterized genome are imprecise since one certain protein can result in very diverse functions depending on its subcellular localization, posttranslational modifications or interacting partners and the identification rate is usually low (Kosová et al. 2011; Cvjetko et al. 2014). Recently, transcriptomic analysis has been widely used to study the expression patterns of genes exposed to different stresses (Díaz-Riquelme et al. 2014; Lee et al. 2014). This technique has great potential for expanding sequence databases for non-model organisms, so that plants can now also be analyzed in functional genomic studies (Neave et al. 2012).
The Allium cepa var. agrogarum L. has various advantages as a classical material for cytological research (Qin et al. 2010), but it has been less investigated at molecular level due to the lack of genomic information. In this study, the effects of Cu (2.0 and 8.0 μM) on root growth of A. cepa var. agrogarum are investigated and protein abundance levels are analyzed by coupling proteomes with transcriptomes. We established an in-house database of A. cepa var. agrogarum roots according to the results of transcriptomic analysis in order to solve the limitations concerning proteome databases. Peptide mass fingerprinting (PMF) and MS/MS data were then submitted to this database for protein identification and functional annotation of the differentially abundant proteins under different concentrations of Cu stress. This study aims to (1) improve the proteomic approach, which could make the protein identification and functional annotation of non-model plants more efficient and accurate; and (2) improve understanding of the mechanisms of Cu toxicity on plant root systems at an early stage at the proteomic level.
Materials and methods
Culture condition and Cu treatment
Healthy and equal-sized onion (A. cepa var. agrogarum) bulbs were chosen for the experiments (average diameter: 2.5 cm). The bulbs did not show either shooting of green leaves or any growth of roots. Dry scales of bulbs were removed prior to experiments. The bulbs were grown in ultrapure water at 27 °C with the base just dipping into water, producing roots reaching about 2.0 cm length. The seedlings were then treated with 2.0 and 8.0 μM Cu solutions (Cu2+, CuSO4 · 5H2O, MW159.6097, AR) for 12 h, respectively. Ultrapure water was used as the media for the control group. In each group, the root length of five seedlings was recorded and the morphology was observed after the 12 h treatments.
Protein extraction
Roots from control and seedlings treated with Cu for 12 h were cut down and immediately frozen with liquid nitrogen. The root samples (2.0 g) were ground respectively, with chilled extraction buffer [50 mM Tris–HCl (pH 8.5), 5 mM EDTA, 100 mM KCl, 2 % (v/v) β-mercaptoethanol, 30 % (v/v) sucrose] and kept at 4 °C for 30 min. They were further ground after adding 5 mL Tris-phenol (pH 8.0). The mixture was placed in centrifuge tubes, mixed at 4 °C for 15 min, and centrifuged at 4 °C (8000 g for 3 min). The supernatant was added at four times the volume of precooled methanol containing 0.1 M ammonium acetate, and then kept still for at least 2 h or overnight. The precipitate was washed 3–5 times with precooled methanol containing 0.1 M ammonium acetate until it was white, then washed 2–3 times with precooled acetone/aqueous (4:1, v/v) and air dried. Lysis buffer [40 mM Tris–HCl (pH 7.5), 7 M Urea, 2 M Thiourea, 1 mM EDTA•Na2, 4 % (w/v) CHAPS, 1 % (w/v) DTT] was added to the precipitate, which was subjected to ultrasound for 10 min, then left standing for 2–4 h at 4 °C to ensure full dissolution in the buffer solution. The supernatant was kept at −20 °C after centrifugation (12,000 g for 5 min). Protein content of each extraction was determined according to Bradford’s method (Bradford 1976) using BSA as a standard. All groups had three biological replicates, and there were three technical replicates for each biological replicate.
Two-dimensional electrophoresis (2-DE) analysis
One mg of total protein from each protein sample was diluted with rehydration solution [7 M urea, 4 % (w/v) CHAPS, 1 % (w/v) DTT, 0.2 % (v/v) IPG buffer, 0.001 % (w/v) bromophenol blue] to 450 μL. Proteins were loaded in a 24 cm, pH 3–10 linear pH gradient IPG strip (Bio-Rad, USA) by passive rehydration for 14 h and then the strips were subjected to isoelectric focusing (20 °C, 50 μA/strip) for 23–24 h. After isoelectric focusing, the strips were equilibrated in an equilibration buffer [6 M urea, 0.375 M Tris–HCl (pH 8.8), 2 % (w/v) SDS, 20 % (v/v) glycerol, 2 % (w/v) DTT] for 15 min, and equilibrated again for another 8 min in the same buffer, except that DTT was replaced by 2.5 % (w/v) iodoacetamide. For the second electrophoretic dimension, the equilibrated IPG strips were placed at the top of 13 % (w/v) SDS-PAGE gels (dimension: 25.6 × 21.2 cm) and sealed with melted 0.5 % (w/v) agarose. The gels were run at 4 W per gel for 40 min, and then at 11 W per gel for 6 h. We performed the analysis using a multicasting gel apparatus employing an Ettan DALTsix Electrophoresis System (GE Healthcare, USA). All protein spots in 2-D gels were visualized using Coomassie Blue R-350 staining. Stained gels were scanned on an UMAX Image-Scanner and Image Master 7.0 software was used for image comparison.
Mass spectrometry analysis
The protein spots with good shape and a significant difference in abundance were excised along the staining edges from 2-D gels for further tryptic digestion and matrix-assisted laser desorption/ionization time-of-flight/time-of-flight mass spectrometry (MALDI-TOF/TOF MS) analysis. The gel spots were de-stained with 30 % (v/v) acetonitrile (ACN) containing 100 mM NH4HCO3 for 15 min. This step was repeated 2–5 times until both gel spots and the de-staining solution were colorless. After washing with ultrapure water twice, the gel spots were dehydrated with 100 % ACN for 5 min and then dried. They were then rehydrated with a 10 ng μL−1 trypsin solution containing 5 % (v/v) ACN and 50 mM (NH4)2CO3 for 45 min on ice and then digested at 37 °C for 9–11 h. The trypsin solution was kept. The digested peptides were redissolved in 20 μL solution containing 33 % (v/v) ACN and 0.1 % (v/v) trifluoroacetic acid (TFA), and then 20 μL solution containing 66 % (v/v) ACN and 0.1 % (v/v) TFA for 30 min each. Working solutions of the two steps were also kept. The above trypsin solution and working solutions were pooled and vacuum freeze-dried until the total volume reached 3–10 μL. Samples were analyzed with an AutoFlex TOF/TOF II mass spectrometer (Bruker Daltonics, Germany). In consideration of massive data calculation, 2–5 peaks were selected to perform MALDI-TOF/TOF MS analysis, which was widely accepted. The working mode was set with positive ion reflection mode, an accelerating voltage of 20 kV, and 150-ns delayed extraction time. The spectrum masses ranging from 700 to 3500 Da were acquired with laser shots at 200/spectrum. A Peptide Mixture-1 kit (Bruker Daltonics) was used for external calibration. The matrix and autolytic peaks of trypsin were used for internal calibration. Monoisotopic mass was analyzed with FlexAnalysis 2.2 (Bruker Daltonics) and automatically collected with a signal to noise ratio >4 and a peak quality index >30. The obtained MS/MS data were used for further protein identification.
Establishment of the in-house database
An experimental sample was obtained by mixing equal parts of the root RNA from the control, 2.0 μM and 8.0 μM Cu treatment groups. A total of 3 μg RNA was used as input material for the RNA sample preparation. The sequencing library was generated using NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to the sample. The library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded sample was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumia) according to the manufacturer’s instructions. After cluster generation, the library preparation was sequenced on an Illumina Hiseq 2000 platform and paired-end reads were generated. The average size of the sequenced RNA reads was 100 bp.
Raw data (raw reads) of fastq format were first processed through in-house perl scripts in which clean data (clean reads) were obtained by removing reads containing adapter, ploy-N and low quality reads from raw data. The downstream analyses were based on clean data with high quality. The left files (read1 files) from the library were pooled into one left.fq file, and right files (read2 files) into one right.fq file. Transcriptome assembly was accomplished based on the left.fq and right.fq using Trinity (Grabherr et al. 2011) with min_kmer_cov set to 2 by default and all other parameters set to the default. The assembled results were called unigenes of the mixed sample.
Gene function was annotated based on the following 7 databases: Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), Pfam (Protein family), KOG/COG (Clusters of Orthologous Groups of proteins), Swiss-Prot (A manually annotated and reviewed protein sequence database), KO (KEGG Ortholog database) and GO (Gene Ontology).
After gene functional annotation, open reading frames (ORF) of the annotated unigenes were recognized, and then the nucleotide sequences of coding regions were translated into amino acid sequences (5′ → 3′) according to the standard codon table.
Restriction enzyme map of the in-house database and protein identification
A restriction enzyme map of the in-house database was simulated by software pFind, a computational solution for mass spectrometry-based proteomics (Fu et al. 2004; Li et al. 2005; Wang et al. 2007). Trypsin was used as the proteolytic enzyme. Peptide length was set to 6–60 bp and peptide mass to 600–10,000 Da. The MS/MS data of the above-mentioned proteins were converted into an mgf file and then submitted to the restriction enzyme map for protein identification using pFind (Fu et al. 2004; Li et al. 2005; Wang et al. 2007). The error was set as 0.3 Da at the first class and 0.8 Da at the second class. The fixed modification was Carbamidomethyl and variable modification was Oxidation [M]/Phospho [S].
Statistical analysis
Analysis of variance of data was performed with SigmaPlot 8.0 software. For statistical analysis, one-way analysis of variance (ANOVA) and t-test were used to determine the significance at P < 0.05.
Results
Macroscopic effects of Cu on root growth
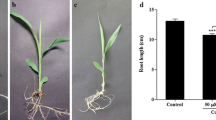
Effects of Cu on root growth of A. cepa var. agrogarum varied with Cu concentration (Fig. 1). After 12 h treatments, 2.0 or 8.0 μM Cu significantly inhibited root growth (P < 0.05). Roots grew to a length of 0.600 cm ± 0.011a cm in the control group, a length of 0.400 cm ± 0.014b cm in the 2.0 μM Cu group and only 0.100 cm ± 0.015c cm in the 8.0 μM Cu group. The effects of Cu on root morphology also varied with the Cu concentration. In media containing 2.0 μM Cu, root morphology was similar to the control roots, except for swelling of the root tips. At 8.0 μM Cu, the roots were stunted, turned soft and some were broken.

Effects of 2.0 μM or 8.0 μM Cu on root growth and morphology of A. cepa var. agrogarum (12 h exposure, n = 5)
Two-dimensional electrophoresis (2-DE) images
The 2-DE combined with MALDI-TOF/TOF MS was employed to investigate variation in the root proteome that occurred after different Cu treatments for 12 h. The protein spot number in the control 2-DE image was 2472 ± 91a, while the numbers in 2.0 and 8.0 μM Cu-treated groups were 2363 ± 24b and 2062 ± 41c respectively, based on three biological replicates for each group and three technical replicates for each biological replicate (P < 0.05) (Fig. 2).

Two-dimensional electrophoresis images of A. cepa var. agrogarum root holoproteins. (a) Control group, (b) 2.0 μM Cu treatment for 12 h, (c) 8.0 μM Cu treatment for 12 h
The 2-DE image of the control group was used as the comparison gel, and the images of the 2.0 μM and 8.0 μM Cu-treated groups were used as the analysis gels. Quantitative spot analysis revealed that 50 protein spots were classified as differentially abundant proteins (P < 0.05). Thirty-two protein spots with good shapes and a significant differential abundance were selected for mass spectrometry analysis and they have been marked in the 2-DE images (Fig. 2).
The in-house database
Transcriptome assembly was accomplished using Trinity, and 87430 unigenes were obtained. All of the 87430 unigenes were submitted to the 7 databases mentioned in the methods for functional annotation. Of these, 24305 unigenes were successfully annotated and their ORFs were identified. The corresponding 24305 amino acid sequences were also obtained (Electronic supplementary material 1). The file header gives the sequence name and other descriptive information, followed by the amino acid sequence. The length of each amino acid sequence is given in Electronic supplementary material 2. In the present investigation, the 24305 amino acid sequences in Electronic supplementary material 1 are considered as the in-house database of A. cepa var. agrogarum roots.
Data comparison results
The result of the restriction enzyme map obtained according to the in-house database of A. cepa var. agrogarum roots is a temporary file. The PMF and MS/MS data of 32 selected proteins were submitted to the restriction enzyme map for protein identification. By data comparison, 27 proteins were successfully matched (Electronic supplementary material 3 and material 4 for details) and the detailed identification of the 27 differentially abundant proteins is given in Table 1.
Enlarged view and functional annotation of the identified 27 proteins
The 2-DE images of the 27 proteins are enlarged to enable clearer observation, and the abundance and significance of difference of the 27 proteins are given in Fig. 3. These proteins were divided into five categories according to their biological functions: defensive response to Cu stress, transcriptional regulation and protein synthesis, cell wall synthesis, cell cycle and DNA replication and other important functions (Table 1 and Fig. 3).


Differentially abundant protein spots involved in different functional categories. The spot numbers were shown on the left. The numbers in the histograms were %Vol of each protein spot which was calculated as a measure of protein abundance. “0” represented no abundance. Values with different letters differed significantly from each other (n = 3, P < 0.05). Bars represented standard errors
Figure 3A shows the proteins involved in defensive responses to Cu stress: antioxidation (proteins 124, 257, 318, 345, 387 and 803) (Fig. 3A-a), detoxification (proteins 291, 318, 387 and 503) (Fig. 3A-b) and osmotic adjustment (proteins 803 and 1934) (Fig. 3A-c). The abundance of all the above proteins in roots exposed to 8.0 μM Cu for 12 h was significantly increased (P < 0.05) when compared with the control, and the abundance of some of these proteins was also increased following 12 h of 2.0 μM Cu treatment when compared to the control (P < 0.05). Protein 803 abundance was increased progressively with the increase in Cu concentration.
Proteins 87, 641, 29, and 740 were involved in transcriptional regulation and protein synthesis (Fig. 3B). The protein 87 abundance in roots exposed to 2.0 μM Cu did not clearly differ from the control, but the abundance in the 8.0 μM Cu-treated group was increased significantly (P < 0.05). Compared with the control, protein 641 treated with 2.0 μM Cu was significantly up-regulated and when treated with 8.0 μM Cu it was dramatically down-regulated (P < 0.05). For proteins 29 and 740, there was no obvious difference in the abundance between the 2.0 μM Cu-treated group and control, but the abundance of the two proteins in roots exposed to 8.0 μM Cu was decreased significantly in comparison with the control (P < 0.05).
Proteins 607, 614, 1554, 819, and 1570 were involved in cell wall synthesis (Fig. 3C). For proteins 607, 614 and 1554, there was no obvious difference in the abundance between the 2.0 μM Cu treatment group and control, while the three proteins were significantly down-regulated in the 8.0 μM Cu treatment group versus control (P < 0.05). Proteins 819 and 1570 showed the same trends as those of protein 641. Proteins 819 and 1554 had no abundance in the 8.0 μM Cu-treated group.
Proteins 659, 665 and 1165 were involved in cell cycle and DNA replication (Fig. 3D). The abundance of proteins 659 and 1165 in roots exposed to 2.0 μM Cu was not different from that in the control group, but it was significantly down-regulated in the 8.0 μM Cu treatment group (P < 0.05). Protein 665 abundance was decreased progressively with increasing Cu concentrations (P < 0.05).
There were also some proteins involved in other important functions (49, 283, 386, 480, 1310, and 1373) (Fig. 3E). Protein 49 was significantly down-regulated after both 2.0 μM and 8.0 μM Cu treatments (P < 0.05). Protein 283 abundance was decreased progressively with increasing Cu concentration (P < 0.05). For protein 386, there was no obvious difference in the abundance between 2.0 μM Cu-treated group and the control, while it exhibited a greater increase in response to 8.0 μM Cu stress (P < 0.05). Abundance of proteins 480, 1310, and 1373 showed the same trends as proteins 659 and 1165.
A Venn diagram was introduced to illustrate the differential abundance of proteins among the control, 2.0 and 8.0 μM Cu treatment groups (Fig. 4).

Venn diagram showing the number of differential abundance of proteins between every two groups and the number of joint differential abundance of proteins. Two represents proteins 49 and 387; Six represents proteins 283, 641, 665, 803, 819 and 1570; Nineteen represents proteins 29, 87, 124, 257, 291, 318, 345, 386, 480, 503, 607, 614, 659, 740, 1165, 1310, 1373, 1554 and 1934
Discussion
Yang et al. (2013) have studied the spatial distribution of toxic elements in soils of Guangdong China. Their results showed that in some places, Cu concentration exceeds 40 mg kg−1, while the Chinese national standard for Cu is 22 mg kg−1 (GB 15618-1995). Heavy metal Cu pollution has been a very serious environmental problem in Guangdong province of China. In order to improve understanding of the toxic effects of Cu on plant growth, we used the Cu solution to conduct the experiments in the laboratory. Different plant species have different tolerances to Cu stress. The roots of A. cepa var. agrogarum are very sensitive to Cu toxicity. In the preliminary tests (Qin et al. 2015), when the roots reached approximately 2 cm in length, they were treated with a series of different concentrations of Cu solutions. In each treatment group, seedlings were observed for root growth and morphology every 12 h. Comparing the results, we found that the effects of Cu on root growth and morphology varied with the Cu concentration and treatment time. After 2.0 μM Cu treatment for 12 h, root growth was inhibited by half compared to the control; after 8.0 μM Cu treatment for 12 h, roots only grew to a length of 0.100 cm ± 0.015c. Among a series of Cu concentrations, the toxic effects were the most obvious in the 2.0 and 8.0 μM Cu treatment groups and were different from each other. Therefore, we chose the two dosages and treatment time to conduct the experiments. In addition, related previous work was introduced by Qin et al. (2015).
In recent years, several studies concerning plant proteome response to Cu stress have been published. Plant proteome response to Cu stress is a diverse process which is dependent on stress intensity, stress duration and plant species. Some protein response pathways are shared by different plant species under Cu stress, while others are distinct.
Reactive oxygen species (ROS), such as O2 · −, H2O2 and OH*, can oxidatively damage biological molecules including DNA, RNA, protein and lipid (Shah et al. 2001). Stress conditions can increase the production of ROS, and meanwhile activate protective enzymatic and non-enzymatic mechanisms to scavenge ROS (Zhang et al. 2009). Our results indicate that the enzymatic system in the roots of A. cepa var. agrogarum plays an important role in ROS defense at an early stage of Cu stress, including proteins 124 Copper/zinc superoxide dismutase (Cu-Zn SOD), 257 Phospholipid hydroperoxide glutathione peroxidase (PHGPx), 318 and 387 Glutathione S-transferase T1 (GSTT1), 345 Glutaredoxin (GRX) and 803 Annexin D1 (Anx D1). The major functions of these proteins are given in Table 2. Coordinated up-regulation of these proteins can scavenge excess ROS to some extent, reducing the extent of oxidative damage to roots. In addition, proteins 318 and 387 were both identified as GSTT1. Analysis of their physicochemical properties and locations in 2D images suggests that protein 318 is possibly a fragment formed by the slight degradation of 387 during the experiment. In addition to antioxidation, proteins 291 Glyoxalase I (GLO I), 503 NADH-cytochrome b5 reductase-like protein (B5R), 318 and 387 GSTT1, whose major functions are given in Table 2, form part of the detoxification system of A. cepa var. agrogarum roots in response to early Cu stress. We assume that after treatment with 8.0 μM Cu, coordinated up-regulation of these proteins can mitigate Cu toxic effects on cells to some extent. Singla-Pareek et al. (2006) demonstrated that overexpression of glyoxalase genes in tobacco and rice led to increased tolerance to heavy metals. Our results are consistent with the findings of Chen et al. (2015) who demonstrated that six significant up-regulated proteins involved in Cu antioxidant defense and detoxification were observed in both rice varieties (Cu-tolerant B1139 and Cu-sensitive B1195). Proteins 1934 Pyrroline-5-carboxylate reductase (P5CR) and 803 Anx D1 are predicted to play an important role in osmotic adjustment (Table 2). In this study, P5CR was not detected in the control and 2.0 μM Cu-treated groups but it was abundantly induced in 8.0 μM Cu-treated roots. Thus a high concentration Cu has a more serious toxic effect on roots.
Transcriptional regulation and protein synthesis are both complex biological processes. Table 2 displays that several crucial proteins associated with the two processes show a differential abundance after 12 h Cu treatments. The higher abundance of protein 87 Nucleoside diphosphate kinase (NDPK) indicates that under high concentration Cu treatment, RNA biosynthesis becomes more active. We suggest that this is due to Cu stimulation of the expression of some genes. Protein 641 was identified as Ran-binding protein 1 (RanBP1). Our results suggest that low concentration Cu treatment is conducive to RNA and protein export so as to actively defend against Cu stress. However, the high concentration Cu treatment hinders the process. Chen et al. (2015) also observed two proteins involved in transcriptional regulation, CHP-rich zinc finger protein-like and putative cold shock protein-1, whose abundance was changed in excess Cu-stressed roots of two rice (Oryza sativa L.) varieties. Proteins 29 and 740 were respectively identified as 40S ribosomal protein S21 (40S RPS21) and Elongation factor 1-beta (EF1β). After 8.0 μM Cu treatment, the abundance of both these proteins was decreased significantly, which would inhibit protein synthesis. The results here are consistent with the previous findings. For example, Zou et al. (2015) suggested that for the marine brown algae Sargassum fusiforme, protein destination-related proteins 26S proteasome beta type 7 subunit and FtsH protease were strongly inhibited under acute Cu stress. Chen et al. (2015) also reported that the abundance of several proteins (translation initiation factor, putative elongation factor EF-2, protein disulfide isomerase, cathepsin B-like cysteine protease, etc.) involved in protein synthesis, modification and degradation was altered in excess Cu-stressed roots of two rice (Oryza sativa L.) varieties.
Proteins involved in cell wall synthesis are as follows and their functions are given in Table 2. Proteins 607, 614 and 1554 were all identified as Rhamnose biosynthetic enzyme 1 (RHM1), and abundance of the three proteins had the same changing trend after Cu treatment. Inhibition of rhamnose biosynthesis would compromise root morphology. In addition, by analyzing the physicochemical properties and locations in 2D images of proteins 607, 614 and 1554, it is suggested that 607 and 614 are possibly formed by different post-translation modifications or genetic isoforms and 1554 may be the isozyme of 607 or 614. Proteins 819 and 1570 were identified as Cinnamoyl-CoA reductase 1 (CCR1) and 5-methyltetrahydropteroyltriglutamate--homocysteine methyltransferase (5-MHM) respectively. Abundance of the two proteins was increased after 2.0 μM Cu treatment for 12 h, which would be beneficial to lignin synthesis. Accordingly, it is considered that roots can prevent cell wall collapse and reduce Cu absorption by increasing the degree of lignification under low concentration Cu. Under the 8.0 μM Cu treatment, these two proteins were significantly down-regulated, and abundance of 819 was completely suppressed, which would inhibit lignin synthesis and lead to a decrease in mechanical strength and density of the cell wall. The results here are in agreement with the observations by Chen et al. (2015) and Song et al. (2013, 2014), and explain the morphological changes of roots elicited by different concentrations of Cu mentioned in the results.
Proteins 659/665 Methylenetetrahydrofolate reductase (MTHFR) and 1165 Proliferation-associated 2 g4 (PA2g4) are involved in the cell cycle and DNA replication (Table 2). Our results showed that MTHFR abundance was decreased under different concentrations of Cu treatments. Previous research has shown that the knock-down of MTHFR could decrease cell survival and result in cell cycle arrest (Sun et al. 2008). Based on the analysis of the physicochemical properties and locations in 2D images of proteins 659 and 665, it is suggested that they are possibly formed by different post-translation modifications or genetic isoforms. Protein 1165 PA2g4 abundance was decreased significantly under 8.0 μM Cu treatment, which would suppress cell proliferation. These results provide further explanations for the phenomena we observed earlier that Cu decreased mitotic index, increased DNA damage and inhibited DNA synthesis (Qin et al. 2015).
Some proteins involved in other important functions were found (Table 2). The abundance of protein 49 Thioredoxin Clot (TRX) in roots was reduced after Cu stress, which would affect its normal full spectrum of different functions. The result here is different from the previous finding of Song et al. (2013) which indicated that excess Cu treatment significantly increased TRX abundance in the rice varieties B1139 and B1195, and they suggested that up-regulation of TRX by Cu stress, along with the antioxidant systems, may had an important role in protection against oxidative stress. Protein 283 was identified as Translationally controlled tumor protein (TCTP). Its abundance was decreased with increasing Cu concentrations, which would affect the stability of microtubules (MTs), a similar observation to another proteomic study (Li et al. 2009). The result here provides further explanation for the phenomenon we observed earlier that exposure to Cu substantially impaired MTs arrangements (Qin et al. 2015). As for protein 386 Triosephosphate isomerase, cytosolic (TPI), its abundance was increased in the 8.0 μM Cu-treated roots. We suggest that the roots prevent Cu damage by increasing available energy (ATP). Contreras et al. (2010) also demonstrated that the energy metabolic pathway related protein was over-expressed in the marine alga Scytosiphon gracilis after Cu stress. Li et al. (2009) suggested that plant energy metabolism was highly affected by Cu stress and the changes of the related proteins possibly re-optimized this system to alleviate Cu stress. Protein 1373 was identified as T-complex protein 1 subunit beta (TCP-1-beta). After 8.0 μM Cu treatment for 12 h, its abundance was decreased dramatically, which presumably inhibits the folding and assembly of proteins to some extent. This result is similar to the observations by Li et al. (2009). In addition, proteins 480 and 1310 were identified respectively as the Glutamine amidotransferase domain (GATase domain) and the K Homology domain (KH domain). Under 8.0 μM Cu treatment, the down-regulation of both domains would be adverse to the integral structure of the proteins mentioned above and thereby disturb their functions.
Establishing the in-house database of A. cepa var. agrogarum roots is highly significant. Firstly, for proteomics research on non-model plants, protein identification has generally been performed by comparison with the protein data banks, so the comparison range is large with poor accuracy and low identification rate. The in-house database was established according to the unigene sequences of A. cepa var. agrogarum roots. Therefore the comparison range is considerably narrowed with an increased accuracy and identification rate. In this study, the identification rate reached 84 %. Secondly, past protein functional annotation of non-model plants is achieved using the known protein functions of other species in the protein data banks. However, protein functional implementation requires a series of posttranslational processing steps. As a result, even though there is a high sequence matching rate between the target protein and a known protein in the protein data banks, they may perform different functions. In contrast, when comparing against our in-house database, the target protein functional annotation is achieved according to its matched unigene function. Because functional annotation at the genetic level is more conservative and stable, it achieves a higher confidence. Thus, the establishment of the in-house database can provide more accurate and intrinsically functional annotations for target proteins. Thirdly, amino acid sequences of the target proteins can be predicted by the in-house database. Although the predicted result is not 100 % correct, it does have a high reference value.
Conclusions
Based on the results of the present research, we draw Fig. 5 to display the early molecular responses to Cu stress in the roots of A. cepa var. agrogarum. Defensive responses and toxic effects are detected after Cu treatments at different concentrations for 12 h. We conclude that under low concentration Cu stress, roots employ some defensive strategies to effectively resist Cu stress. However, under high concentration Cu stress, although there are also certain defensive responses, they are not sufficient to resist the Cu stress. As a result, Cu causes damage to several important biological processes. Moreover, the in-house database, established according to the unigene sequences of A. cepa var. agrogarum roots, can provide more accurate and intrinsic functional annotations for target proteins and elevate protein identification rate. The establishment of the in-house database is without doubt a big improvement for proteomics research of non-model plants.

Schematic diagram for early molecular events in Cu-induced responses in the roots of A. cepa var. agrogarum. (a) Defensive responses, (b) Toxic effects
References
Adams E, Frank L (1980) Metabolism of proline and the hydroxyprolines. Annu Rev Biochem 49:1005–1061
Ahsan N, Renaut J, Komatsu S (2009) Recent developments in the application of proteomics to the analysis of plant responses to heavy metals. Proteomics 9:2602–2621
Andrés-Colás N, Perea-García A, Puig S, Peňarrubia L (2010) Deregulated copper transport affects Arabidopsis development especially in the absence of environmental cycles. Plant Physiol 153:170–184
Arcuri F, Papa S, Carducci A, Romagnoli R, Liberatori S, Riparbelli MG, Sanchez JC, Tosi P, del Vecchio MT (2004) Translationally controlled tumor protein (TCTP) in the human prostate and prostate cancer cells: expression, distribution, and calcium binding activity. Prostate 60:130–140
Avis JM, Clarke PR (1996) Ran, a GTPase involved in nuclear process: its regulators and effectors. J Cell Sci 109:2423–2427
Bona E, Marsano F, Cavaletto M, Berta G (2007) Proteomic characterization of copper stress response in Cannabis sativa roots. Proteomics 7:1121–1130
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Burkhead JL, Reynolds KAG, Abdel-Ghany SE, Cohu CM, Pilon M (2009) Copper homeostasis. New Phytol 182:799–816
Chen C, Song YF, Zhuang K, Li L, Xia Y, Shen ZG (2015) Proteomic analysis of copper-binding proteins in excess copper-stressed roots of two rice (Oryza sativa L.) varieties with different Cu tolerances. PLoS One 10:e0125367
Contreras L, Moenne A, Gaillard F, Potin P, Correa JA (2010) Proteomic analysis and identification of copper stress-regulated proteins in the marine alga Scytosiphon gracilis (Phaeophyceae). Aquat Toxicol 96:85–89
Cvjetko P, Zovko M, Balen B (2014) Proteomics of heavy metal toxicity in plants. Arh Hig Rada Toksikol 65:1–18
Díaz-Riquelme J, Martínez-Zapater JM, Carmona MJ (2014) Transcriptional analysis of tendril and inflorescence development in Grapevine (Vitis vinifera L.). PLoS One 9:e92339
Dorion S, Clendenning A, Jeukens J, Salas JJ, Parveen N, Haner AA, Law RD (2012) A large decrease of cytosolic triosephosphate isomerase in transgenic potato roots affects the distribution of carbon in primary metabolism. Planta 236:1177–1190
Doucey MA, Bender FC, Hess D, Hofsteenge J, Bron C (2006) Caveolin-1 interacts with the chaperone complex TCP-1 and modulates its protein folding activity. Cell Mol Life Sci 63:939–948
Fu Y, Yang Q, Sun RX, Li DQ, Zeng R, Ling CX, Gao W (2004) Exploiting the kernel trick to correlate fragment ions for peptide identification via tandem mass spectrometry. Bioinformatics 20:1948–1954
Fujimori T, Takigami H (2014) Pollution distribution of heavy metals in surface soil at an informal electronic-waste recycling site. Environ Geochem Health 36:159–168
Gao Y, Chen XB, Zhang ZY (2007) Advances in research on lignin biosynthesis and its molecular regulation. Biotechnol Bull 2:47–51
Giri PK, Jing-Song F, Shanmugam MK, Ding JL, Sethi G, Swaminathan K, Sivaraman J (2012) NMR structure of Carcinoscorpius rotundicauda thioredoxin-related protein 16 and its role in regulating transcription factor NF-κB activity. J Biol Chem 287:29417–29428
Gorecka KM, Konopka-Postupolska D, Hennig J, Buchet R, Pikula S (2005) Peroxidase activity of annexin 1 from Arabidopsis thaliana. Biochem Bioph Res Commun 336:868–875
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD, Chen ZH, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Haensch R, Mendel RR (2009) Physiological functions of mineral micronutrients (Cu, Zn, Mn, Fe, Ni, Mo, B, Cl). Curr Opin Plant Biol 12:259–266
Hollingworth D, Candel AM, Nicastro G, Martin SR, Briata P, Gherzi R, Ramos A (2012) KH domains with impaired nucleic acid binding as a tool for functional analysis. Nucleic Acids Res 40:6873–6886
Hossain Z, Komatsu S (2013) Contribution of proteomic studies towards understanding plant heavy metal stress response. Front Plant Sci 3(article 310):1–12
Hwang J, Oh CS, Kang BC (2013) Translation elongation factor 1B (eEF1B) is an essential host factor for Tobacco mosaic virus infection in plants. Virology 439:105–114
Izuta S, Kitahara M, Hamaguchi T (2004) Regulation of eukaryotic DNA replication by proliferation associated protein, PA2G4, in vitro. Nucleic Acids Symp Ser 48:285–286
Khedr AHA, Abbas MA, Wahid AAA, Quick WP, Abogadallah GM (2003) Proline induces the expression of salt stress responsive proteins and may improve the adaptation of Pancratium maritimum L. to salt-stress. J Exp Bot 54:2553–2562
Kim TH, Lee BR, Jung WJ, Kim KY, Avice JC, Ourry A (2004) De novo protein synthesis in relation to ammonia and proline accumulation in water stressed white clover. Funct Plant Biol 31:847–855
Kopczewski T, Kuźniak E (2013) Redox signals as a language of interorganellar communication in plant cells. Cent Eur J Biol 8:1153–1163
Kosová K, Vítámvás P, Prášil IT, Renaut J (2011) Plant proteome changes under abiotic stress — contribution of proteomics studies to understanding plant stress response. J Proteomics 74:1301–1322
Lauvergeat V, Lacomme C, Lacombe E, Lasserre E, Roby D (2001) Two cinnamoyl-CoA reductase (CCR) genes from Arabidopsis thaliana are differentially expressed during development and in response to infection with pathogenic bacteria. Phytochemistry 57:1187–1195
Lee J, Lee H, Noh EK, Park M, Park H, Kim JH, Kim IC, Yim JH (2014) Expression analysis of transcripts responsive to osmotic stress in Deschampsia antarctica Desv. Genes Genom 36:283–291
Lee S, Lee EJ, Yang EJ, Lee JE, Park AR, Song WH, Park OK (2004) Proteomic identification of annexins, calcium-dependent membrane binding proteins that mediate osmotic stress and abscisic acid signal transduction in Arabidopsis. Plant Cell 16:1378–1391
Li DQ, Fu Y, Sun RX, Ling CX, Wei YG, Zhou H, Zeng R, Yang Q, He S, Gao W (2005) pFind: a novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics 21:3049–3050
Li F, Shi JY, Shen CF, Chen GC, Hu SP, Chen YX (2009) Proteomic characterization of copper stress response in Elsholtzia splendens roots and leaves. Plant Mol Biol 71:251–263
Li GZ, Peng XQ, Xuan HM, Wei LT, Yang YY, Guo TC, Kang GZ (2013) Proteomic analysis of leaves and roots of common wheat (Triticum aestivum L.) under copper-stress conditions. J Proteome Res 12:4846–4861
Mirlean N, Roisenberg A, Chies JO (2007) Metal contamination of vineyard soils in wet subtropics (Southern Brazil). Environ Pollut 149:10–17
Mostofa MG, Fujita M (2013) Salicylic acid alleviates copper toxicity in rice (Oryza sativa L.) seedlings by up-regulating antioxidative and glyoxalase systems. Ecotoxicology 22:959–973
Mota R, Pereira SB, Meazzini M, Fernandes R, Santos A, Evans CA, Philippis RD, Wright PC, Tamagnini P (2015) Effects of heavy metals on Cyanothece sp. CCY 0110 growth, extracellular polymeric substances (EPS) production, ultrastructure and protein profiles. J Proteomics 120:75–94
Neave MJ, Streten-Joyce C, Nouwens AS, Glasby CJ, McGuinness KA, Parry DL, Gibb KS (2012) The transcriptome and proteome are altered in marine polychaetes (Annelida) exposed to elevated metal levels. J Proteomics 75:2721–2735
Nishi R, Hashimoto H, Uchimiya H, Kato A (1993) The primary structure of two proteins from the small ribosomal subunit of rice. Biochim Biophys Acta Gene Struct Expr 1216:113–114
Oka T, Nemoto T, Jigami Y (2007) Functional analysis of Arabidopsis thaliana RHM2/MUM4, a multidomain protein involved in UDP-D-glucose to UDP-L-rhamnose conversion. J Biol Chem 282:5389–5403
Ou CY, Stevenson RE, Brown VK, Schwartz CE, Allen WP, Khoury MJ, Rozen R, Oakley GP Jr, Adams MJ Jr (1996) 5, 10 methylenetetrahydrofolate reductase genetic polymorphism as a risk factor for neural tube defects. Am J Med Genet 63:610–614
Parker CC, Parker ML, Smith AC, Waldron KW (2001) Pectin distribution at the surface of potato parenchyma cells in relation to cell-cell adhesion. J Agric Food Chem 49:4364–4371
Qin R, Jiao YQ, Zhang SS, Jiang WS, Liu DH (2010) Effects of aluminum on nucleoli in root tip cells and selected physiological and biochemical characters in Allium cepa var. agrogarum L. BMC Plant Biol 10:225
Qin R, Wang CY, Chen D, Björn LO, Li SS (2015) Copper induced root growth inhibition of Allium cepa var. agrogarum L. involves disturbances in cell division and DNA damage. Environ Toxicol Chem 34:1045–1055
Qin XR, Zhang MJ, Wu L (2012) Purification and characterization of Cu, Zn superoxide dismutase from pumpkin (Cucurbita moschata) pulp. Eur Food Res Technol 235:1049–1054
Quintero-Reyes IE, Garcia-Orozco KD, Sugich-Miranda R, Arvizu-Flores AA, Velazquez-Contreras EF (2012) Shrimp oncoprotein nm23 is a functional nucleoside diphosphate kinase. J Bioenerg Biomembr 44:325–331
Ravanel S, Block MA, Rippert P, Jabrin S, Curien G, Rebeille F, Douce R (2004) Methionine metabolism in plants: chloroplasts are autonomous for de novo methionine synthesis and can import S-adenosylmethionine from the cytosol. J Biol Chem 279:22548–22557
Saunders KM, Harrison JJ, Butler ECV, Hodgson DA, McMinn A (2013) Recent environmental change and trace metal pollution in World Heritage Bathurst Harbour, southwest Tasmania, Australia. J Paleolimnol 50:471–485
Shah K, Kumar RG, Verma A, Dubey RS (2001) Effect of cadmium on lipid peroxidation, superoxide anion generation and activities of antioxidant enzymes in growing rice seedlings. Plant Sci 161:1135–1144
Singla-Pareek SL, Yadav SK, Pareek A, Reddy MK, Sopory SK (2006) Transgenic tobacco overexpressing glyoxalase pathway enzymes grow and set viable seeds in zinc spiked soils. Plant Physiol 140:613–623
Song YF, Cui J, Zhang HX, Wang GP, Zhao FJ, Shen ZG (2013) Proteomic analysis of copper stress responses in the roots of two rice (Oryza sativa L.) varieties differing in Cu tolerance. Plant Soil 366:647–658
Song YF, Zhang HX, Chen C, Wang GP, Zhuang K, Cui J, Shen ZG (2014) Proteomic analysis of copper-binding proteins in excess copper-stressed rice roots by immobilized metal affinity chromatography and two-dimensional electrophoresis. Biometals 27:265–276
Su YP, Lu XD, Shen SD, Zhang CY (2011) GATL 12 is essential for chloroplast biogenesis in Arabidopsis. Chin Bull Bot 46:379–385
Sugimoto M, Sakamoto W (1997) Putative phospholipid hydroperoxide glutathione peroxidase gene from Arabidopsis thaliana induced by oxidative stress. Genes Genet Syst 72:311–316
Sun DF, Weng YR, Chen YX, Lu R, Wang X, Fang JY (2008) Knock-down of methylenetetrahydrofolate reductase reduces gastric cancer cell survival: an in vitro study. Cell Biol Int 32:879–887
Tan YF, O’Toole N, Taylor NL, Millar AH (2010) Divalent metal ions in plant mitochondria and their role in interactions with proteins and oxidative stress-induced damage to respiratory function. Plant Physiol 152:747–761
Wagner U, Edwards R, Dixon DP, Mauch F (2002) Probing the diversity of the Arabidopsis glutathione S-transferase gene family. Plant Mol Biol 49:515–532
Wang LH, Li DQ, Fu Y, Wang HP, Zhang JF, Yuan ZF, Sun RX, Zeng R, He SM, Gao W (2007) pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun Mass Spectrom 21:2985–2991
Wang YT, Björn LO (2014) Heavy metal pollution in Guangdong Province, China, and the strategies to manage the situation. Front Environ Sci 2(article 9):1–12
Yan SP, Tang ZC, Su WA, Sun WN (2005) Proteomic analysis of salt stress-responsive proteins in rice root. Proteomics 5:235–244
Yang CL, Guo RP, Yue L, Zhou K, Wu ZF (2013) Environmental quality assessment and spatial pattern of potentially toxic elements in soils of Guangdong Province, China. Environ Earth Sci 70:1903–1910
Yruela I (2009) Copper in plants: acquisition, transport and interactions. Funct Plant Biol 36:409–430
Zhang SS, Zhang HM, Qin R, Jiang WS, Liu DH (2009) Cadmium induction of lipid peroxidation and effects on root tip cells and antioxidant enzyme activities in Vicia faba L. Ecotoxicology 18:814–823
Zou HX, Pang QY, Zhang AQ, Lin LD, Li N, Yan XF (2015) Excess copper induced proteomic changes in the marine brown algae Sargassum fusiforme. Ecotoxicol Environ Saf 111:271–280
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31070242), the Leading Scientists Project of Guangdong Province, the Guangdong Pearl River Scholar Funded Scheme (2012), the Research Fund for the Doctoral Program of Higher Education of China (20114407110006), the Science and Technology Program of Guangzhou, China (2014J4100053) and the Scientific Research Foundation of Graduate School of South China Normal University (2012kyjj114). The authors are grateful to Dr. Rachel Naele (QIMR Berghofer Medical Research Institute, Australia), Dr. Andy McLeod (The University of Edinburgh, Edinburgh, UK) and Prof. John Richard Schrock (Emporia State University, Kansas, USA) for language revision.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
We declare that we have no conflict of interest.
Additional information
Responsible Editor: Henk Schat.
Rong Qin and Chanjuan Ning contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic supplementary material 1
(PDF 48292 kb)
Electronic supplementary material 2
(PDF 5493 kb)
Electronic supplementary material 3
(PDF 273 kb)
Electronic supplementary material 4
(PDF 143 kb)
Rights and permissions
About this article

Cite this article
Qin, R., Ning, C., Björn, L.O. et al. Proteomic analysis of Allium cepa var. agrogarum L. roots under copper stress. Plant Soil 401, 197–212 (2016). https://doi.org/10.1007/s11104-015-2741-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11104-015-2741-9