
Abstract
Deschampsia antarctica Desv., the only monocot species thriving in the tough conditions of the Antarctic region, is surrounded by regions with snow and ice where available amount of liquid water is very limited. Considering the species as an invaluable resource for the identification of genes associated with tolerance to osmotic stress, we have characterized the physiological response and the changes in gene expression induced by PEG treatment using a cDNA macroarray. Since relative chlorophyll contents of leaves decreased to 69 %, proline levels were changed significantly, and expression levels of dehydrin and late embryogenesis abundant protein were significantly up-regulated at 1 day after PEG treatment, we used corresponding plant samples as the material for gene expression analysis. As a result of hybridization and analysis of cDNA macroarray, we identified 44 and 20 transcripts that were up-regulated and down-regulated by PEG treatment respectively, and they were divided into several functional categories. Most of the up-regulated transcripts are involved in ‘Response to stimulus’, ‘Amino acid metabolism’, and ‘Respiration’. On the other hand, most of the down-regulated transcripts are involved in ‘Photosynthesis’ and ‘Translation’. Among the up-regulated transcripts, ∆-1-pyrroline-5-carboxylate synthase 2 gene has shown the highest expression level at 1 day after PEG treatment suggesting its possible role as a main player for proline biosynthesis in D. antarctica under osmotic stress. Collectively, these data suggest that the transcripts differentially expressed under osmotic stress may represent candidate genes for dissecting molecular mechanism of adaptation to extreme environment for plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Antarctic has the most extreme environment for terrestrial plants due to large amount of ice cover, low temperature, extreme photoperiod, and low annual precipitation (Alberdi et al. 2002). Deschampsia antarctica, the Antarctic hairgrass, is the only natural grass species growing in the maritime Antarctic (Edwards and Lewis-Smith 1988). Since the natural habitat of D. antarctica is surrounded by snow and ice, available amount of liquid water is very limited. Moreover, strong and desiccant wind lower chances of water uptake from the air or the soil. Accordingly, D. antarctica might be an ideal model to study physiological adaptation and response of plants to water stresses.
Previous transcriptome studies have adopted various methods to understand the molecular mechanism for plant stress responses and to discover novel stress-responsive genes. In case of monocot plants under water stress, cDNA macroarray of 2,500 maize expressed sequence tags (ESTs) was used to investigate gene expression in maize kernel in response to water stress (Andjelkovic and Thompson 2006). Suppression subtractive hybridization cDNA library construction and clone sequencing enabled identification of functional candidate genes for drought tolerance in rice (Fu et al. 2007) and in silico screening of stress responsive genes in common wheat resulted in tissue expression mapping of a large number of differential ESTs (Mochida et al. 2006). Microarray is also powerful owing to its high sensitivity and high-throughput capacity. Customized microarray composed of 1,654 cDNA clones was used to profile barley transcriptome under dehydration shock and drought stress (Talamè et al. 2007), and Affimetrix 50 K GeneChip was used for genome-wide gene expression profiling of drought responsiveness in rice (Wang et al. 2011). In recent years, transcriptome of Sorghum bicolor in response to osmotic stress was unveiled using next generation sequencing technology of RNA-seq (Dugas et al. 2011). Despite such a remarkable development of technology, cDNA macroarray still has advantages when working with mid-scale experiment with low cost.
We have obtained over 2,000 ESTs from D. antarctica, and compared gene expression profiles between Antarctic field and greenhouse grown plant samples in previous study (Lee et al. 2008). However the cause of differential gene expression could not be specified because of complex state of Antarctic environment. The aim of this study is to isolate the genes differentially expressed under PEG treatment using cDNA macroarray, thereby providing more information to understand the genetic control of response to adaptation of D. antarctica under osmotic stress, and to Antarctic environment.
Materials and methods
Plant materials, growth conditions and stress treatment
Deschampsia antarctica Desv. (Poaceae) plants were collected in the near of the Korean King Sejong Antarctic Station (62°14′29′′S; 58°44′18′′W), on the Barton Peninsula of King George Island and then transported to the laboratory for in vitro culture at Korea Polar Research Institute, where they were cultivated in a growing chamber on MS-sucrose (2 %) agar medium at 15 °C with 16 h light/8 h dark cycle with a light intensity of 150 μmol m−2 s−1. For PEG treatment, five seedlings of 5 cm tillers grown in liquid medium were transferred to osmotic test solution for the indicated times with continuous shaking. Osmotic test solutions were prepared as −0.6 MPa by dissolving PEG6000 in MS medium using the formulas of Michel and Kaufmann (1973). Experiments were replicated biologically three times.
Physiological response analysis
Fresh weight (FW) was measured immediately after harvesting and dry weight (DW) was determined after drying the tissue for 10 min at 105 °C and then at 80 °C until constant mass was obtained. Water content (WC, %) was calculated as follows: WC = 100 × (FW − DW)/FW. Total chlorophyll content in leaves was analyzed according to Lichtenthaler methodology (Lichtenthaler and Wellburn 1983). Briefly, 0.1 g of leaves was homogenized with 2 ml of 80 % acetone, maintaining the mortar on ice. Samples were centrifuged at 4 °C for 10 min at 10,000 g. Then 50 ml of the supernatant was mixed with 1 ml of 80 % acetone and absorbance was determined at 663.6 and 646.6 nm. Total chlorophyll content was calculated using the formula: Chla+b = 17.76A646.6 + 17.34A663.6 (Porra et al. 1989).
cDNA macroarray preparation and hybridization
All plasmid DNA from 1,199 recombinant clones from previous study (Lee et al. 2008) were stored in 96-well plates. The cDNA inserts were amplified by PCR machine using PCR primers specific to own plasmid backbone. The PCR reactions contained 5 μl of 10× PCR buffer, 5 μM of each T3 and T7 primer, 4 μl of dNTP mix (2.5 mM each), 1 unit of Taq DNA polymerase (5 U/μl, Biomedic, Korea), 1 ng of plasmid DNA, and sterile water up to 50 μl. Thermocycling condition was as follows: an initial denaturation at 94 °C for 5 min followed by 30 cycles of 94 °C for 10 s, 55 °C for 30 s, and 72 °C for 2 min, and a final extension at 72 °C for 7 min. The PCR products were electrophoresed on a 1 % agarose gel to confirm the amplification quality and quantity. One microliter of purified PCR products was dotted onto Hybond-N+ nylon membranes (Amersham, Pscataway, NJ, USA) using a robotic device (CAS-1200, Corbett Robotics, Australia). Two identical membranes with cDNA arrays were prepared for 2 different screening probes labeled by digoxigenin (DIG) using the DIG labeling and detection kit (Roche, Indianapolis, IN, USA). Hybridization and detection were carried out following the method described by manufacturer. Each membrane was stripped after hybridization and used for repeated experiment with different probe.
cDNA macroarray data acquisition and analysis
Hybridization images were acquired using LAS-3000 and data analysis was performed with GenePix pro software (Axon Instruments Inc., Foster City, CA, USA). Signal values were initially normalized during the image scanning process to adjust the average ratios between two channels. The overall intensity of the hybridized slide was then normalized by GenePix pro software. Those spots flagged ‘‘Bad’’ or ‘‘Not Found’’ were removed and only those spots that showed fluorescent intensity levels above the threefold background in at least one experiment were input to Acuity 4 software for further analysis. Signal values for each spot were calculated, using the median intensity of pixels minus the median local background for each channel. Student’s t test for paired observations was applied to determine whether genes showed significant differential expression values (P value cutoff at 0.05) from each treatment. In the present study, only those transcripts with a experimental/control ratio <0.34 or >3 in both independent experiments were considered to be differentially expressed.
Real-time quantitative RT-PCR (qPCR) analysis
Total RNA was extracted from leaves of plant samples and purified using the RNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA), according to the manufacturer’s instructions. Two microgram of each RNA sample was reverse transcribed to cDNA with MMLV reverse transcriptase (Enzynomics, Korea) using random hexamers. Oligo primer molecules and dNTPs were eliminated by using PCR purification kit according to the manufacturer’s instructions (Intron biotech, Korea). The cDNA concentration was checked using ND-1000 spectrophotometer (NanoDrop Technologies, Montchanin, DE, USA). PCR reaction using SYBR Green I technology on Rotor-Gene 6000 Real-time PCR cycler (Qiagen) was performed. A master mix for each PCR run was prepared with SYBR Premix Ex Taq II (TaKaRa, Japan). Two nanogram of cDNAs were added for 10 μl reaction. The primer concentration was 0.3 μM each for forward and reverse primers (Table 1), and two reference genes were used as internal control; DaACT1 forward primer 5′-GCTCCAACAAATCCAACCCAACCA-3′ and reverse primer 5′-CTGCATGTGCAAGTGCCAAGTACA-3′, DaEF1a1 forward primer 5′-TTTGTCCACTGCTACACTCGTGGT-3′ and reverse primer 5′-TCGAAGGCTGACGGACATAACCAA-3′ (Lee et al. 2010). PCR cycling with an annealing temperature of 55 °C were performed in four replicates from the same RNA preparation and the mean value was considered. PCR product sizes were checked on a 3 % agarose gel and all corresponded to the expected size and melting curves showed a single amplified product.
Phylogenetic analysis
The sequence generated in this study and eighteen ∆-1-pyrroline-5-carboxylate synthase (P5CS) amino acid sequences retrieved from the GenBank database were proofread, all downstream analysis were performed using MEGA5.2 (Tamura et al. 2011). Phylogenetic trees were inferred from the data sets by neighbor-joining method. The evolutionary distances were computed using the Poisson correction method and all positions containing gaps and missing data were eliminated. Supports for internal branches in three analyses were tested by the bootstrap analyses of 1,000 replications.
Results and discussion
Physiological parameters changes upon PEG treatment
After PEG treatment, chlorophyll and proline contents of D. antarctica plants were measured as markers for physiological changes by osmotic stress. Relative chlorophyll contents of leaves decreased from 84 % at 4 h to 69 % at 1 day, and to 54 % at 2 day after treatment, as compared with the water controls (Fig. 1a). The proline levels started to elevate at 12 h and showed significant change at 24 h after the treatment (Fig. 1b). This can be indicative of the fact that proline is accumulated as the water stress is magnified.

Physiological effects of PEG induced osmotic stress on a chlorophyll contents, b proline contents, and c expression levels of osmotic stress-responsive genes
As an additional molecular parameter for plants subjected to PEG treatment and the corresponding water control, expression levels of dehydrin (DaDHN1) and late embryogenesis abundant protein (DaLEA) were investigated for quantifying stress levels indirectly. Expression of DaDHN1 was increased by 2.5-fold at 1 day, in the PEG treatment plants relative to the water control. It has shown continuous increase up to ninefold at 2 day and 5.5-fold at 3 day. Expression of DaLEA was increased by fivefold at 1 day, 14-fold at 2 day, and 21-fold at 3 day (Fig. 1c). Based on these results, we considered the time of 1 day after PEG treatment as a critical point when significant physiological and molecular changes occur, and used the corresponding plant sample as an experimental target for analyzing differential gene expression by PEG treatment for D. antarctica.
Identification of differentially expressed genes by PEG treatment
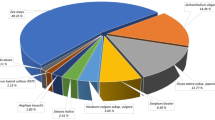
cDNA macroarrays were hybridized with probes from D. antarctica plants 1 day after PEG treatment and from untreated plants, prepared as described in Materials and Methods section. An average signal of duplicate spots was used for the analysis and normalization of signal intensities with respect to that of the exogenously added control gene was carried out. A first criterion for selection of genes of interest was according to statistically significant difference in expression levels between treatment and control conditions. For the chosen genes we calculated the ratio of signal intensity from the experimental conditions. By analyzing two independent experiments carried out, transcripts that had a ratio (stressed/control) of more than threefold in each experiment were identified as differentially expressed genes. We identified 44 and 20 transcripts that were up-regulated and down-regulated by PEG treatment respectively, and they were divided in several categories, according to the functional categories of the Arabidopsis proteins (Fig. 2). For the up-regulated transcripts, the largest categories were ‘Not assigned’, and ‘Response to stimulus’, and ‘Amino acid metabolism’ were followed. For the down-regulated transcripts, the two largest groups were ‘Photosynthesis’ and ‘Translation’.

Distribution of transcripts up-regulated (a) and down-regulated (b) by PEG treatment based on the gene function categories
Amino acid metabolism
Among the drought-regulated genes, those involved in the metabolism of proline, and aromatic amino acid families were up-regulated. A gene for proline metabolism, encoding ∆-1-pyrroline-5-carboxylate synthase (P5CS), was highly expressed by PEG treatment, and a gene encoding acetylornithine aminotransferase in the metabolism of spermidine was up-regulated (Table 2). We also found that a gene 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase, for the biosynthesis of l-methionine via salvage pathway was down-regulated (Table 3).
Osmolytes like proline and spermidine can enhance resistance of the plant organs to drought stress in various ways. First, accumulation of osmolytes lowers the osmotic potential of drought-stressed tissues, allowing them to maintain more water. Secondly, osmolytes are strong water structure formers. They can substitute for water by forming hydrogen bonds with polar residues on the surface of proteins and maintain the three dimensional structure (Hoekstra et al. 2001). Furthermore, osmolytes protect macromolecules from oxidative damage during stress by scavenging ROS (Smirnoff 1998). It has also been suggested that spermidine may play a regulatory role in signal transduction that leads to enhanced resistance to stress (Kasukabe et al. 2004).
Proline biosynthesis
Proline, the most widely studied osmolytes, is synthesized from glutamic acid or ornithine (Verslues and Sharma 2010). In the pathway from ornithine, P5CS is a rate limiting enzyme for proline biosynthesis from glutamic acid, and its expression increases during drought or osmotic stress (Kishor et al. 1995).
The P5CS gene has been isolated as two copies from many plants species. In Arabidopsis thaliana, the AthP5CS1 and AthP5CS2 genes show different temporal and spatial expression patterns. While AthP5CS1 gene seems to be ubiquitously expressed in almost all organs and rapidly induced by stress, AthP5CS2 is expressed in dividing cells and in response to incompatible interactions (Strizhov et al. 1997). In Medicago truncatula P5CS1 encodes a developmental ‘housekeeping’ enzyme, while P5CS2 acts as a shoot-specific osmo-regulated isoform (Verdoy et al. 2006). In rice, OsP5CS1 gene is inducible by salt, dehydration, cold, and abscisic acid (ABA), whereas OsP5CS2 is inducible by NaCl and mannitol (Hur et al. 2004). In sorghum, both SbP5CS1 and SbP5CS2 genes are inducible by drought, salt, and MeJA, but with different temporal expression patterns and with different amplitudes (Su et al. 2011). In D. antarctica, second P5CS gene sequence was obtained by previous high-throughput transcriptome analysis (Lee et al. 2013). Based on the homology to rice P5CS, the genes with GenBank accession JX470539 and FL686618 were designated as DaP5CS1 and DaP5CS2.
In order to analyze the evolutionary relationship of DaP5CS proteins and other species’ P5CS, the phylogenetic tree was deduced from amino acid sequence by MEGA 5.2 using a neighbor-joining method (Fig. 3a). The tree could be divided into two major groups, separating P5CS proteins of monocots from dicots. Two separate clusters were obtained for monocot species. One cluster contained all the sequences homologous to DaP5CS1 while the second cluster contained all the sequences homologous to DaP5CS2. This pattern was not observed for the P5CS duplication in taxonomic orders of dicots. In fact, P5CS1 and P5CS2 formed independent clusters within taxonomic orders used, but they are grouped based on taxonomic orders, as in Brassicales, and in Fabales, which is different pattern with that of monocot.

Phylogenetic relationship of P5CS amino acid sequences within different taxa and the PEG responsive expression analysis of P5CS genes from D. antarctica. a Deduced amino acid sequences were aligned with ClustalW and a neighbor-joining tree was generated via MEGA 5.2. Sources and GenBank accession numbers of P5CS sequences used are D. antarctica (in this study, DaP5CS1 and DaP5CS2), A. thaliana (AtP5CS1, AAB87129; AtP5CS2, AEE79408), B. distachyon (BdP5CS1, XP_003568327; BdP5CS2, XP_003564608), O. sativa (OsP5CS1, BAF17637; OsP5CS2, NP_001044802), S. bicolor (SbP5CS1, ACU65226; SbP5CS2, ACU65227), and Z. mays (ZmP5CS1, ABI21839; ZmP5CS2, NP_001147111). T. aestivum (TaP5CS, BAD97364), Brassica napus (BnP5CSA, AAK01360; BnP5CSB, AAK01361), M. sativa (MsP5CS1, CAA67069; MsP5CS2, CAA67070), M. truncatula (MtP5CS1, CAC82184; MtP5CS2, AET87351; MtP5CS3, AET35478). b qPCR analysis data for the expression of two P5CS genes from D. antarctica plants at indicated time points after PEG treatment. Relative quantification was calculated by comparing levels of gene expression in the PEG treated plants (grey bar) with the non-treated plants (control, white bar) after normalization using DaACT1 and DaEF1a1 as internal references
QPCR was performed to determine the expression patterns of the two P5CS genes responsive to PEG treatment. Both genes were activated by water deficit (Fig. 3b). The transcript levels of DaP5CS1 gene began to increase at 8 h then continued to increase, peaking at 48 h. The DaP5CS2 transcript showed significant increase above fourfold at 24 h and declined to twofold at 48 h revealing differences in transcriptional control of the closely related genes in D. antarctica under osmotic stress. The expression levels of DaP5CS2 were strongly induced at 24 h after PEG treatment when the significant elevation of proline levels occurred, whereas the DaP5CS1 was more subtly induced. Therefore, we conclude that the DaP5CS2 might be a critical player for proline biosynthesis under osmotic stress.
Response to stimulus
Several biotic and abiotic stress responsive genes were up-regulated by osmotic stress in D. antarctica. The major stress responsive genes up-regulated were involved in ‘Defense response’, such as genes encoding late embryogenesis abundant (LEA) protein, heat shock protein (HSP), flavonoid biosynthetic enzyme (cinnamoyl CoA reductase), and genes previously annotated as stress responsive ones at transcription level (Table 2).
Often, drought damages cells in a manner similar to that of pathogens and insects. Consequently, there is an overlap in the responses to drought and biotic stresses. Up-regulation of genes encoding elicitor-responsive protein 3 (>5-fold), disease resistance protein RPM1 (>5-fold), brown planthopper susceptibility protein (>4-fold), cysteine protease inhibitor 3 (>3-fold), and subtilisin-chymotrypsin inhibitor (>3-fold) in D. antarctica under osmotic stress is in agreement with other studies that showed increased expression of genes in this group in response to osmotic stresses (Kawasaki et al. 2001; Singh et al. 2009). Proteases like subtilisin-chymotrypsin are the enzymes that catalyze the hydrolytic cleavage of specific peptide bonds when the pathogens invade their hosts. In plants, various protease inhibitors are induced in response to injury or attack by insects or pathogens as defense strategies (Habib and Fazili 2007).
A dehydrin gene was highly up-regulated by sixfold under osmotic stress comparing to untreated control plant sample (Table 2). Dehydrin gene are known to be induced by drought and low temperature stress in various plant species (Chaves et al. 2003). Dehydrins have a highly conserved lysine-rich domain, which is predicted to be involved in hydrophobic interactions and water exclusion, leading to stabilization of proteins and macromolecular complexes (Close 1996). Even though it is clear that dehydrins are involved in a range of responses to abiotic stressors, the exact mechanism by which dehydrin synthesis is enhanced and its functions in vivo remain poorly understood (Cellier et al. 2000).
One of the risks of drought stress is unfolding or misfolding of proteins. Plants, including barley, express HSP genes during drought to maintain proper folding of polypeptides (Wang et al. 2003). Likewise, the HSP family gene was up-regulated by fivefold in drought-stressed D. antarctica plant (Table 2). HSPs bind unfolded proteins, prevent intermolecular interactions, and stabilize the three dimensional structure. Refolding certain proteins cannot be accomplished by chaperons alone. Chaperonins, such as chn60, facilitate folding of these proteins by assisting chaperones (Wang et al. 2003). This may indicate that the role of HSP is to prevent molecular damage in D. antarctica under environmental stresses.
Besides, various genes previously revealed as environmental stress responsive ones were up-regulated, such as genes encoding low temperature and salt responsive protein (>4-fold), cold acclimation protein isoform 3 (>4-fold), early responsive do dehydration 15 (>3-fold), and salt stress-responsive protein (>3-fold), indicating that the experiments of this work had been conducted reasonably.
Respiration
None of the typical glycolysis genes were up-regulated and expression of pyruvate kinase was rather down-regulated in D. antarctica under osmotic stress (Table 3). But interestingly, a gene for alternative oxidase was up-regulated (Table 2). This finding is in agreement with previous studies that showed increased activity of the alternative pathway during drought (Giraud et al. 2008). During drought, metabolic processes are slower and the small amount of ATP produced during glycolysis and the alternative pathway may be sufficient to sustain maintenance and repair processes. More importantly, AOX reduces accumulation of ROS due to inhibition of cytochrome c oxidase during stress (Umbach et al. 2005).
Down-regulated genes
Several genes involved in the photosynthesis were down-regulated in D. antarctica by PEG treatment, while none of the photosynthesis genes was up-regulated (Table 3). This phenomenon is consistent with previous studies (Seki et al. 2002; Chaves et al. 2007), expression of genes involved in photosynthesis was primarily down-regulated by osmotic stress. This suggests the necessity to investigate the tolerance mechanism to prevent damage to the photosynthetic machinery from the harsh antarctic environment, like the stabilization of chloroplast by unsaturation of membrane lipids (Moon et al. 1995). In addition, several genes involved in ‘Translation’ process, including ribosomal protein genes and elongation initiation factor, and peroxidase 5 gene were down-regulated.
In conclusion, studies on differential expression of transcripts in D. antarctica by PEG treatment have revealed the presence of many genes that are responsive to osmotic stress. Furthermore, elucidating the mechanism for osmotic stress tolerance specific to antarctic plants, these findings may have important implications in the identification of target genes or metabolic products for improving osmotic tolerance of other plants through genetic engineering.
References
Alberdi M, Bravo LA, Guitiérrez AH, Gidekel M, Corcuera LJ (2002) Ecophysiology of Antarctic vascular plants. Physiol Plant 115:479–486
Andjelkovic V, Thompson R (2006) Changes in gene expression in maize kernel in response to water and salt stress. Plant Cell Rep 25:71–79
Cellier F, Conéjéro G, Casse F (2000) Dehydrin transcript fluctuations during a day/night cycle in drought-stressed sunflower. J Exp Bot 51:299–304
Chaves MM, Maroco JP, Pereira JS (2003) Understanding plant responses to drought—from genes to the whole plant. Funct Plant Biol 30:239–264
Chaves MM, Flexas J, Pinheiro C (2007) Photosynthesis under drought and salt stress: regulation mechanisms from whole plant to cell. Ann Bot 103:551–560
Close TJ (1996) Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiol Plant 97:795–803
Dugas DV, Monaco MK, Olsen A, Klein RR, Kumari S, Ware D, Klein PE (2011) Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genom 12:514
Edwards JA, Lewis-Smith RI (1988) Photosynthesis and respiration of Colobanthus quitensis and Deschampsia antarctica from the maritime Antarctic. BAS Bull 81:43–63
Fu BY, Xiong JH, Zhu LH, Zhao XQ, Xu HX, Gao YM, Li YS, Xu JL, Li ZK (2007) Identification of functional candidate genes for drought tolerance in rice. Mol Genet Genomics 278:599–609
Giraud E, Ho LH, Clifton R, Carroll A, Estavillo G, Tan YF, Howell KA, Ivanova A, Pogson BJ, Millar AH, Whelan J (2008) The absence of ALTERNATIVE OXIDASE1a in Arabidopsis results in acute sensitivity to combined light and drought stress. Plant Physiol 147:595–610
Habib H, Fazili KM (2007) Plant protease inhibitors: a defense strategy in plants. Biotechnol Mol Biol Rev 2:68–85
Hoekstra FA, Golovina EA, Buitink J (2001) Mechanisms of plant desiccation tolerance. Trends Plant Sci 6:431–438
Hur J, Jung KH, Lee CH, An GH (2004) Stress-inducible OsP5CS2 gene is essential for salt and cold tolerance in rice. Plant Sci 167:417–426
Kasukabe Y, He L, Nada K, Misawa S, Ihara I, Tachibana S (2004) Overexpression of spermidine synthase enhances tolerance to multiple environmental stresses and up-regulates the expression of various stress-regulated genes in transgenic Arabidopsis thaliana. Plant Cell Physiol 45:712–722
Kawasaki S, Borchert C, Deyholos M, Wang H, Brazille S, Kawai K, Galbraith D, Bohnert HJ (2001) Gene expression profiles during the initial phase of salt stress in rice. Plant Cell 13:889–905
Kishor KPB, Hong Z, Miao GH, Hu CAA, Verma DPS (1995) Overexpression of Δ-pyrroline-5-carboxylate synthetase increases proline production and confers osmotolerance in transgenic plants. Plant Physiol 108:1387–1394
Lee H, Cho HH, Kim IC, Yim JH, Lee HK, Lee YK (2008) Expressed sequence tag analysis of Antarctic hairgrass Deschampsia antarctica from King George Island, Antarctica. Mol Cells 25:258–264
Lee H, Kim JH, Park M, Kim I-C, Yim JH, Lee HK (2010) Reference genes validation for qPCR normalization in Deschampsia antarctica during abiotic stresses. Antarct Sci 22:477–484
Lee J, Noh EK, Choi H-S, Shin SC, Park H, Lee H (2013) Transcriptome sequencing of the Antarctic vascular plant Deschampsia antarctica Desv. under abiotic stress. Planta 237:823–836
Lichtenthaler H, Wellburn A (1983) Determinations of total carotenoids and chlorophylls a and b of leaf extracts in different solvents. Biochem Soc Trans 11:591–592
Michel BE, Kaufmann MR (1973) The osmotic potential of polyethylene glycol 6000. Plant Physiol 51:914–916
Mochida K, Kawaura K, Shimosaka E, Kawakami N, Shin-I T, Kohara Y, Yamazaki Y, Ogihara Y (2006) Tissue expression map of a large number of expressed sequence tags and its application to in silico screening of stress response genes in common wheat. Mol Genet Genomics 276:304–312
Moon BY, Higashi S, Gombos Z, Murata N (1995) Unsaturation of the membrane lipids of chloroplasts stabilizes the photosynthetic machinery against low-temperature photoinhibition in transgenic tobacco plants. Proc Natl Acad Sci USA 92:6219–6223
Porra RJ, Thompson WA, Kriedemann PE (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim Biophys Acta 975:384–394
Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, Kamiya A, Nakajima M, Enju A, Sakurai T, Satou M, Akiyama K, Taji T, Yamaguchi-Shinozaki K, Carninci P, Kawai J, Hayashizaki Y, Shinozaki K (2002) Monitoring the expression profiles of 7,000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J 31:279–292
Singh A, Sahi C, Grover A (2009) Chymotrypsin protease inhibitor gene family in rice: genomic organization and evidence for the presence of a bidirectional promoter shared between two chymotrypsin protease inhibitor genes. Gene 428:9–19
Smirnoff N (1998) Plant resistance to environmental stress. Curr Opin Biotechnol 9:214–219
Strizhov N, Abrahám E, Okrész L, Blickling S, Zilberstein A, Schell J, Koncz C, Szabados L (1997) Differential expression of two P5CS genes controlling proline accumulation during salt-stress requires ABA and is regulated by ABA1, ABI1 and AXR2 in Arabidopsis. Plant J 12:557–569
Su M, Li XF, Ma XY, Peng XJ, Zhao AG, Cheng LQ, Chen SY, Liu GS (2011) Cloning two P5CS genes from bioenergy sorghum and their expression profiles under abiotic stresses and MeJA treatment. Plant Sci 181:652–659
Talamè V, Ozturk NZ, Bohnert HJ, Tuberosa R (2007) Barley transcript profiles under dehydration shock and drought stress treatments: a comparative analysis. J Exp Bot 58:229–240
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Umbach AL, Fiorani F, Siedow JN (2005) Characterization of transformed Arabidopsis with altered alternative oxidase levels and analysis of effects on reactive oxygen species in tissues. Plant Physiol 139:1806–1820
Verdoy D, De la Pena TC, Redondo FJ, Lucas MM, Pueyo JJ (2006) Transgenic Medicago truncatula plants that accumulate proline display nitrogen-fixing activity with enhanced tolerance to osmotic stress. Plant Cell Environ 29:1913–1923
Verslues PE, Sharma S (2010) Proline metabolism and its implications for plant-environment interaction. Arabidopsis Book 8:e014
Wang W, Vinocur B, Altman A (2003) Plant responses to drought, salinity and extreme temperatures: towards genetic engineering for stress tolerance. Planta 218:1–14
Wang D, Pan Y, Zhao X, Zhu L, Fu B, Li Z (2011) Genome-wide temporal–spatial gene expression profiling of drought responsiveness in rice. BMC Genom 12:149
Acknowledgments
This work was supported by Korea Polar Research Institute (Grants PE13020 and PE13040).
Conflict of interest
The authors have declared that no conflicts of interest exist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, J., Lee, H., Noh, E.K. et al. Expression analysis of transcripts responsive to osmotic stress in Deschampsia antarctica Desv.. Genes Genom 36, 283–291 (2014). https://doi.org/10.1007/s13258-013-0166-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-013-0166-5