
Abstract
The past decade has witnessed significant advances in our understanding of skeletal homeostasis and the mechanisms that mediate the loss of bone in primary and secondary osteoporosis. Recent breakthroughs have primarily emerged from identifying disease–causing mutations and phenocopying human bone disease in rodents. Notably, using genetically–modified rodent models, disrupting the reciprocal relationship with tropic pituitary hormone and effector hormones, we have learned that pituitary hormones have independent roles in skeletal physiology, beyond their effects exerted through target endocrine glands. The rise of follicle–stimulating hormone (FSH) in the late perimenopause may account, at least in part, for the rapid bone loss when estrogen is normal, while low thyroid–stimulating hormone (TSH) levels may contribute to the bone loss in thyrotoxicosis. Admittedly speculative, suppressed levels of adrenocorticotropic hormone (ACTH) may directly exacerbate bone loss in the setting of glucocorticoid–induced osteoporosis. Furthermore, beyond their established roles in reproduction and lactation, oxytocin and prolactin may affect intergenerational calcium transfer and therefore fetal skeletal mineralization, whereas elevated vasopressin levels in chronic hyponatremic states may increase the risk of bone loss.. Here, we discuss the interaction of each pituitary hormone in relation to its role in bone physiology and pathophysiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
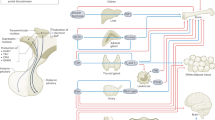
Ubiquity and distributed functions of pituitary hormones and their receptors
The pituitary gland orchestrates diverse physiological processes by secreting hormones that target various endocrine and non–endocrine tissues. These ancient hormones have acquired distributed somatic and sensory functions throughout evolution, and in mammals, have been shown recently to exhibit a complex array of actions. Pituitary hormones exert their effects via G–protein–coupled receptors (GPCRs) within the rhodopsin–like receptor class. Thyroid–stimulating hormone (TSH), follicle–stimulating hormone (FSH), and luteinizing hormone (LH) have originated from a common ancestral hormone, thyrostimulin, with their receptor evolving to accommodate distinct hormone functions in vertebrates [1].
A thyroid–stimulating hormone receptor (TSHR) family gene with a mammalian intron–exon structure is expressed widely in coelenterates, with a primitive nervous system, but without endocrine glands. In bony fish, TSHRs are expressed in abundance in the thyroid, but are also found in ovaries and several other tissues, including the heart, muscle, and brain [2]. TSHRs are also expressed in bone and bone cells, namely, calvaria–derived primary osteoblasts, preosteoblasts, human osteoblast–like cells, differentiated osteoclasts, and osteoclast precursors [2,3,4,5,6]. Furthermore, TSH secretion is not limited to pituitary glands: immune cells, including macrophages and lymphocytes, produce a splice variant of TSH, TSH–βv, which is regulated differently from pitutary–derived TSH [7,8,9,10,11].
Similarly to TSHR, the expression of FSHRs is not confined to gonadal tissue, but is widely distributed, including in bone tissue, as demonstrated by in vivo imaging in live mice using a near–infrared fluorophore conjugated to FSH [12, 13]. Apart from the adenal gland, the receptor for ACTH, melanocortin receptor 2 (MC2R), is also expressed widely in tissues that include the brain, immune cells, adipose tissue, bone cells, and the pituitary itself. ACTH has therefore has been demonstrated to exert a variety of biological activities in addition to its primary role in regulating glucocorticoid hormone production [14, 15].
As for the posterior pituitary hormones, oxytocin (OXT) and vasopressin (AVP) genes have evolved from the ancestral mesotocin and vasotocin genes [16, 17]. The resulting nonapeptides have emerged during the expansion of mammals around 100–200 million years ago and subsequently acquired a diverse range of functions. Following gene duplication events in nematodes, the encoded OXT– and AVP–like peptides have become involved in behavioral adaptation as a means to ensure reproductive success. During this evolutionary period, the single receptor for OXT and three distinct GPCRs for AVP also evolved from ancestral mesotocin and vasotocin receptors to undertake widely distributed roles in modern mammals [18]. Both OXT and AVP act directly on bone through their respective GPCRs, the OXTR and AVPR1A [19,20,21,22]. In addition to its neurohypophyseal origin, OXT is produced by both human and murine osteoblasts and is regulated by estrogen [21, 23, 24].
The skeletal effect of growth hormone
Studies of GH and the human skeleton
While there is considerable clarity the skeletal phenotype of patients with GH deficiency or excess, interpreting human data is challenging due to the heterogeneity of the study designs and subjects, differences in the etiology, onset age, duration, concomitant pituitary hormone deficiencies, and hormone supplementation. GH deficiency is generally associated with a low bone mineral density (BMD), but the timing of onset––whether before or after the age of achieving peak bone mass––and sex have a significant impact on BMD accrual. Patients with childhood–onset GH deficiency typically exhibit Z-scores in the range of −1 to −2, whereas adult–onset patients are less affected (Z-scores typically between -1 and 0) [25,26,27,28,29]. Patients with adult–onset GH deficiency thus show 17% and 10% higher BMD at lumbar spine (LS) and hip, respectively, compared with childhood–onset disease [30]. These findings may be confounded by differences in body (bone) size. For example, a study using peripheral quantitative computed tomography (pQCT) in adult patients with childhood–onset GH deficiency showed only a mild decrease in cortical BMD (~ 2%) in the radius and normal trabecular BMD [31]. In the radius, cortical BMD is lower than in the control group, while trabecular bone is not affected [31]––suggesting that the GH/IGF-1 axis exerts differential effects on trabecular and cortical bone. There is also a sex–dependent difference in patients with GH deficiency. In adult men, BMD is lower at the lumbar spine (Z –2.03 vs. –0.57) and hip (Z –1.04 vs. –0.24); women, however, did not show any difference despite lower bone turnover markers [32].
Conversely, GH excess in patients with acromegaly is associated with increased BMD in cortical–bone–rich areas, such as the femur, but not in trabecular-bone-rich vertebrae [33]. Conflicting data also exist where only 8% of patients display osteopenia at the femur compared with ~ 20% at the lumbar spine [34]––this difference might be related to concomitant hypogonadism. Most patients with osteopenia had concomitant hypogonadism and bone loss at the lumbar spine was associated with a longer duration of hypogonadism [34,35,36]. Similarly, when female patients were stratified by menstrual status, BMD at the lumbar spine, but not at other sites, was significantly higher in menstruating patients compared to controls [37].
What appears to to be clear from human data is that GH excess causes high bone turnover. Bone biopsies on patients with acromegaly show increased bone turnover [38, 39]. Serum osteocalcin and urinary hydroxyproline are elevated in acromegalic patients [33]. Likewise high bone turnover can also be seen in patients with GH deficiency after GH replacement therapy. Serum bone Gla protein (BGP) and bone-specific alkaline phosphatase (B-AP) were found to be lower in patients with childhood–onset GH deficiency, but all bone turnover markers, namely BGP, C–telopeptide (CTX), procollagen type 1 N-terminal peptide (P1NP), and B-AP, increased after 3 and 6 months of GH supplementation [40, 41]. This increased bone turnover appears to be associated with a net anabolic effect on bone mass. GH replacement in adults with isolated GH deficiency increased spinal trabecular BMD by 5% after 6 months [42]. However, the response to GH replacement seemed to differ depending on the onset of the condition, wherein childhood–onset patients showed a more robust increase in osteocalcin compared to adult–onset disease [43].
There is also a relationship between GH action and PTH action. GH has anti–phosphaturic action, which leads to PTH secretion [44]. Untreated GH deficiency is also complicated with PTH insensitivity. 24–hour monitoring of PTH, phosphorus and bone turnover markers in GH–deficient patients showed elevated PTH with low serum phosphorus, together with suppressed P1NP and CTX levels [45]. A subgroup of patients with low BMD had suppressed nephrogenous cyclic AMP (NcAMP), reflecting reduced renal response to PTH action [46]. Conversely, GH treatment improved PTH sensitivity, resulting in decreased PTH levels and increased phosphate reabsorption [47, 48].
Mechanisms of GH action.
GH acts on bone primarily by secreting insulin–like growth factors (IGFs) 1 and 2. Several genetically–engineered mouse models have thus been employed to dissect the IGF effects. Given the critical physiological importance of IGF-1 and IGF-2, mice with mutations in IGF-1R do not survive after birth. Surviving mice with disruptions in IGF-1 or -2 exhibit a 40% smaller body size compared with wild type mice [49, 50]. Interestingly, double mutants of IGF-1 and IGF-1R do not differ from IGF-1R single mutant mice, but mice with mutations in both IGF-1 and IGF-2 display a further decrease in body size compared with mice with mutations in either gene––suggesting an additive effect [50]. The importance of IGF-1 action in skeletal growth is indirectly demonstrated by disrupting the JAK/STAT signaling pathway. Disrupting Stat5b or both Stat5a and 5b, but not 5a alone, causes decreased IGF-1 levels, as well as reduced body growth [51,52,53].
Given that, in addition to its secretion from the liver, IGF-1 is also produced locally by osteoblasts [54], there has been renewed interest in understanding the endocrine vs. paracrine skeletal actions of IGF-1. This becomes even more complex when considering the role of binding proteins. IGF binding protein-3 (IGFBP-3) and the acid labile subunit (ALS), which binds to ~ 80% of circulating IGF-1, prolong the half–life of IGF-1. Furthermore, IGFBP-3 expression in osteoblasts scavenges IGF-1 locally and, in doing so, affects its bioavailability [54, 55]. While liver–specific IGF-1 deficient mice, namely Alb-Cre+;Igf1fl/fl mice, achieved a ~ 75% decrease in serum IGF-1 levels, skeletal growth remained unaltered, suggesting that the relatively small quantity of IGF-1 from extrahepatic tissues may, in fact, be sufficient for normal growth [56]. Subsequent studies with double mutant mice obtained by crossing Alb-Cre+;Igf1fl/fl mice with ALS-deficient mice showed a further decrease in IGF-1 to ~ 10% of normal levels––which, together with a concommitant rise in GH due to negative feedback––resulted in only a ~ 30% reduction in body size that was rescued by IGF-1 treatment with ~ 6% gain. Of note, these mice did not show compensatory upregulation of IGF-1 expression in bone [55].
While systemic IGF-1 is required for skeletal growth, local IGF-1 also seems to play a critical role in bone growth and remodeling. Chondrocyte–specific Col2α1-Cre+;Igf1fl/fl mice display reduced body and femoral length and total body BMD [57]. Furthermore, ablation of the IGF-1R in osteoblasts using Osteocalcin-Cre results in mice of normal size and weight, but with reduced bone remodeling. Namely, both osteoblast and osteoclast number was decreased, together with impaired trabecular and periosteal bone formation [58, 59]. Furthermore, the anabolic action of PTH was less prominent in these mice, suggesting that IGF-1 action is required for the osteoblastic action of PTH action [58]. Conversely, transgenic mice overexpressing IGF-1 locally (and not systemically) through an OC–IGF-1 chimeric fusion gene showed higher trabecular bone volume and thickness, as well as increased bone formation rate, without changes in body size [60].
In addition to local IGF-1 and IGF-2, tissue-specific expression of IGF binding protein (IGFBP) also exerts skeletal effects by sequestering bioavailable IGF-1. For example, upregulating the expressioj of IGFBP-4, which is abundant in bone, reduced bone formation and bone turnover by ~ 50% [61]. Of note is that IGFBP-5 levels were reduced in these transgenic mice, suggesting a compensatory mechanism to maintain IGF-1 bioavailability [61]. Similarly, IGFBP-5 overexpression resulted in decreased trabecular bone volume and reduced osteoblast bone formation [62].
Finally, a question arises––does GH display actions on bone that are independent of IGF-1/2. Notably, GHR–deficient mice showed runting, decreased bone volume and density, as well as reduced bone turnover [52, 63, 64]. However, these mice also had significantly lower levels of IGF-1 [64], and, importantly, IGF-1 treatment rescued the impaired bone growth and remodeling [52]. Similarly, mutant mice obtained by crossing Ghr−/− mice with mice overexpressing IGF-1 showed restored body length and normalized bone area and density [63]–– together suggesting that IGF-1 may be required for the actions of GH on the skeleton. However, there is evidence that GH may have an independent action, specifically on linear growth. Double knock-out mice (Ghr−/−;Igf1−/− mice) were noted to be shorter (~ 50%) compared with Ghr−/− (~ 25–35%) and Igf-1−/− mice (~ 35–45%), respectively [65]. In a hypogonadal state, such as in ovariectomized mice, sensitivity to GH increases, which appears to preserve bone through periosteal bone formation in the setting of systemic IGF-1 deficiency [66]. In rats, GH treatment also raises PTH levels, and those rats have heavier parathyroid glands [67], suggesting an indirect effect of GH exerted via PTH action.
TSH protects bone
Correlative, interventional and genetic studies
It is known since the time of von Recklinghausen, that patients with hyperthyroidism are at a higher risk of developing osteoporosis and sustaining fracture [68, 69]. Furthermore, it is well established that thyroid hormone stimulates bone resorption mainly via the thyroid hormone receptor (TR) α1 [70,71,72,73,74]. However, it is also clear that patients with subclinical hyperthyroidism, in whom T4/T3 levels are normal but TSH is suppressed are also at a high risk for osteoporosis and fracture. Thus the question arises––whether TSH has direct effects on bone. In 2003, we discovered that TSH directly affects bone mass in mice, in the broader perspective, as the first conclusive evidence for the action of any pituitary hormone beyond its traditional unitary target [75].
Multiple observational studies have since shown strong correlations between low TSH levels and high bone turnover, low bone density, and a high fracture risk, importantly, independently of thyroid hormone levels [76]. In euthyroidal subjects, low TSH levels are associated with adverse skeletal phenotypes, including lower BMD or a higher risk of fracture [77,78,79,80,81,82,83,84]. Patients with subclinical hyperthyroidism, where TSH is suppressed and thyroid hormones are normal, show increased risk of osteoporosis and of fracture. For example, the Study of Osteoporotic Fracture (SOF), a large prospective study of postmenopausal women from the U.S., has documented that women with low TSH (< 0.1 mIU/L) have a higher risk of fracture [Hip (HR = 3.6, 1.0–12.9), vertebral (4.5, 1.3–15.6) and nonvertebral fracture (2.3, 0.8–6.8)] compared with women with normal TSH levels (0.5–5.5 mIU/L) [78]. A large female cohort from a health promotion center in Korea also noted almost threefold increase in the risk of osteoporosis in patients with TSH < 0.5 mIU/L compared with TSH 2.8–5 mIU/L [79]. Conversely, thyroid hormone supplementation in patients with subclinical hypothyroidism increased bone turnover, as reflected by elevated serum alkaline phosphatase and CTX, and urine deoxypyridinoline after 24 weeks. At 48 weeks of treatment, lumbar spine areal BMD decreased 1.2% compared with the placebo–treated group [85]. A recent study using peripheral quantitative computed tomography, however, did not show any difference in volumetric BMD or bone geometry parameter in levothyroxine–treated group vs. placebo [86]. Unlike subclinical hyperthyroidism, multiple epidemiology studies did not observe BMD changes or increased risk of fracture in patients with subclinical hypothyroidism [87,88,89,90].
Intervention using recombinant human TSH (rhTSH) suggest direct anti–resorptive and pro–anabolic actions of TSH based on acute changes in bone turnover markers. In patients with thyroid cancer, who underwent rhTSH–stimulated whole body scan, serum CTX and urinary CTX and NTX were decreased [91, 92]. Conversely, B-ALP and P1NP increased after rhTSH injection [92, 93]. Finally, the Rotterdam Study and other studies from the U.K. and China have shown that individuals harboring gain–of–function TSHRD727E variants had increased bone density [83, 94, 95]. However, a study utilizing a Mendelian randomization did not find any effect of genetic variants on bone density [96].
Mechanistic studies
The independent role of TSH in the skeletal metabolism was revealed primarily through studies in genetically modified mice. Most notably, Tshr haploinsufficient mice with normal thyroid glands and unaltered thyroid function displayed significant bone loss [75]––in essence, separating the effect of reduced TSHR signaling from thyroid hormone action. Similarly, homozygotic Tshr–/– mice displayed low BMD despite being maintained in a euthyroid state using on thyroid hormone replacement from the birth [75, 97]. Furthermore, and importantly, when hyperthyroidism was induced by implanting T4 pellets, hyperthyroid Tshr–/– mice lost more bone than hyperthyroid wild type littermates, suggesting that TSHR signaling affords skeletal protection against thyroid hormone excess [98].
The decreased bone mass in Tshr–/– mice was primarily associated with increased osteoclastogenesis. In line with the observed decreases in bone resorption markers in humans after rhTSH injection, osteoclastogenesis was suppressed in mice upon exposure to an agonist anti–TSHR antibody [99, 100]. We find that the anti–resorptive action of TSH is mediated, in part, through the suppression of pro–inflammatory cytokines. TNFα, a well–known osteoclastogenic cytokine [101], was upregulated in Tshr−/− mice, and an anti–TNFα neutralizing antibody reversed the increased osteoclastogenesis [102]. Similarly, the bone loss with increased osteoclast differentiation in Tshr deficient mice was not noted in compound mutants with reduced or absent Tnfa expression, such as in Tshr−/−;Tnfα−/− mice [97]. The inhibition of Tnfa expression by TSH was medicated through the high–mobility group box proteins (HMGB). Notably, TSH downregulated HMGB–1 and –2, in addition to attenuating JNK1/2 and IκBα phosphorylation and c-jun and p65 nuclear translocation [75, 103]. TSHR overexpression also decreased NFκB binding in response to RANKL and TNFα [102].
TSH is anabolic in addition to being anti–resorptive. In in vivo studies, intermittent low–dose rhTSH injections in wild type and ovariectomized mice increased bone formation and bone mass [4, 100]. TSH also promoted osteoblast differentiation and proliferation by activating protein kinase Cδ and upregulating the non–canonical WNT components FRZ and WNT5a in embryonic stem cell cultures [104]. However, in bone marrow stromal cell cultures TSH was found to downregulate osteoblast differentiation genes, as well as the VEGF receptor FLK-1 and the WNT co–receptor LRP5 [75].
Finally, it is notable that CD11b + and other immune cells express the splice variant TSHβv [7]. This expression of local TSH could, in fact, amplify the physiological effects of TSH on bone remodeling. Interestingly, immune cell–derived TSHβv is regulated differently from pituitary–derived TSH. TSHβv injection into mice increased T4 and T3 levels, yet T3 injection or TSH releasing hormone (TRH) treatment did not alter TSHβv expression in peripheral blood leukocytes, spleen, thyroid, or the pituitary gland [105]. Instead, pro–inflammatory cytokines upregulated intrathyroidal TSHβv expression, and trafficked immune cells expressing TSHβv to the thyroid gland [106, 107].
FSH directly causes bone loss
Human evidence
The independent skeletal effect of FSH can be inferred by observing specific groups clinically, such as women in the perimenopausal transition, patients with Turner syndrome, or even elderly cohorts. Notably, rapid and substantial bone loss occurs around three years prior to the final menstrual period, coinciding with relatively normal serum estrogen levels and escalating FSH levels [108, 109]. This rapid bone loss cannot conceivably be explained by low estrogen––which led us to evaluate FSH as a possible culprit.
Furthermore, a set of evolving epidemiologic studies have suggested that FSH is a stronger predictor of BMD loss than estrogen. Notably, the Study of Women’s Health Across the Nation (SWAN), a longitudinal cohort of 2375 perimenopausal women (42–52 years old) showed a rise in FSH over 4 years predicted a decline in BMD [109]. The significant association between high serum FSH and bone resorption markers or BMD, particularly within the highest quartiles of serum FSH, was also noted in perimenopausal Chinese women [110,111,112,113,114]. The Bone Turnover Range of Normality (BONTURNO) Study also showed that the women with serum levels of FSH > 30 IU/l had higher bone turnover markers than age-matched women with FSH levels of < 30 IU/l [115]. Likewise, in NHANES III, high serum FSH was correlated with high bone turnover markers and low BMD [116]. Similarly, the AGES–Reykjavik Study of Older Adults from Iceland showed an inverse correlation between serum FSH and BMD in elderly women [116, 117].
Comparing patients with hypergonadotropic vs. hypogonadotropic hypogonadism, such as functional hypothalamic amenorrhea, has added another line of compelling evidence for the role of FSH in causing bone loss. Patients with amenorrhea with high levels of FSH (> 40 IU/L) had lower BMD than patients with lower FSH levesl (< 40 IU/L) [118], and unlike postmenopausal bone loss, functional hypothalamic amenorrhea is associated with mild to moderate bone loss [119]. Likewise, Turner syndrome is also characterized by hypergonadotropic hypogonadism with ovarian insufficiency. The biphasic pattern of elevated FSH during infancy and at puberty has been consistently observed in patients with monosomy (45, X) and in patients without spontaneous puberty [120, 121]. Expectedly, Turner syndrome patients have higher risk of osteoporosis and fracture, despite being on hormone replacement therapy. The areal BMD after correction for height and weight, and volumentric BMD are both low with increased bone resorption markers and decreased bone formation markers [122,123,124]. An observational study has further shown an inverse correlation between FSH levels and BMD Z-score in postpubertal Turner syndrome patients [125]. In addition, peripheral blood mononuclear cells (PBMCs) cultures from Turner syndrome patients with high FSH levels showed an increased number of TRAP–positive osteoclasts after M-CSF and RANKL treatment, and monocytes of those patients expressed higher levels of c-fos, RANK and TNF-α [123]–-all being surrogate markers of increased bone resorption.
In an interventional study, postmenopausal women were treated with a GnRH agonist, leuprolide acetate, or placebo, with both groups receiving the aromatase inhibitor, letrozole, to eliminate variations in endogenous estrogen levels as a confounder [126]. In the GnRH group, while suppression of FSH secretion did not reduce the levels of resorption markers, serum P1NP, a bone formation marker, was increased significantly by ~ 15% from baseline. The multiplicity of actions of the GnRH agonist on other hormones, such as LH, or the action of GnRH itself, might account for the unexpected action on resorption. But it is clear that lowering serum FSH does increase P1NP, a validated bone formation marker. This is in line with bone–forming actions of FSH inhibition in rodent models (below).
Finally, it is worth noting that women harboring an activating FSHRN680S variant display lower bone mass and high bone resorption markers [127]. Additionally, digenic combinations between wild type genotype of the 3’UTR and IVS4 markers for the CYP19A1 (aromatase) gene, and the BMP15 and FSHR genes have been described as being osteoprotective [128].
Mechanistic studies
Supporting the strong epidemiologic and genetic evidence for a role of FSH in bone mass regulation in humans is compelling evidence from rodents. We found earlier that haploinsufficient Fshb+/– mice displayed increased bone mass with no loss of ovarian function [5]. Furthermore, the injection of FSH or FSH antagonist in mice exacerbated and protected, respectively, the bone loss from ovariectomy [129, 129, 130, 130]. Likewise, the injection of the ovotoxin 4-vinylcyclohexene diepoxide in rats to mimic the human perimenopausal transition resulted in 10% bone loss during the high FSH––normal estrogen window [131].
FSH binding to bone tissue was shown in vivo using a near–infrared fluorophore conjugated to FSH [12]. FSH promotes osteoclast formation in all species studied through a distinct FSHR isoform lacking exon 9 [5, 111, 112, 132,133,134]. Unlike granulosa cells, FSHRs in CD11b+ osteoclast precursors and osteoclasts are coupled with an inhibitory Gαi protein. Thus, the effect of FSH on osteoclasts was attenuated in Gαi−/−cells [5]. FSHR activation in osteoclasts induces the nuclear localization of c-Fos and activates Erk1/2 and Akt phosphorylation [5].
FSH also acts indirectly to promote osteoclastogenesis by augmenting inflammatory pathways. For example, FSH was found to enhances the expression of RANK [135], as well as IL-1β, TNFα and IL-6 [101, 136]. In humans, FSH levels were positively associated with serum cytokine levels [136, 137]. The pro–osteoclastogenic response to FSH was not seen in mice lacking immunoreceptor tyrosine–based activation motif (ITAM) adapter signaling molecules [134], suggesting an interaction with immune receptors. Finally, FSHRs were found to be expressed on human osteoblast precursors and stromal cells isolated from mice [138]. Our anti–FSH antibody increased a number of osteoblast precursors, upregulated osteoblastogenic genes expression, and promoted bone formation [132, 138, 139].
FSH effects on adiposity
The FSHR is also expressed in fat tissue and adipocytes [140,141,142]. Full–length FSHR was identified in adipose tissues in mice, boar, and human by qPCR and Sanger sequencing, and immunostaining [141, 143, 144]. Like in osteoclasts and monocytes, FSHRs in adipocytes are coupled to Gαi protein, and, by decreasing intracellular cAMP levels [144], downregulate cAMP–mediated β3-adrenergic receptor signaling. This results in downregulation of UCP1–mediated beiging [144, 145]. In addition, FSH induces lipid synthesis by upregulating peroxisome proliferator-activated receptor gamma (PPARγ), CCAAT enhancer binding proteins (C/EBPα), lipoprotein lipase (LPL), and fatty acid synthase (FAS) [141, 144].
In loss–of–function experiments, attenuating the action of FSH, either genetically in Fshr+/– mice or pharmacologically using our FSH–blocking antibodies in high–fat–diet–treated or ovariectomized mice, stimulated UCP1 expression and mitochondrial biogenesis, resulting in increased energy expenditure and reduced fat mass in a prevention setting [5, 138, 144]. This suggests that FSH blockade in vivo could be a potential avenue for not only increasing bone mass, but also reducing body fat. Importantly, FSH blockade did not cause satiety; instead there were trends to increased food intake, despite which, the mice lost weight [144].
There is evidence from human studies that FSH is associated with increased adiposity. The SWAN study has shown that elevated FSH levels are positively correlated with waist circumference and fat mass during the perimenopausal transition, while estrogen levels remain stable [146, 147]. At menopause, when FSH levels remain high and estrogen levels plummet, serum FSH continues to serve as a better predictor of high fat mass than estrogen. The AGES–Reykjavik study shows that elderly women in the highest FSH quartile had higher bone marrow adiposity [117], and a Chinese cohort study reported that subjects with high FSH had high BMI [141]. Finally, there is also a positive association between FSH and fat mass in men. A population study conducted in China revealed a positive correlation between FSH levels and BMI in older men [141]. In patients with prostate cancer, surgical orchiectomy causing acute hypogonadal transition (low testosterone and high FSH) displayed greater weight gain and fat mass both in subcutaneous and visceral fat compartments, compared with patients who received GnRH agonist treatment causing secondary hypogonadism (low testosterone and low FSH) [148]. The evidence together suggests that FSH might contribute to obesity both in men and women.
FSH effects on cognition
Alzheimer’s disease (AD) disproportionately affects women in terms of life-time risk, rate of progression and symptom burden, suggesting that the female sex could be a major risk factor in developing AD, particularly after menopause [149,150,151,152,153]. It has been widely believed that estrogen deficiency may underlie the preponderance of AD in postmenopausal women. However, the evidence regarding the relationship between hormone replacement and AD is mixed with improvements, no effects or even deterioration of cognition [154,155,156,157,158]. In contrast, FSH levels in cerebrospinal fluid (CSF) increase almost threefold in postmenopausal women compared with premenopausal women [159], and FSHRs are abundant in the hippocampus, an AD–vulnerable brain region [160,161,162]. The SWAN and the Penn Ovarian Aging Study showed that cognitive performance specifically in cognitive processing speed, verbal encoding, and verbal episodic memory, declined during the perimenopause when FSH starts rising prior to the decline of estrogen [163,164,165]. Furthermore, FSH levels were positively correlated with the risk of dementia in both men [166, 167] and women [168]. Furthermore, women aged 40–65, gonadotropin, particularly FSH levels, were associated with higher Aβ load and lower gray matter volume in AD–prone brain regions [169]. Finally, GnRH agonist treatment in patietns with AD showed improvement in the cognitive functions [170, 171].
We documented FSHR expression mainly in AD–vulnerable regions of mouse and human brains––these regions, namely the entorhinal cortex and the granular layer of the dentate gyrus of the hippocampus are involved in learning behaviors and memory [160, 161]. In a study using AD–prone 3xTg transgenic mice, we found that elevated FSH levels after ovariectomy or intraperitoneal FSH injections caused a rapid accumulation of Tau and Aβ with impaired spatial learning and memory retrieval. Consistent with the hypothesis that FSH promotes AD–like pathology and memory loss, the downregulation of the Fshr in the hippocampus or injection of our FSH–blocking antibody attenuated the memory loss and neuropathology in ovariectomized 3xTg mice [162]. Importantly, we found that Fshr deletion on a 3xTg background resulted in a gene–dose dependent rescue of spatial and recognition memory in 3xTg;Fshr+/− and 3xTg;Fshr−/− mice compared with 3xTg;Fshr+/+ mice [172]. FSH administration also exacerbated the AD–like pathology in ApoE4 knock–in mice, suggesting that FSH might have an additive effect on the known propensity of the ApoE4 phenotype in AD pathogenesis [173]. In a study using Ts65Dn mice recapitulating Down syndrome, a condition associated with high FSH and cognitive impairment, GnRH therapy improved cognitive performance by normalizing FSH levels [174].
Genetically, a polymorphism in the FSHR (FSHRA307,S680/A307,S680) has been linked to a lower risk of AD in women (OR = 0.36, 0.15–0.85) [175]. Individuals with Down syndrome also have an increased risk of developing AD, with males having a threefold higher risk compared to females [176]. Male patients with Down syndrome have been found to have higher gonadotropin levels, despite having normal testosterone levels [177, 178].
FSH as a therapeutic target for human diseases
Based on preclinical and human studies, FSH has become an actionable target for osteoporosis, obesity and AD. Our original polyclonal and monoclonal antibodies target a computationally–defined 13–mer epitope acids (LVYKDPARPKIQK) within the receptor–binding domain of human FSHβ[13, 138, 138, 144, 179]. These antibodies increase bone mass in both wild type and ovariectomized mice through both anti–resorptive and anabolic actions [13, 138, 138, 139, 179]. In addition, blocking FSH action prevented fat gain and induced beiging in all fat depots in mice on a high–fat diet or post–ovariectomy [144]. In studies from other labs, vaccination with tandem repeats of the same epitope in mice also prevented fat accrual and induced beiging [180]. Boars vaccinated with these tandem repeats gained less fat compared with castrated pigs where FSH levels are high [143]. Blocking FSH action also prevented the onset of AD–like neuropathology––Aβ plaque and neurofibrillary tangle formation––and memory loss in the ovariectomized 3xTg mouse [162]. Antibody treatment also reduced neuronal apoptosis while increasing dendritic spine and synapse number, and restoring spatial memory in the Morris Water Maze test [162].
Our lead humanized candidate, Hu6, was chosen from a panel of 30 humanized FSH antibody clones as one that displayed the highest binding affinity (KD) of 7.53 nM [139]. Using HEK cells overexpressing the FSHR, we found that Hu6, directly prevented FSH binding to the FSHR, and in doing so, attenuated osteoclastogenesis in a bone marrow cell culture assay, and inhibited the expression of beiging genes in vitro in 3T3-L1 adipocytes [139]. We also found that Hu6 stimulated bone formation in Thermo mice, as well as in C57BL/6 mice that has been ovariectomized and allowed to lose bone––consistent with an anabolic action [181]. We have also developed a clinical–grade formulation and performed a range of biophysical tests. We find that Hu6 displays thermal, monomeric, colloidal, structural and accelerated stability, as well as acceptable levels of viscosity, clarity and turbidity at an ultra–high concentration of up to 150 mg/mL in formulation––which is suitable for human use [182, 183]. Finally, studies in African green monkeys have shown no evidence of acute effects on vital signs, serum chemistries or blood counts [181].
ACTH action on bone
ACTH has a direct effect on bone remodeling independent of glucocorticoids. We now know that prolonged glucocorticoid use in humans results in suppressed osteoblast differentiation, increased osteoclastogenesis, and osteocyte apoptosis [184]. But it remains unclear whether suppressed ACTH in this setting has any direct skeletal action, given that glucocorticoids are required for osteoblast differentiation in vitro [185], and ACTH receptor, MC2R, is expressed in both osteoblasts and osteoclasts [15, 186].
An observational study comparing patients with ACTH–independent adrenal Cushing’s syndrome (low ACTH and high cortisol) with ACTH–dependent Cushing disease (high ACTH and high cortisol) showed greater bone loss in the former group [187]. Mc2r−/− mice exhibited high bone formation, increased cortical bone mass, and unaltered trabecular microstructure, but this mice were confounded by adrenal insufficiency [188]. However, a cleat protective effect of ACTH was observed in a rabbit model of glucocorticoid–induced osteonecrosis. ACTH injections reduced necrotic surfaces, upregulated the osteoblastogenesis gene program, as well as Vegf and Tgfb in vitro [189,190,191].
Oxytocin, prolactin and pregancy– and lactation–induced bone loss
During the pregnancy, ~ 80% of mineral that is pat of the fetal skeleton accrues during the third trimester of pregnancy, with intergenerational calcium transfer from mother at a rate of 300–350 mg/day. After delivery, the neonate requires 200 mg of calcium daily from milk during the first 6 months and 120 mg from milk during the second 6 months [192]. Maternal skeletal adaptations to meet these needs leads to enhanced bone resorption and a negative calcium balance in the mother, particularly during the late pregnancy when fetal demand is at the peak [193,194,195]. For this reason, although it is uncommon, vertebral fracture and low bone mass have been reported during pregnacny and lactation [192, 196].
OXT and prolactin, both of which regulate lactation and parturition, may play a role in intergenerational calcium transfer during pregnancy and lactation along with other calcitropic hormones, such as parathyroid hormone–related protein (PTH-rP) [197]. Both Oxt−/− and Oxtr−/− mice showed age–dependent bone loss with reduced bone formation and impaired osteoblastogenesis [21]. Likewise, ovariectomized mice and rats had decreased OXT levels, and OXT injection reveresed bone loss [19]. Transcriptomic analysis identified the oxytiocin receptor (OXTR) pathway as a potential regulator of osteogenesis, wherein OXT and carbetocin (an OXT analogue) promoted osteoblast differentiation at the expense of adiopogenesis in multipotent adipose–derived stem (hMADS) cells [19]. Consistently, osteoblast–specific deletion in Col2.3–Cre+;Oxtrfl/fl mice resulted in low bone mass and reduced bone formation, whereas osteoclast-specific deletion in Acp5-Cre+;Oxtrfl/fl mice increased bone mass and reduced resorption [198]––– together suggesting anabolic and pro–resorptive actions of OXT. More importantly, with osteoblast–selective OXTR deletion, the normal maternal bone loss that occurs during pregnancy and lactation was attenuated and Oxt–/– fetuses showed decreased mineralization in trabecular bone [199], suggesting a role for OXT in the increased bone turnover required for intergenerational calcium transfer and fetal skeletal mineralization [24, 198].
Regarding prolactin action, an in vivo study using dams measured bone turnover after prolactin administration during pregnancy. Newborn pups from treated dams showed a ~ 30% decrease in alkaline phosphatase and reduced bone formation, without changes in serum calcium or PTH levels [200]. However, global prolactin receptor–deficient (Prlr–/–) mice showed lower BMD in both sexes. Bone formation parameters were suppressed based on dynamic histomorphometry. This observation might be confounded by hypogonadism, particularly as estrogen levels in female Prlr–/– mice were significantly lower compared with wild type littermates, although testosterone levels in males did not differ [201]. Conversely, hyperprolactinemia, which was induced by anterior pituitary transplantation, exacerbated bone resorption in the context of ovariectomy, potentially through decreased osteoprotegerin (OPG) expression in osteoblasts [202]. The mechanisms by which the rise of OXT and prolactin during pregnancy and lactation affect the maternal skeleton and how a potentially hypophyseal–bone interaction might lead to pregnancy– and lactation–associated osteoporosis need further study.
Vasopressin, water balance and bone
Chronic hyponatremia is associated with osteoporosis and fracture [203,204,205]. Vasopressin (AVP), a key regulator of serum osmolality and fluid status, has been implicated in bone remodeling based on animal studies. Avpr1α−/− mice had high bone mass with increased bone formation and decreased resorption [20, 22]. AVP or AVPR-1a antagonist (SR49059) reduced or increased bone mass, respectively, suggesting that AVP negatively regulates skeletal remodeling [22]. However, blocking AVPR2, the primary receptor in the kidney, by tolvaptan did not affect skeleton [20]. Since the skeleton is the largest reservoir for sodium, it is expected that sodium and calcium resorption from bone are co–regulated, and high AVP levels may contribute to bone loss in the setting of chronic hyponatremia [206, 207].
The broader context
Discoveries that pituitary hormones, once thought to have single functions in regulating endocrine glands, have direct actions on bone, fat, and brain are of considerable physiological importance. These studies have been complicated by intricate feedback loops and dominant sex steroid effects. An evolving understanding of their independent role nonetheless continues to highlight multiple clinical implications. Furthermore, pituitary hormones and their receptors might be a potential therapeutic targets, in essence, highlighting the significance of undersatanding integrative physiology towards innovative drug development. In addition to the FSH–blocking antibody, described above, TSH has been shown to stimulate adipogenesis, adipocyte beiging, and lipolysis in various animal models, which may have clinical implications in altering body composition in patients with thyroid disorders [2, 208,209,210,211]. Likewise, oxytocin besides its central–mediated anorexigenic effects [212], also triggers peripheral beiging and thermogenesis [198, 213,214,215]. Clinical trials investigating the use of OXT for treating obesity in adults and adolescents are thus currently in progress [216].
Data availability
No datasets were generated or analysed during the current study.
References
Kleinau G, Krause G (2009) Thyrotropin and homologous glycoprotein hormone receptors: structural and functional aspects of extracellular signaling mechanisms. Endocr Rev 30:133–151
Davies T, Marians R, Latif R (2002) The TSH receptor reveals itself. J Clin Invest 110:161–164
Inoue M, Tawata M, Yokomori N, Endo T, Onaya T (1998) Expression of thyrotropin receptor on clonal osteoblast-like rat osteosarcoma cells. Thyroid 8:1059–1064
Sampath TK, Simic P, Sendak R, Draca N, Bowe AE, O’Brien S, Schiavi SC, McPherson JM, Vukicevic S (2007) Thyroid-stimulating hormone restores bone volume, microarchitecture, and strength in aged ovariectomized rats. J Bone Miner Res 22:849–859
Sun L, Peng Y, Sharrow AC, Iqbal J, Zhang Z, Papachristou DJ, Zaidi S, Zhu LL, Yaroslavskiy BB, Zhou H, Zallone A, Sairam MR, Kumar TR, Bo W, Braun J, Cardoso-Landa L, Schaffler MB, Moonga BS, Blair HC, Zaidi M (2006) FSH directly regulates bone mass. Cell 125:247–260
Tsai JA, Janson A, Bucht E, Kindmark H, Marcus C, Stark A, Zemack HR, Torring O (2004) Weak evidence of thyrotropin receptors in primary cultures of human osteoblast-like cells. Calcif Tissue Int 74:486–491
Baliram R, Chow A, Huber AK, Collier L, Ali MR, Morshed SA, Latif R, Teixeira A, Merad M, Liu L, Sun L, Blair HC, Zaidi M, Davies TF (2013) Thyroid and bone: macrophage-derived TSH-beta splice variant increases murine osteoblastogenesis. Endocrinology 154:4919–4926
Baliram R, Latif R, Morshed SA, Zaidi M, Davies TF (2016) T3 Regulates a human macrophage-derived TSH-beta splice variant: implications for human bone biology. Endocrinology 157:3658–3667
Harbour DV, Kruger TE, Coppenhaver D, Smith EM, Meyer WJ 3rd (1989) Differential expression and regulation of thyrotropin (TSH) in T cell lines. Mol Cell Endocrinol 64:229–241
Smith EM, Phan M, Kruger TE, Coppenhaver DH, Blalock JE (1983) Human lymphocyte production of immunoreactive thyrotropin. Proc Natl Acad Sci U S A 80:6010–6013
Vincent BH, Montufar-Solis D, Teng BB, Amendt BA, Schaefer J, Klein JR (2009) Bone marrow cells produce a novel TSHbeta splice variant that is upregulated in the thyroid following systemic virus infection. Genes Immun 10:18–26
Feng Y, Zhu S, Antaris AL, Chen H, Xiao Y, Lu X, Jiang L, Diao S, Yu K, Wang Y, Herraiz S, Yue J, Hong X, Hong G, Cheng Z, Dai H, Hsueh AJ (2017) Live imaging of follicle stimulating hormone receptors in gonads and bones using near infrared II fluorophore. Chem Sci 8:3703–3711
Ji Y, Liu P, Yuen T, Haider S, He J, Romero R, Chen H, Bloch M, Kim SM, Lizneva D, Munshi L, Zhou C, Lu P, Iqbal J, Cheng Z, New MI, Hsueh AJ, Bian Z, Rosen CJ, Sun L, Zaidi M (2018) Epitope-specific monoclonal antibodies to FSHbeta increase bone mass. Proc Natl Acad Sci U S A 115:2192–2197
Mountjoy KG, Robbins LS, Mortrud MT, Cone RD (1992) The cloning of a family of genes that encode the melanocortin receptors. Science 257:1248–1251
Zhong Q, Sridhar S, Ruan L, Ding KH, Xie D, Insogna K, Kang B, Xu J, Bollag RJ, Isales CM (2005) Multiple melanocortin receptors are expressed in bone cells. Bone 36:820–831
Knobloch HS, Grinevich V (2014) Evolution of oxytocin pathways in the brain of vertebrates. Front Behav Neurosci 8:31
Yamashita K, Kitano T (2013) Molecular evolution of the oxytocin-oxytocin receptor system in eutherians. Mol Phylogenet Evol 67:520–528
Gimpl G, Fahrenholz F (2001) The oxytocin receptor system: structure, function, and regulation. Physiol Rev 81:629–683
Elabd C, Basillais A, Beaupied H, Breuil V, Wagner N, Scheideler M, Zaragosi LE, Massiera F, Lemichez E, Trajanoski Z, Carle G, Euller-Ziegler L, Ailhaud G, Benhamou CL, Dani C, Amri EZ (2008) Oxytocin controls differentiation of human mesenchymal stem cells and reverses osteoporosis. Stem Cells 26:2399–2407
Sun L, Tamma R, Yuen T, Colaianni G, Ji Y, Cuscito C, Bailey J, Dhawan S, Lu P, Calvano CD, Zhu LL, Zambonin CG, Di Benedetto A, Stachnik A, Liu P, Grano M, Colucci S, Davies TF, New MI, Zallone A, Zaidi M (2016) Functions of vasopressin and oxytocin in bone mass regulation. Proc Natl Acad Sci U S A 113:164–169
Tamma R, Colaianni G, Zhu LL, DiBenedetto A, Greco G, Montemurro G, Patano N, Strippoli M, Vergari R, Mancini L, Colucci S, Grano M, Faccio R, Liu X, Li J, Usmani S, Bachar M, Bab I, Nishimori K, Young LJ, Buettner C, Iqbal J, Sun L, Zaidi M, Zallone A (2009) Oxytocin is an anabolic bone hormone. Proc Natl Acad Sci U S A 106:7149–7154
Tamma R, Sun L, Cuscito C, Lu P, Corcelli M, Li J, Colaianni G, Moonga SS, Di Benedetto A, Grano M, Colucci S, Yuen T, New MI, Zallone A, Zaidi M (2013) Regulation of bone remodeling by vasopressin explains the bone loss in hyponatremia. Proc Natl Acad Sci U S A 110:18644–18649
Colaianni G, Di Benedetto A, Zhu LL, Tamma R, Li J, Greco G, Peng Y, Dell’Endice S, Zhu G, Cuscito C, Grano M, Colucci S, Iqbal J, Yuen T, Sun L, Zaidi M, Zallone A (2011) Regulated production of the pituitary hormone oxytocin from murine and human osteoblasts. Biochem Biophys Res Commun 411:512–515
Colaianni G, Sun L, Di Benedetto A, Tamma R, Zhu LL, Cao J, Grano M, Yuen T, Colucci S, Cuscito C, Mancini L, Li J, Nishimori K, Bab I, Lee HJ, Iqbal J, Young WS 3rd, Rosen C, Zallone A, Zaidi M (2012) Bone marrow oxytocin mediates the anabolic action of estrogen on the skeleton. J Biol Chem 287:29159–29167
Beshyah SA, Freemantle C, Thomas E, Rutherford O, Page B, Murphy M, Johnston DG (1995) Abnormal body composition and reduced bone mass in growth hormone deficient hypopituitary adults. Clin Endocrinol (Oxf) 42:179–189
Holmes SJ, Economou G, Whitehouse RW, Adams JE, Shalet SM (1994) Reduced bone mineral density in patients with adult onset growth hormone deficiency. J Clin Endocrinol Metab 78:669–674
Kaufman JEAN-MARC, Taelman PAUL, Vermeulen A, Vandeweghe M (1992) Bone mineral status in growth hormone-deficient males with isolated and multiple pituitary deficiencies of childhood onset. J Clin Endocrinol Metab 74:118–123
Rosén T, Hansson T, Granhed H, Szucs J, Bengtsson BA (1993) Reduced bone mineral content in adult patients with growth hormone deficiency. Acta Endocrinol (Copenh) 129:201–206
Toogood AA, Adams JE, O’Neill PA, Shalet SM (1997) Elderly patients with adult-onset growth hormone deficiency are not osteopenic. J Clin Endocrinol Metab 82:1462–1466
Yang H, Chen M, Xu H, Zhen Y, Zhang Y, Wang L, Duan L, Gong F, Zhu H, Pan H (2024) Bone mineral density in adults growth hormone deficiency with different ages of onset: a real-world retrospective study. Endocrine. https://doi.org/10.1007/s12020-024-03786-4
Murray RD, Adams JE, Shalet SM (2006) A densitometric and morphometric analysis of the skeleton in adults with varying degrees of growth hormone deficiency. J Clin Endocrinol Metab 91:432–438
Hitz MF, Jensen JE, Eskildsen PC (2006) Bone mineral density in patients with growth hormone deficiency: does a gender difference exist? Clin Endocrinol (Oxf) 65:783–791
Kotzmann H, Bernecker P, Hübsch P, Pietschmann P, Woloszczuk W, Svoboda T, Geyer G, Luger A (1993) Bone mineral density and parameters of bone metabolism in patients with acromegaly. J Bone Miner Res 8:459–465
Kayath MJ, Vieira JG (1997) Osteopenia occurs in a minority of patients with acromegaly and is predominant in the spine. Osteoporos Int 7:226–230
Bolanowski M, Daroszewski J, Medraś M, Zadrozna-Sliwka B (2006) Bone mineral density and turnover in patients with acromegaly in relation to sex, disease activity, and gonadal function. J Bone Miner Metab 24:72–78
Scillitani A, Battista C, Chiodini I, Carnevale V, Fusilli S, Ciccarelli E, Terzolo M, Oppizzi G, Arosio M, Gasperi M, Arnaldi G, Colao A, Baldelli R, Ghiggi MR, Gaia D, Di Somma C, Trischitta V, Liuzzi A (2003) Bone mineral density in acromegaly: the effect of gender, disease activity and gonadal status. Clin Endocrinol (Oxf) 58:725–731
Scillitani A, Chiodini I, Carnevale V, Giannatempo GM, Frusciante V, Villella M, Pileri M, Guglielmi G, Di Giorgio A, Modoni S, Fusilli S, Di Cerbo A, Liuzzi A (1997) Skeletal involvement in female acromegalic subjects: the effects of growth hormone excess in amenorrheal and menstruating patients. J Bone Miner Res 12:1729–1736
Aloia JF, Roginsky MS, Jowsey J, Dombrowski CS, Shukla KK, Cohn SH (1972) Skeletal metabolism and body composition in acromegaly. J Clin Endocrinol Metab 35:543–551
Halse J, Melsen F, Mosekilde L (1981) Iliac crest bone mass and remodelling in acromegaly. Acta Endocrinol (Copenh) 97:18–22
Nielsen HK, Jørgensen JO, Brixen K, Christiansen JS (1991) Serum osteocalcin and bone isoenzyme alkaline phosphatase in growth hormone-deficient patients: dose-response studies with biosynthetic human GH. Calcif Tissue Int 48:82–87
Sartorio A, Conti A, Monzani M, Morabito F, Faglia G (1993) Growth hormone treatment in adults with GH deficiency: effects on new biochemical markers of bone and collagen turnover. J Endocrinol Invest 16:893–898
O’Halloran DJ, Tsatsoulis A, Whitehouse RW, Holmes SJ, Adams JE, Shalet SM (1993) Increased bone density after recombinant human growth hormone (GH) therapy in adults with isolated GH deficiency. J Clin Endocrinol Metab 76:1344–1348
Attanasio AF, Lamberts SW, Matranga AM, Birkett MA, Bates PC, Valk NK, Hilsted J, Bengtsson BA, Strasburger CJ (1997) Adult growth hormone (GH)-deficient patients demonstrate heterogeneity between childhood onset and adult onset before and during human GH treatment. Adult Growth Hormone Deficiency Study Group. J Clin Endocrinol Metab 82:82–88
Gertner JM, Horst RL, Broadus AE, Rasmussen H, Genel M (1979) Parathyroid function and vitamin D metabolism during human growth hormone replacement. J Clin Endocrinol Metab 49:185–188
Ahmad AM, Hopkins MT, Fraser WD, Ooi CG, Durham BH, Vora JP (2003) Parathyroid hormone secretory pattern, circulating activity, and effect on bone turnover in adult growth hormone deficiency. Bone 32:170–179
Broadus AE, Mahaffey JE, Bartter FC, Neer RM (1977) Nephrogenous cyclic adenosine monophosphate as a parathyroid function test. J Clin Invest 60:771–783
Ahmad AM, Hopkins MT, Thomas J, Durham BH, Fraser WD, Vora JP (2004) Parathyroid responsiveness to hypocalcemic and hypercalcemic stimuli in adult growth hormone deficiency after growth hormone replacement. Am J Physiol Endocrinol Metab 286:E986–E993
White HD, Ahmad AM, Durham BH, Peter R, Prabhakar VK, Corlett P, Vora JP, Fraser WD (2007) PTH circadian rhythm and PTH target-organ sensitivity is altered in patients with adult growth hormone deficiency with low BMD. J Bone Miner Res 22:1798–1807
DeChiara TM, Efstratiadis A, Robertson EJ (1990) A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345:78–80
Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A (1993) Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75:59–72
Davey HW, Xie T, McLachlan MJ, Wilkins RJ, Waxman DJ, Grattan DR (2001) STAT5b is required for GH-induced liver IGF-I gene expression. Endocrinology 142:3836–3841
Sims NA, Clément-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, Goffin V, Coschigano K, Gaillard-Kelly M, Kopchick J, Baron R, Kelly PA (2000) Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest 106:1095–1103
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, Brown M, Bodner S, Grosveld G, Ihle JN (1998) Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93:841–850
Ernst M, Rodan GA (1990) Increased activity of insulin-like growth factor (IGF) in osteoblastic cells in the presence of growth hormone (GH): positive correlation with the presence of the GH-induced IGF-binding protein BP-3. Endocrinology 127:807–814
Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D (2002) Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest 110:771–781
Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D (1999) Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A 96:7324–7329
Govoni KE, Lee SK, Chung YS, Behringer RR, Wergedal JE, Baylink DJ, Mohan S (2007) Disruption of insulin-like growth factor-I expression in type IIalphaI collagen-expressing cells reduces bone length and width in mice. Physiol Genomics 30:354–362
Wang Y, Nishida S, Boudignon BM, Burghardt A, Elalieh HZ, Hamilton MM, Majumdar S, Halloran BP, Clemens TL, Bikle DD (2007) IGF-I receptor is required for the anabolic actions of parathyroid hormone on bone. J Bone Miner Res 22:1329–1337
Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL (2002) Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem 277:44005–44012
Zhao G, Monier-Faugere MC, Langub MC, Geng Z, Nakayama T, Pike JW, Chernausek SD, Rosen CJ, Donahue LR, Malluche HH, Fagin JA, Clemens TL (2000) Targeted overexpression of insulin-like growth factor I to osteoblasts of transgenic mice: increased trabecular bone volume without increased osteoblast proliferation. Endocrinology 141:2674–2682
Zhang M, Faugere MC, Malluche H, Rosen CJ, Chernausek SD, Clemens TL (2003) Paracrine overexpression of IGFBP-4 in osteoblasts of transgenic mice decreases bone turnover and causes global growth retardation. J Bone Miner Res 18:836–843
Devlin RD, Du Z, Buccilli V, Jorgetti V, Canalis E (2002) Transgenic mice overexpressing insulin-like growth factor binding protein-5 display transiently decreased osteoblastic function and osteopenia. Endocrinology 143:3955–3962
De Jesus K, Wang X, Liu JL (2009) A general IGF-I overexpression effectively rescued somatic growth and bone deficiency in mice caused by growth hormone receptor knockout. Growth Factors 27:438–447
Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA (2004) Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol 180:247–255
Lupu F, Terwilliger JD, Lee K, Segre GV, Efstratiadis A (2001) Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev Biol 229:141–162
Fritton JC, Emerton KB, Sun H, Kawashima Y, Mejia W, Wu Y, Rosen CJ, Panus D, Bouxsein M, Majeska RJ, Schaffler MB, Yakar S (2010) Growth hormone protects against ovariectomy-induced bone loss in states of low circulating insulin-like growth factor (IGF-1). J Bone Miner Res 25:235–246
Lancer SR, Bowser EN, Hargis GK (1976) The effect of growth hormone on parathyroid function in rats. Endocrinology 98:1289–1293
Vestergaard P, Mosekilde L (2003) Hyperthyroidism, bone mineral, and fracture risk–a meta-analysis. Thyroid 13:585–593
von Recklinghausen FD (1891) Die fibröse oder deformierende Ostitis, die Osteomalacie und die osteoplastische Carcinose in ihren gegenseitigen Beziehungen. In: Virchow Rudolf (ed) Virchow Festschrift. George Reimer, Berlin
Abu EO, Bord S, Horner A, Chatterjee VK, Compston JE (1997) The expression of thyroid hormone receptors in human bone. Bone 21:137–142
Bassett JH, O’Shea PJ, Sriskantharajah S, Rabier B, Boyde A, Howell PG, Weiss RE, Roux JP, Malaval L, Clement-Lacroix P, Samarut J, Chassande O, Williams GR (2007) Thyroid hormone excess rather than thyrotropin deficiency induces osteoporosis in hyperthyroidism. Mol Endocrinol 21:1095–1107
Britto JM, Fenton AJ, Holloway WR, Nicholson GC (1994) Osteoblasts mediate thyroid hormone stimulation of osteoclastic bone resorption. Endocrinology 134:169–176
Rizzoli R, Poser J, Bürgi U (1986) Nuclear thyroid hormone receptors in cultured bone cells. Metabolism 35:71–74
Sato K, Han DC, Fujii Y, Tsushima T, Shizume K (1987) Thyroid hormone stimulates alkaline phosphatase activity in cultured rat osteoblastic cells (ROS 17/2.8) through 3,5,3’-triiodo-L-thyronine nuclear receptors. Endocrinology 120:1873–1881
Abe E, Marians RC, Yu W, Wu XB, Ando T, Li Y, Iqbal J, Eldeiry L, Rajendren G, Blair HC, Davies TF, Zaidi M (2003) TSH is a negative regulator of skeletal remodeling. Cell 115:151–162
Kim SM, Ryu V, Miyashita S, Korkmaz F, Lizneva D, Gera S, Latif R, Davies TF, Iqbal J, Yuen T, Zaidi M (2021) Thyrotropin, hyperthyroidism, and bone mass. J Clin Endocrinol Metab 106:e4809–e4821
Abrahamsen B, Jorgensen HL, Laulund AS, Nybo M, Brix TH, Hegedus L (2014) Low serum thyrotropin level and duration of suppression as a predictor of major osteoporotic fractures-the OPENTHYRO register cohort. J Bone Miner Res 29:2040–2050
Bauer DC, Ettinger B, Nevitt MC, Stone KL, Group Study of Osteoporotic Fractures Research (2001) Risk for fracture in women with low serum levels of thyroid-stimulating hormone. Ann Intern Med 134:561–568
Kim DJ, Khang YH, Koh JM, Shong YK, Kim GS (2006) Low normal TSH levels are associated with low bone mineral density in healthy postmenopausal women. Clin Endocrinol (Oxf) 64:86–90
Lee SJ, Kim KM, Lee EY, Song MK, Kang DR, Kim HC, Youm Y, Yun YM, Park HY, Kim CO, Rhee Y (2016) Low normal TSH levels are associated with impaired BMD and hip geometry in the elderly. Aging Dis 7:734–743
Morris MS (2007) The association between serum thyroid-stimulating hormone in its reference range and bone status in postmenopausal American women. Bone 40:1128–1134
Noh HM, Park YS, Lee J, Lee W (2015) A cross-sectional study to examine the correlation between serum TSH levels and the osteoporosis of the lumbar spine in healthy women with normal thyroid function. Osteoporos Int 26:997–1003
van der Deure WM, Uitterlinden AG, Hofman A, Rivadeneira F, Pols HA, Peeters RP, Visser TJ (2008) Effects of serum TSH and FT4 levels and the TSHR-Asp727Glu polymorphism on bone: the Rotterdam study. Clin Endocrinol (Oxf) 68:175–181
Waring AC, Harrison S, Fink HA, Samuels MH, Cawthon PM, Zmuda JM, Orwoll ES, Bauer DC, Study Osteoporotic Fractures in Men (2013) A prospective study of thyroid function, bone loss, and fractures in older men: The MrOS study. J Bone Miner Res 28:472–479
Meier C, Beat M, Guglielmetti M, Christ-Crain M, Staub JJ, Kraenzlin M (2004) Restoration of euthyroidism accelerates bone turnover in patients with subclinical hypothyroidism: a randomized controlled trial. Osteoporos Int 15:209–216
Büchi AE, Feller M, Netzer S, Blum MR, Gonzalez Rodriguez E, Collet TH, Del Giovane C, van Heemst D, Quinn T, Kearney PM, Westendorp RGJ, Gussekloo J, Mooijaart SP, Hans D, Bauer DC, Rodondi N, Aeberli D (2022) Bone geometry in older adults with subclinical hypothyroidism upon levothyroxine therapy: a nested study within a randomized placebo controlled trial. Bone 161:116404
Garin MC, Arnold AM, Lee JS, Robbins J, Cappola AR (2014) Subclinical thyroid dysfunction and hip fracture and bone mineral density in older adults: the cardiovascular health study. J Clin Endocrinol Metab 99:2657–2664
Lee JS, Buzková P, Fink HA, Vu J, Carbone L, Chen Z, Cauley J, Bauer DC, Cappola AR, Robbins J (2010) Subclinical thyroid dysfunction and incident hip fracture in older adults. Arch Intern Med 170:1876–1883
Segna D, Bauer DC, Feller M, Schneider C, Fink HA, Aubert CE, Collet TH, da Costa BR, Fischer K, Peeters RP, Cappola AR, Blum MR, van Dorland HA, Robbins J, Naylor K, Eastell R, Uitterlinden AG, Rivadeneira Ramirez F, Gogakos A, Gussekloo J, Williams GR, Schwartz A, Cauley JA, Aujesky DA, Bischoff-Ferrari HA, Rodondi N (2018) Association between subclinical thyroid dysfunction and change in bone mineral density in prospective cohorts. J Intern Med 283:56–72
Siru R, Alfonso H, Chubb SAP, Golledge J, Flicker L, Yeap BB (2018) Subclinical thyroid dysfunction and circulating thyroid hormones are not associated with bone turnover markers or incident hip fracture in older men. Clin Endocrinol (Oxf) 89:93–99
Karga H, Papaioannou G, Polymeris A, Papamichael K, Karpouza A, Samouilidou E, Papaioannou P (2010) The effects of recombinant human TSH on bone turnover in patients after thyroidectomy. J Bone Miner Metab 28:35–41
Mazziotti G, Sorvillo F, Piscopo M, Cioffi M, Pilla P, Biondi B, Iorio S, Giustina A, Amato G, Carella C (2005) Recombinant human TSH modulates in vivo C-telopeptides of type-1 collagen and bone alkaline phosphatase, but not osteoprotegerin production in postmenopausal women monitored for differentiated thyroid carcinoma. J Bone Miner Res 20:480–486
Martini G, Gennari L, De Paola V, Pilli T, Salvadori S, Merlotti D, Valleggi F, Campagna S, Franci B, Avanzati A, Nuti R, Pacini F (2008) The effects of recombinant TSH on bone turnover markers and serum osteoprotegerin and RANKL levels. Thyroid 18:455–460
Albagha OME, Natarajan R, Reid DM, Ralston SH (2005) The D727E polymorphism of the human thyroid stimulating hormone receptor is associated with bone mineral density and bone loss in women from the UK. J Bone Miner Res 20:S341–S441
Liu RD, Chen RX, Li WR, Huang YL, Li WH, Cai GR, Zhang H (2012) The Glu727 allele of thyroid stimulating hormone receptor gene is associated with osteoporosis. N Am J Med Sci 4:300–304
van Vliet NA, Noordam R, van Klinken JB, Westendorp RG, Bassett JD, Williams GR, van Heemst D (2018) Thyroid stimulating hormone and bone mineral density: evidence from a two-sample mendelian randomization study and a candidate gene association study. J Bone Miner Res 33:1318–1325
Sun L, Zhu LL, Lu P, Yuen T, Li J, Ma R, Baliram R, Moonga SS, Liu P, Zallone A, New MI, Davies TF, Zaidi M (2013) Genetic confirmation for a central role for TNFalpha in the direct action of thyroid stimulating hormone on the skeleton. Proc Natl Acad Sci U S A 110:9891–9896
Baliram R, Sun L, Cao J, Li J, Latif R, Huber AK, Yuen T, Blair HC, Zaidi M, Davies TF (2012) Hyperthyroid-associated osteoporosis is exacerbated by the loss of TSH signaling. J Clin Invest 122:3737–3741
Ma R, Morshed S, Latif R, Zaidi M, Davies TF (2011) The influence of thyroid-stimulating hormone and thyroid-stimulating hormone receptor antibodies on osteoclastogenesis. Thyroid 21:897–906
Sun L, Vukicevic S, Baliram R, Yang G, Sendak R, McPherson J, Zhu LL, Iqbal J, Latif R, Natrajan A, Arabi A, Yamoah K, Moonga BS, Gabet Y, Davies TF, Bab I, Abe E, Sampath K, Zaidi M (2008) Intermittent recombinant TSH injections prevent ovariectomy-induced bone loss. Proc Natl Acad Sci U S A 105:4289–4294
Iqbal J, Sun L, Kumar TR, Blair HC, Zaidi M (2006) Follicle-stimulating hormone stimulates TNF production from immune cells to enhance osteoblast and osteoclast formation. Proc Natl Acad Sci U S A 103:14925–14930
Hase H, Ando T, Eldeiry L, Brebene A, Peng Y, Liu L, Amano H, Davies TF, Sun L, Zaidi M, Abe E (2006) TNFalpha mediates the skeletal effects of thyroid-stimulating hormone. Proc Natl Acad Sci U S A 103:12849–12854
Yamoah K, Brebene A, Baliram R, Inagaki K, Dolios G, Arabi A, Majeed R, Amano H, Wang R, Yanagisawa R, Abe E (2008) High-mobility group box proteins modulate tumor necrosis factor-alpha expression in osteoclastogenesis via a novel deoxyribonucleic acid sequence. Mol Endocrinol 22:1141–1153
Baliram R, Latif R, Berkowitz J, Frid S, Colaianni G, Sun L, Zaidi M, Davies TF (2011) Thyroid-stimulating hormone induces a Wnt-dependent, feed-forward loop for osteoblastogenesis in embryonic stem cell cultures. Proc Natl Acad Sci U S A 108:16277–16282
Liu C, Miao J, Liu X, Zhao Z, Kou T, Liu J, Wang R, Li L, Dong Q (2019) HPT axis-independent TSHβ splice variant regulates the synthesis of thyroid hormone in mice. Mol Med Rep 19:4514–4522
Montufar-Solis D, Klein JR (2016) Splenic leukocytes traffic to the thyroid and produce a novel TSHβ isoform during acute listeria monocytogenes infection in mice. PLoS ONE 11:e0146111
van der Poll T, Romijn JA, Wiersinga WM, Sauerwein HP (1990) Tumor necrosis factor: a putative mediator of the sick euthyroid syndrome in man. J Clin Endocrinol Metab 71:1567–1572
Randolph JF Jr, Sowers M, Gold EB, Mohr BA, Luborsky J, Santoro N, McConnell DS, Finkelstein JS, Korenman SG, Matthews KA, Sternfeld B, Lasley BL (2003) Reproductive hormones in the early menopausal transition: relationship to ethnicity, body size, and menopausal status. J Clin Endocrinol Metab 88:1516–1522
Sowers MR, Greendale GA, Bondarenko I, Finkelstein JS, Cauley JA, Neer RM, Ettinger B (2003) Endogenous hormones and bone turnover markers in pre- and perimenopausal women: SWAN. Osteoporos Int 14:191–197
Cheung E, Tsang S, Bow C, Soong C, Yeung S, Loong C, Cheung CL, Kan A, Lo S, Tam S, Tang G, Kung A (2011) Bone loss during menopausal transition among southern Chinese women. Maturitas 69:50–56
Wang B, Song Y, Chen Y, Wang ES, Zheng D, Qu F, Zhou JH (2015) Correlation analysis for follicle-stimulating hormone and C-terminal cross-linked telopetides of type i collagen in menopausal transition women with osteoporosis. Int J Clin Exp Med 8:2417–2422
Wang J, Zhang W, Yu C, Zhang X, Zhang H, Guan Q, Zhao J, Xu J (2015) Follicle-Stimulating hormone increases the risk of postmenopausal osteoporosis by stimulating osteoclast differentiation. PLoS ONE 10:e0134986
Wu XY, Wu XP, Xie H, Zhang H, Peng YQ, Yuan LQ, Su X, Luo XH, Liao EY (2010) Age-related changes in biochemical markers of bone turnover and gonadotropin levels and their relationship among Chinese adult women. Osteoporos Int 21:275–285
Xu ZR, Wang AH, Wu XP, Zhang H, Sheng ZF, Wu XY, Xie H, Luo XH, Liao EY (2009) Relationship of age-related concentrations of serum FSH and LH with bone mineral density, prevalence of osteoporosis in native Chinese women. Clin Chim Acta 400:8–13
Adami S, Bianchi G, Brandi ML, Giannini S, Ortolani S, DiMunno O, Frediani B, Rossini M, Bonturno Study Group (2008) Determinants of bone turnover markers in healthy premenopausal women. Calcif Tissue Int 82:341–347
Gallagher CM, Moonga BS, Kovach JS (2010) Cadmium, follicle-stimulating hormone, and effects on bone in women age 42–60 years, NHANES III. Environ Res 110:105–111
Veldhuis-Vlug AG, Woods GN, Sigurdsson S, Ewing SK, Le PT, Hue TF, Vittinghoff E, Xu K, Gudnason V, Sigurdsson G, Kado DM, Eiriksdottir G, Harris T, Schafer AL, Li X, Zaidi M, Rosen CJ, Schwartz AV (2021) Serum FSH is associated with BMD, bone marrow adiposity, and body composition in the AGES-Reykjavik study of older adults. J Clin Endocrinol Metab 106:e1156–e1169
Devleta B, Adem B, Senada S (2004) Hypergonadotropic amenorrhea and bone density: new approach to an old problem. J Bone Miner Metab 22:360–364
Podfigurna-Stopa A, Pludowski P, Jaworski M, Lorenc R, Genazzani AR, Meczekalski B (2012) Skeletal status and body composition in young women with functional hypothalamic amenorrhea. Gynecol Endocrinol 28:299–304
Chrysis D, Spiliotis BE, Stene M, Cacciari E, Davenport ML (2006) Gonadotropin secretion in girls with turner syndrome measured by an ultrasensitive immunochemiluminometric assay. Horm Res 65:261–266
Hagen CP, Main KM, Kjaergaard S, Juul A (2010) FSH, LH, inhibin B and estradiol levels in turner syndrome depend on age and karyotype: longitudinal study of 70 Turner girls with or without spontaneous puberty. Hum Reprod 25:3134–3141
Davies MC, Gulekli B, Jacobs HS (1995) Osteoporosis in turner’s syndrome and other forms of primary amenorrhoea. Clin Endocrinol (Oxf) 43:741–746
Faienza MF, Brunetti G, Ventura A, Piacente L, Messina MF, De Luca F, Ciccarelli M, Oranger A, Mori G, Natale MP, Gigante M, Ranieri E, Gesualdo L, Colucci S, Cavallo L, Grano M (2015) Mechanisms of enhanced osteoclastogenesis in girls and young women with turner’s syndrome. Bone 81:228–236
Gravholt CH, Lauridsen AL, Brixen K, Mosekilde L, Heickendorff L, Christiansen JS (2002) Marked disproportionality in bone size and mineral, and distinct abnormalities in bone markers and calcitropic hormones in adult turner syndrome: a cross-sectional study. J Clin Endocrinol Metab 87:2798–2808
Ikegawa K, Koga E, Itonaga T, Sakakibara H, Kawai M, Hasegawa Y (2024) Factors associated with low bone mineral density in Turner syndrome: a multicenter prospective observational study. Endocr J. https://doi.org/10.1507/endocrj.EJ23-0628
Drake MT, McCready LK, Hoey KA, Atkinson EJ, Khosla S (2010) Effects of suppression of follicle-stimulating hormone secretion on bone resorption markers in postmenopausal women. J Clin Endocrinol Metab 95:5063–5068
Rendina D, Gianfrancesco F, De Filippo G, Merlotti D, Esposito T, Mingione A, Nuti R, Strazzullo P, Mossetti G, Gennari L (2010) FSHR gene polymorphisms influence bone mineral density and bone turnover in postmenopausal women. Eur J Endocrinol 163:165–172
Mendoza N, Quereda F, Presa J, Salamanca A, Sanchez-Borrego R, Vazquez F, Martinez Astorquiza T (2012) Estrogen-related genes and postmenopausal osteoporosis risk. Climacteric 15:587–593
Liu S, Cheng Y, Fan M, Chen D, Bian Z (2010) FSH aggravates periodontitis-related bone loss in ovariectomized rats. J Dent Res 89:366–371
Liu S, Cheng Y, Xu W, Bian Z (2010) Protective effects of follicle-stimulating hormone inhibitor on alveolar bone loss resulting from experimental periapical lesions in ovariectomized rats. J Endod 36:658–663
Lukefahr AL, Frye JB, Wright LE, Marion SL, Hoyer PB, Funk JL (2012) Decreased bone mineral density in rats rendered follicle-deplete by an ovotoxic chemical correlates with changes in follicle-stimulating hormone and inhibin A. Calcif Tissue Int 90:239–249
Robinson LJ, Tourkova I, Wang Y, Sharrow AC, Landau MS, Yaroslavskiy BB, Sun L, Zaidi M, Blair HC (2010) FSH-receptor isoforms and FSH-dependent gene transcription in human monocytes and osteoclasts. Biochem Biophys Res Commun 394:12–17
Sun L, Zhang Z, Zhu LL, Peng Y, Liu X, Li J, Agrawal M, Robinson LJ, Iqbal J, Blair HC, Zaidi M (2010) Further evidence for direct pro-resorptive actions of FSH. Biochem Biophys Res Commun 394:6–11
Wu Y, Torchia J, Yao W, Lane NE, Lanier LL, Nakamura MC, Humphrey MB (2007) Bone microenvironment specific roles of ITAM adapter signaling during bone remodeling induced by acute estrogen-deficiency. PLoS ONE 2:e586
Cannon JG, Kraj B, Sloan G (2011) Follicle-stimulating hormone promotes RANK expression on human monocytes. Cytokine 53:141–144
Cannon JG, Cortez-Cooper M, Meaders E, Stallings J, Haddow S, Kraj B, Sloan G, Mulloy A (2010) Follicle-stimulating hormone, interleukin-1, and bone density in adult women. Am J Physiol Regul Integr Comp Physiol 298:R790–R798
Gertz ER, Silverman NE, Wise KS, Hanson KB, Alekel DL, Stewart JW, Perry CD, Bhupathiraju SN, Kohut ML, Van Loan MD (2010) Contribution of serum inflammatory markers to changes in bone mineral content and density in postmenopausal women: a 1-year investigation. J Clin Densitom 13:277–282
Zhu LL, Blair H, Cao J, Yuen T, Latif R, Guo L, Tourkova IL, Li J, Davies TF, Sun L, Bian Z, Rosen C, Zallone A, New MI, Zaidi M (2012) Blocking antibody to the beta-subunit of FSH prevents bone loss by inhibiting bone resorption and stimulating bone synthesis. Proc Natl Acad Sci U S A 109:14574–14579
Gera S, Sant D, Haider S, Korkmaz F, Kuo TC, Mathew M, Perez-Pena H, Xie H, Chen H, Batista R, Ma K, Cheng Z, Hadelia E, Robinson C, Macdonald A, Miyashita S, Williams A, Jebian G, Miyashita H, Gumerova A, Ievleva K, Smith P, He J, Ryu V, DeMambro V, Quinn MA, Meseck M, Kim SM, Kumar TR, Iqbal J, New MI, Lizneva D, Rosen CJ, Hsueh AJ, Yuen T, Zaidi M (2020) First-in-class humanized FSH blocking antibody targets bone and fat. Proc Natl Acad Sci U S A 117:28971–28979
Cui H, Zhao G, Liu R, Zheng M, Chen J, Wen J (2012) FSH stimulates lipid biosynthesis in chicken adipose tissue by upregulating the expression of its receptor FSHR. J Lipid Res 53:909–917
Liu XM, Chan HC, Ding GL, Cai J, Song Y, Wang TT, Zhang D, Chen H, Yu MK, Wu YT, Qu F, Liu Y, Lu YC, Adashi EY, Sheng JZ, Huang HF (2015) FSH regulates fat accumulation and redistribution in aging through the Galphai/Ca(2+)/CREB pathway. Aging Cell 14:409–420
Sponton CH, Kajimura S (2017) Burning fat and building bone by FSH blockade. Cell Metab 26:285–287
Han X, Meng F, Cao X, Du X, Bu G, Kong F, Huang A, Zeng X (2021) FSH promotes fat accumulation by activating PPARgamma signaling in surgically castrated, but not immunocastrated, male pigs. Theriogenology 160:10–17
Liu P, Ji Y, Yuen T, Rendina-Ruedy E, DeMambro VE, Dhawan S, Abu-Amer W, Izadmehr S, Zhou B, Shin AC, Latif R, Thangeswaran P, Gupta A, Li J, Shnayder V, Robinson ST, Yu YE, Zhang X, Yang F, Lu P, Zhou Y, Zhu LL, Oberlin DJ, Davies TF, Reagan MR, Brown A, Kumar TR, Epstein S, Iqbal J, Avadhani NG, New MI, Molina H, van Klinken JB, Guo EX, Buettner C, Haider S, Bian Z, Sun L, Rosen CJ, Zaidi M (2017) Blocking FSH induces thermogenic adipose tissue and reduces body fat. Nature 546:107–112
Cero C, Lea HJ, Zhu KY, Shamsi F, Tseng YH, Cypess AM (2021) beta3-Adrenergic receptors regulate human brown/beige adipocyte lipolysis and thermogenesis. JCI Insight. 6
Randolph JF Jr, Sowers M, Bondarenko IV, Harlow SD, Luborsky JL, Little RJ (2004) Change in estradiol and follicle-stimulating hormone across the early menopausal transition: effects of ethnicity and age. J Clin Endocrinol Metab 89:1555–1561
Sowers M, Zheng H, Tomey K, Karvonen-Gutierrez C, Jannausch M, Li X, Yosef M, Symons J (2007) Changes in body composition in women over six years at midlife: ovarian and chronological aging. J Clin Endocrinol Metab 92:895–901
Ostergren PB, Kistorp C, Fode M, Bennedbaek FN, Faber J, Sonksen J (2019) Metabolic consequences of gonadotropin-releasing hormone agonists vs orchiectomy: a randomized clinical study. BJU Int 123:602–611
Brookmeyer R, Gray S, Kawas C (1998) Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health 88:1337–1342
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. a meta-analysis. APOE and Alzheimer Disease meta analysis consortium. JAMA 278:1349–1356
Fisher DW, Bennett DA, Dong H (2018) Sexual dimorphism in predisposition to Alzheimer’s disease. Neurobiol Aging 70:308–324
Koran MEI, Wagener M, Hohman TJ, Neuroimaging IA (2017) Sex differences in the association between AD biomarkers and cognitive decline. Brain Imaging Behav 11:205–213
Laws KR, Irvine K, Gale TM (2016) Sex differences in cognitive impairment in Alzheimer’s disease. World J Psychiatry 6:54–65
Asthana S, Baker LD, Craft S, Stanczyk FZ, Veith RC, Raskind MA, Plymate SR (2001) High-dose estradiol improves cognition for women with AD: results of a randomized study. Neurology 57:605–612
Doraiswamy PM, Bieber F, Kaiser L, Krishnan KR, Reuning-Scherer J, Gulanski B (1997) The Alzheimer’s disease assessment scale: patterns and predictors of baseline cognitive performance in multicenter Alzheimer’s disease trials. Neurology 48:1511–1517
Espeland MA, Rapp SR, Shumaker SA, Brunner R, Manson JE, Sherwin BB, Hsia J, Margolis KL, Hogan PE, Wallace R, Dailey M, Freeman R, Hays J, Study Women’s Health Initiative Memory (2004) Conjugated equine estrogens and global cognitive function in postmenopausal women: women’s health initiative memory study. JAMA 291:2959–2968
Paganini-Hill A, Henderson VW (1996) Estrogen replacement therapy and risk of Alzheimer disease. Arch Intern Med 156:2213–2217
Shumaker SA, Legault C, Kuller L, Rapp SR, Thal L, Lane DS, Fillit H, Stefanick ML, Hendrix SL, Lewis CE, Masaki K, Coker LH, Study Women’s Health Initiative Memory (2004) Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s health initiative memory study. JAMA 291:2947–2958
Molnar G, Kassai-Bazsa Z (1997) Gonadotropin, ACTH, prolactin, sexual steroid and cortisol levels in postmenopausal women’s cerebrospinal fluid (CSF). Arch Gerontol Geriatr 24:269–280
Lei ZM, Rao CV, Kornyei JL, Licht P, Hiatt ES (1993) Novel expression of human chorionic gonadotropin/luteinizing hormone receptor gene in brain. Endocrinology 132:2262–2270
Ryu V, Gumerova A, Korkmaz F, Kang SS, Katsel P, Miyashita S, Kannangara H, Cullen L, Chan P, Kuo T, Padilla A, Sultana F, Wizman SA, Kramskiy N, Zaidi S, Kim SM, New MI, Rosen CJ, Goosens KA, Frolinger T, Haroutunian V, Ye K, Lizneva D, Davies TF, Yuen T, Zaidi M (2022) Brain atlas for glycoprotein hormone receptors at single-transcript level. Elife. https://doi.org/10.7554/eLife.79612
Xiong J, Kang SS, Wang Z, Liu X, Kuo TC, Korkmaz F, Padilla A, Miyashita S, Chan P, Zhang Z, Katsel P, Burgess J, Gumerova A, Ievleva K, Sant D, Yu SP, Muradova V, Frolinger T, Lizneva D, Iqbal J, Goosens KA, Gera S, Rosen CJ, Haroutunian V, Ryu V, Yuen T, Zaidi M, Ye K (2022) FSH blockade improves cognition in mice with Alzheimer’s disease. Nature 603:470–476
Epperson CN, Sammel MD, Freeman EW (2013) Menopause effects on verbal memory: findings from a longitudinal community cohort. J Clin Endocrinol Metab 98:3829–3838
Greendale GA, Huang MH, Wight RG, Seeman T, Luetters C, Avis NE, Johnston J, Karlamangla AS (2009) Effects of the menopause transition and hormone use on cognitive performance in midlife women. Neurology 72:1850–1857
Greendale GA, Karlamangla AS, Maki PM (2020) The menopause transition and cognition. JAMA 323:1495–1496
Bowen RL, Isley JP, Atkinson RL (2000) An association of elevated serum gonadotropin concentrations and Alzheimer disease? J Neuroendocrinol 12:351–354
Li Y, Li S, Xu S, Yu H, Tang L, Liu X, Wang X, Zhang Y, Zhang K, Mi S, Chen M, Cui H (2020) Association of androgens and gonadotropins with amnestic mild cognitive impairment and probable Alzheimer’s disease in Chinese elderly men. J Alzheimers Dis 78:277–290
Short RA, Bowen RL, O’Brien PC, Graff-Radford NR (2001) Elevated gonadotropin levels in patients with Alzheimer disease. Mayo Clin Proc 76:906–909
Nerattini M, Rubino F, Jett S, Andy C, Boneu C, Zarate C, Carlton C, Loeb-Zeitlin S, Havryliuk Y, Pahlajani S, Williams S, Berti V, Christos P, Fink M, Dyke JP, Brinton RD, Mosconi L (2023) Elevated gonadotropin levels are associated with increased biomarker risk of Alzheimer’s disease in midlife women. Front Dement. https://doi.org/10.3389/frdem.2023.1303256
Gregory CW, Bowen RL (2005) Novel therapeutic strategies for Alzheimer’s disease based on the forgotten reproductive hormones. Cell Mol Life Sci 62:313–319
Swayzer DV, Gerriets V (2024) Leuprolide. In: StatPearls, Treasure Island
Frolinger T, Korkmaz F, Sims S, Sen F, Sultana F, Laurencin V, Cullen L, Pallapati AR, Liu A, Rojekar S, Pevnev G, Cheliadinova U, Vasilyeva D, Burganova G, Macdonald A, Saxena M, Goosens K, Rosen C, Barak O, Lizneva D, Gumerova A, Ye K, Ryu V, Yuen T, Zaidi M (2024) Gene-dose-dependent reduction fshr expression improves spatial memory deficits in Alzheimer’s mice. Res Sq. https://doi.org/10.21203/rs.3.rs-3964789/v1
Xiong J, Kang SS, Wang M, Wang Z, Xia Y, Liao J, Liu X, Yu SP, Zhang Z, Ryu V, Yuen T, Zaidi M, Ye K (2023) FSH and ApoE4 contribute to Alzheimer’s disease-like pathogenesis via C/EBPbeta/delta-secretase in female mice. Nat Commun 14:6577
Manfredi-Lozano M, Leysen V, Adamo M, Paiva I, Rovera R, Pignat JM, Timzoura FE, Candlish M, Eddarkaoui S, Malone SA, Silva MSB, Trova S, Imbernon M, Decoster L, Cotellessa L, Tena-Sempere M, Claret M, Paoloni-Giacobino A, Plassard D, Paccou E, Vionnet N, Acierno J, Maceski AM, Lutti A, Pfrieger F, Rasika S, Santoni F, Boehm U, Ciofi P, Buee L, Haddjeri N, Boutillier AL, Kuhle J, Messina A, Draganski B, Giacobini P, Pitteloud N, Prevot V (2022) GnRH replacement rescues cognition in down syndrome. Science 377:eabq4515
Corbo RM, Gambina G, Broggio E, Scacchi R (2011) Influence of variation in the follicle-stimulating hormone receptor gene (FSHR) and age at menopause on the development of Alzheimer’s disease in women. Dement Geriatr Cogn Disord 32:63–69
Schupf N, Kapell D, Nightingale B, Rodriguez A, Tycko B, Mayeux R (1998) Earlier onset of Alzheimer’s disease in men with down syndrome. Neurology 50:991–995
Campbell WA, Lowther J, McKenzie I, Price WH (1982) Serum gonadotrophins in down’s syndrome. J Med Genet 19:98–99
Hestnes A, Stovner LJ, Husoy O, Folling I, Fougner KJ, Sjaastad O (1991) Hormonal and biochemical disturbances in down’s syndrome. J Ment Defic Res 35(Pt 3):179–193
Zhu LL, Tourkova I, Yuen T, Robinson LJ, Bian Z, Zaidi M, Blair HC (2012) Blocking FSH action attenuates osteoclastogenesis. Biochem Biophys Res Commun 422:54–58
Han X, Guan Z, Xu M, Zhang Y, Yao H, Meng F, Zhuo Y, Yu G, Cao X, Du X, Bu G, Kong F, Huang A, Zeng X (2020) A novel follicle-stimulating hormone vaccine for controlling fat accumulation. Theriogenology 148:103–111
Gera S, Kuo TC, Gumerova AA, Korkmaz F, Sant D, DeMambro V, Sudha K, Padilla A, Prevot G, Munitz J, Teunissen A, van Leent MMT, Tgjm Post JC, Fernandes J, Netto F, Sultana E, Shelly S, Rojekar P, Kumar L, Cullen J, Chatterjee A, Pallapati S, Miyashita H, Kannangara M, Bhongade P, Sengupta K, Ievleva V, Muradova R, Batista C, Robinson A, Macdonald S, Hutchison M, Saxena M, Meseck J, Caminis J, Iqbal MI, New V, Ryu SM, Kim JJ, Cao N, Zaidi ZA, Fayad D, Lizneva CJ, Rosen TY, Zaidi M (2022) FSH-blocking therapeutic for osteoporosis. Elife. https://doi.org/10.7554/eLife.78022
Rojekar S, Pallapati AR, Gimenez-Roig J, Korkmaz F, Sultana F, Sant D, Haeck CM, Macdonald A, Kim SM, Rosen CJ, Barak O, Meseck M, Caminis J, Lizneva D, Yuen T, Zaidi M (2023) Development and biophysical characterization of a humanized FSH-blocking monoclonal antibody therapeutic formulated at an ultra-high concentration. Elife. https://doi.org/10.7554/eLife.88898
Sant D, Rojekar S, Gera S, Pallapati AR, Gimenez-Roig J, Kuo TC, Padilla A, Korkmaz F, Cullen L, Chatterjee J, Shelly E, Meseck M, Miyashita S, Macdonald A, Sultana F, Barak O, Ryu V, Kim SM, Robinson C, Rosen CJ, Caminis J, Lizneva D, Haider S, Yuen T, Zaidi M (2023) Optimizing a therapeutic humanized follicle-stimulating hormone-blocking antibody formulation by protein thermal shift assay. Ann N Y Acad Sci 1521:67–78
Compston J (2018) Glucocorticoid-induced osteoporosis: an update. Endocrine 61:7–16
Hofbauer LC, Rauner M (2009) Minireview: live and let die: molecular effects of glucocorticoids on bone cells. Mol Endocrinol 23:1525–1531
Isales CM, Zaidi M, Blair HC (2010) ACTH is a novel regulator of bone mass. Ann N Y Acad Sci 1192:110–116
Minetto M, Reimondo G, Osella G, Ventura M, Angeli A, Terzolo M (2004) Bone loss is more severe in primary adrenal than in pituitary-dependent cushing’s syndrome. Osteoporos Int 15:855–861
Sato T, Iwata T, Usui M, Kokabu S, Sugamori Y, Takaku Y, Kobayashi T, Ito K, Matsumoto M, Takeda S, Xu R, Chida D (2020) Bone phenotype in melanocortin 2 receptor-deficient mice. Bone Rep 13:100713
Sadeghi F, Vahednia E, Naderi Meshkin H, Kerachian MA (2020) The effect of adrenocorticotropic hormone on alpha-2-macroglobulin in osteoblasts derived from human mesenchymal stem cells. J Cell Mol Med 24:4784–4790
Tourkova IL, Liu L, Sutjarit N, Larrouture QC, Luo J, Robinson LJ, Blair HC (2017) Adrenocorticotropic hormone and 1,25-dihydroxyvitamin D(3) enhance human osteogenesis in vitro by synergistically accelerating the expression of bone-specific genes. Lab Invest 97:1072–1083
Zaidi M, Sun L, Robinson LJ, Tourkova IL, Liu L, Wang Y, Zhu LL, Liu X, Li J, Peng Y, Yang G, Shi X, Levine A, Iqbal J, Yaroslavskiy BB, Isales C, Blair HC (2010) ACTH protects against glucocorticoid-induced osteonecrosis of bone. Proc Natl Acad Sci U S A 107:8782–8787
Kovacs CS (2016) Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiol Rev 96:449–547
Purdie DW, Aaron JE, Selby PL (1988) Bone histology and mineral homeostasis in human pregnancy. Br J Obstet Gynaecol 95:849–854
Tolstykh EI, Degteva MO, Kozheurov VP, Burmistrov DS (1998) Strontium transfer from maternal skeleton to the fetus estimated on the basis of the Techa river data. Radiat Prot Dosimetry 79:307–310
Yamaga A, Taga M, Minaguchi H, Sato K (1996) Changes in bone mass as determined by ultrasound and biochemical markers of bone turnover during pregnancy and puerperium: a longitudinal study. J Clin Endocrinol Metab 81:752–756
Kyvernitakis I, Reuter TC, Hellmeyer L, Hars O, Hadji P (2018) Subsequent fracture risk of women with pregnancy and lactation-associated osteoporosis after a median of 6 years of follow-up. Osteoporos Int 29:135–142
Athonvarangkul D, Wysolmerski JJ (2023) Crosstalk within a brain-breast-bone axis regulates mineral and skeletal metabolism during lactation. Front Physiol 14:1121579
Sun L, Lizneva D, Ji Y, Colaianni G, Hadelia E, Gumerova A, Ievleva K, Kuo TC, Korkmaz F, Ryu V, Rahimova A, Gera S, Taneja C, Khan A, Ahmad N, Tamma R, Bian Z, Zallone A, Kim SM, New MI, Iqbal J, Yuen T, Zaidi M (2019) Oxytocin regulates body composition. Proc Natl Acad Sci U S A 116:26808–26815
Liu X, Shimono K, Zhu LL, Li J, Peng Y, Imam A, Iqbal J, Moonga S, Colaianni G, Su C, Lu Z, Iwamoto M, Pacifici M, Zallone A, Sun L, Zaidi M (2009) Oxytocin deficiency impairs maternal skeletal remodeling. Biochem Biophys Res Commun 388:161–166
Coss D, Yang L, Kuo CB, Xu X, Luben RA, Walker AM (2000) Effects of prolactin on osteoblast alkaline phosphatase and bone formation in the developing rat. Am J Physiol Endocrinol Metab 279:E1216–E1225
Clement-Lacroix P, Ormandy C, Lepescheux L, Ammann P, Damotte D, Goffin V, Bouchard B, Amling M, Gaillard-Kelly M, Binart N, Baron R, Kelly PA (1999) Osteoblasts are a new target for prolactin: analysis of bone formation in prolactin receptor knockout mice. Endocrinology 140:96–105
Seriwatanachai D, Thongchote K, Charoenphandhu N, Pandaranandaka J, Tudpor K, Teerapornpuntakit J, Suthiphongchai T, Krishnamra N (2008) Prolactin directly enhances bone turnover by raising osteoblast-expressed receptor activator of nuclear factor kappaB ligand/osteoprotegerin ratio. Bone 42:535–546
Kinsella S, Moran S, Sullivan MO, Molloy MG, Eustace JA (2010) Hyponatremia independent of osteoporosis is associated with fracture occurrence. Clin J Am Soc Nephrol 5:275–280
Murthy K, Ondrey GJ, Malkani N, Raman G, Hodge MB, Marcantonio AJ, Verbalis JG (2019) The effects of hyponatremia on bone density and fractures: a systematic review and meta-analysis. Endocr Pract 25:366–378
Upala S, Sanguankeo A (2016) Association between hyponatremia, osteoporosis, and fracture: a systematic review and meta-analysis. J Clin Endocrinol Metab 101:1880–1886
Sejling AS, Pedersen-Bjergaard U, Eiken P (2012) Syndrome of inappropriate ADH secretion and severe osteoporosis. J Clin Endocrinol Metab 97:4306–4310
Sejling AS, Thorsteinsson AL, Pedersen-Bjergaard U, Eiken P (2014) Recovery from SIADH-associated osteoporosis: a case report. J Clin Endocrinol Metab 99:3527–3530
Draman MS, Stechman M, Scott-Coombes D, Dayan CM, Rees DA, Ludgate M, Zhang L (2017) The role of thyrotropin receptor activation in adipogenesis and modulation of fat phenotype. Front Endocrinol (Lausanne) 8:83
Endo T, Kobayashi T (2012) Expression of functional TSH receptor in white adipose tissues of hyt/hyt mice induces lipolysis in vivo. Am J Physiol Endocrinol Metab 302:E1569–E1575
Lu M, Lin RY (2008) TSH stimulates adipogenesis in mouse embryonic stem cells. J Endocrinol 196:159–169
Lundback V, Kulyte A, Dahlman I, Marcus C (2020) Adipose-specific inactivation of thyroid stimulating hormone receptors in mice modifies body weight, temperature and gene expression in adipocytes. Physiol Rep 8:e14538
Blevins JE, Schwartz MW, Baskin DG (2004) Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol 287:R87–R96
Blevins JE, Graham JL, Morton GJ, Bales KL, Schwartz MW, Baskin DG, Havel PJ (2015) Chronic oxytocin administration inhibits food intake, increases energy expenditure, and produces weight loss in fructose-fed obese rhesus monkeys. Am J Physiol Regul Integr Comp Physiol 308:R431–R438
Noble EE, Billington CJ, Kotz CM, Wang C (2014) Oxytocin in the ventromedial hypothalamic nucleus reduces feeding and acutely increases energy expenditure. Am J Physiol Regul Integr Comp Physiol 307:R737–R745
Yuan J, Zhang R, Wu R, Gu Y, Lu Y (2020) The effects of oxytocin to rectify metabolic dysfunction in obese mice are associated with increased thermogenesis. Mol Cell Endocrinol 514:110903
Wronski ML, Plessow F, Kerem L, Asanza E, O’Donoghue ML, Stanford FC, Bredella MA, Torriani M, Soukas AA, Kheterpal A, Eddy KT, Holmes TM, Deckersbach T, Vangel M, Holsen LM, Lawson EA (2022) A randomized, double-blind, placebo-controlled clinical trial of 8-week intranasal oxytocin administration in adults with obesity: rationale, study design, and methods. Contemp Clin Trials 122:106909
Bassett JH, Williams AJ, Murphy E, Boyde A, Howell PG, Swinhoe R, Archanco M, Flamant F, Samarut J, Costagliola S, Vassart G, Weiss RE, Refetoff S, Williams GR (2008) A lack of thyroid hormones rather than excess thyrotropin causes abnormal skeletal development in hypothyroidism. Mol Endocrinol 22:501–512
Gothe S, Wang Z, Ng L, Kindblom JM, Barros AC, Ohlsson C, Vennstrom B, Forrest D (1999) Mice devoid of all known thyroid hormone receptors are viable but exhibit disorders of the pituitary-thyroid axis, growth, and bone maturation. Genes Dev 13:1329–1341
Wang J, Zhou J, Bondy CA (1999) Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J 13:1985–1990
Bikle D, Majumdar S, Laib A, Powell-Braxton L, Rosen C, Beamer W, Nauman E, Leary C, Halloran B (2001) The skeletal structure of insulin-like growth factor I-deficient mice. J Bone Miner Res 16:2320–2329
Funding
Work at Icahn School of Medicine at Mount Sinai carried out at the Center for Translational Medicine and Pharmacology was supported by R01 AG071870 to M.Z., T.Y. and S-M.K.; R01 AG074092 and U01 AG073148 to T.Y. and M.Z.; and U19 AG060917 to M.Z and C-J.R. M.Z. also thanks the Harrington Discovery Institute for the Innovator-Scholar Award toward the development of anti-FSH antibody.
Author information
Authors and Affiliations
Contributions
S-M.K and M.Z wrote the main manuscript text and prepared Table
1. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
M.Z. is inventor on issued and pending patents on the use of FSH as a target for osteoporosis, obesity and Alzheimer’s disease. M.Z., T.Y. and S.R. are inventors on a pending patent on the formulation of a monoclonal antibody against FSH. The patents will be held by Icahn School of Medicine at Mount Sinai, and M.Z, T.Y. and S.R. would be recipients of royalties, per institutional policy, should the patents be granted. The other authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
